4.2. Intensity-Dependent Photomechanical Measurements
The sample is pumped with 488-nm wavelength light with a square wave intensity profile and the photomechanical stress response is measured as a function of time for three CTA volume fractions in the apparatus described in
Section 3.2. The DR1 concentration is fixed in all fibers at 0.5% by weight. The stress is measured as a function of the intensity amplitude of the square wave separately for vertical and horizontal polarization of the pump light to determine the photomechanical stress response tensor. The black points in
Figure 10 show typical smoothed data for a 60-s period square wave and the solid curve is a fit to a single exponential given by Equations (
33) and (
34) as described below.
We note that the stress in
Figure 10 changes sign, so the material transitions form a stretched state to a compressed state relative to its equilibrium dimensions. As a result, buckling may take place beyond the transition at high stress.
Figure 10 is representative of data for the highest pump intensities. However, most of the intensities reported here are at levels below the buckling point. Even those above the buckling point are observed to follow the trends, so buckling does not appear to be an issue for samples of this geometry. Higher pre-stress than those used is avoided to prevent sample damage or inducing anisotropies.
The data in
Figure 10 behave as a single saturated exponentials in both the pump on and off regions. We require that the response in the two regions join together continuously but the time constants are permitted to be different. Putting these criteria together leads to the model
and
where
and
are the amplitudes and time constants and
—the time durations of the beam being on and the beam being off. These types of fits are commonly used [
49].
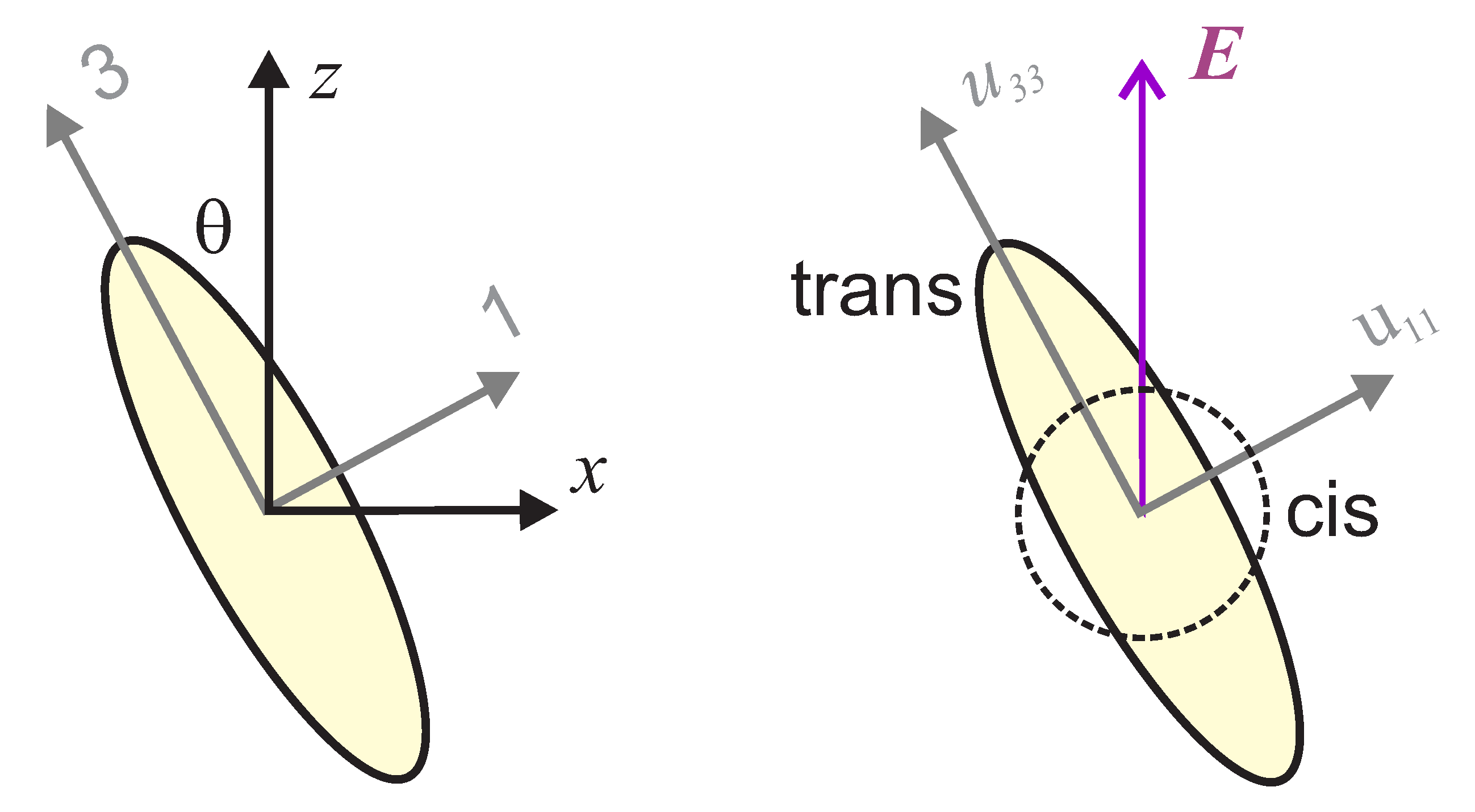
Depending on the material, light can induce an expansion or contraction. Since an increase in the length of a sample leads to a decrease in the stress for a pre-stressed sample, we define the photomechanical stress response to be of the form,
where
is the stress,
the photomechanical stress response, and
I the intensity absorbed by the sample. With this definition,
is positive when the length of the sample increases [
49,
50].
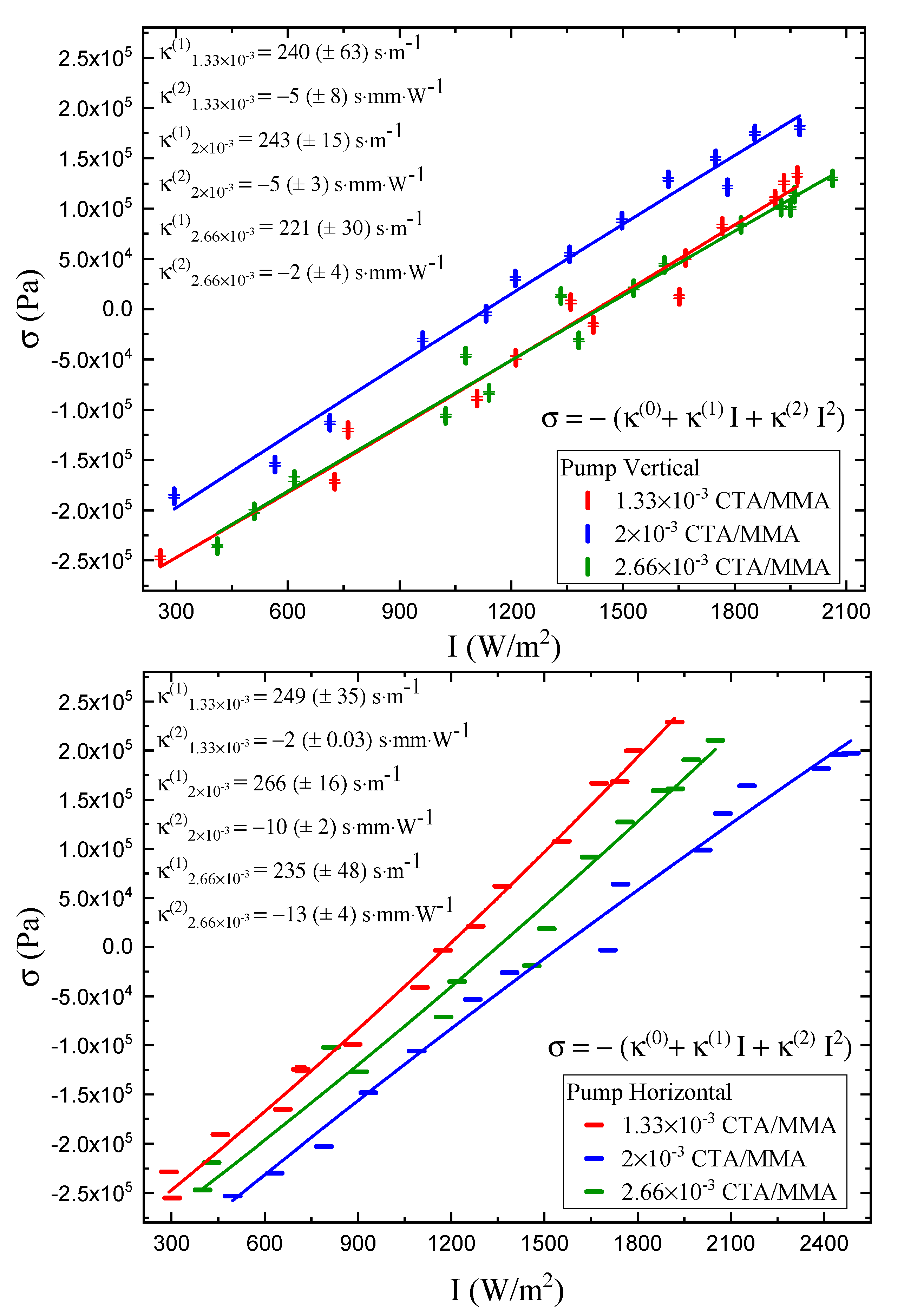
Figure 10 shows typical stress data as a function of time and fits to the theory given by Equations (
33) and (
34). Equation (
35) assumes that the stress function
can be expanded in a series and that the first two terms are sufficient to approximate its behavior.
Here we define the linear intensity-dependent photomechanical response,
, which quantifies the linear photomechanical response of a material that is bathed in light of intensity
. This quantifies the change in stress for a small added intensity
to the large background intensity
. It can be determined from a measurement of the stress
as a function of
I, yielding
We can view the normal linear response as being the limiting case of when . As such, provides another parameter that can be varied to test hypotheses of what makes a large photomechanical response.
The data for vertically and horizontally polarized pump beams are separately fit to Equations (
33) and (
34).
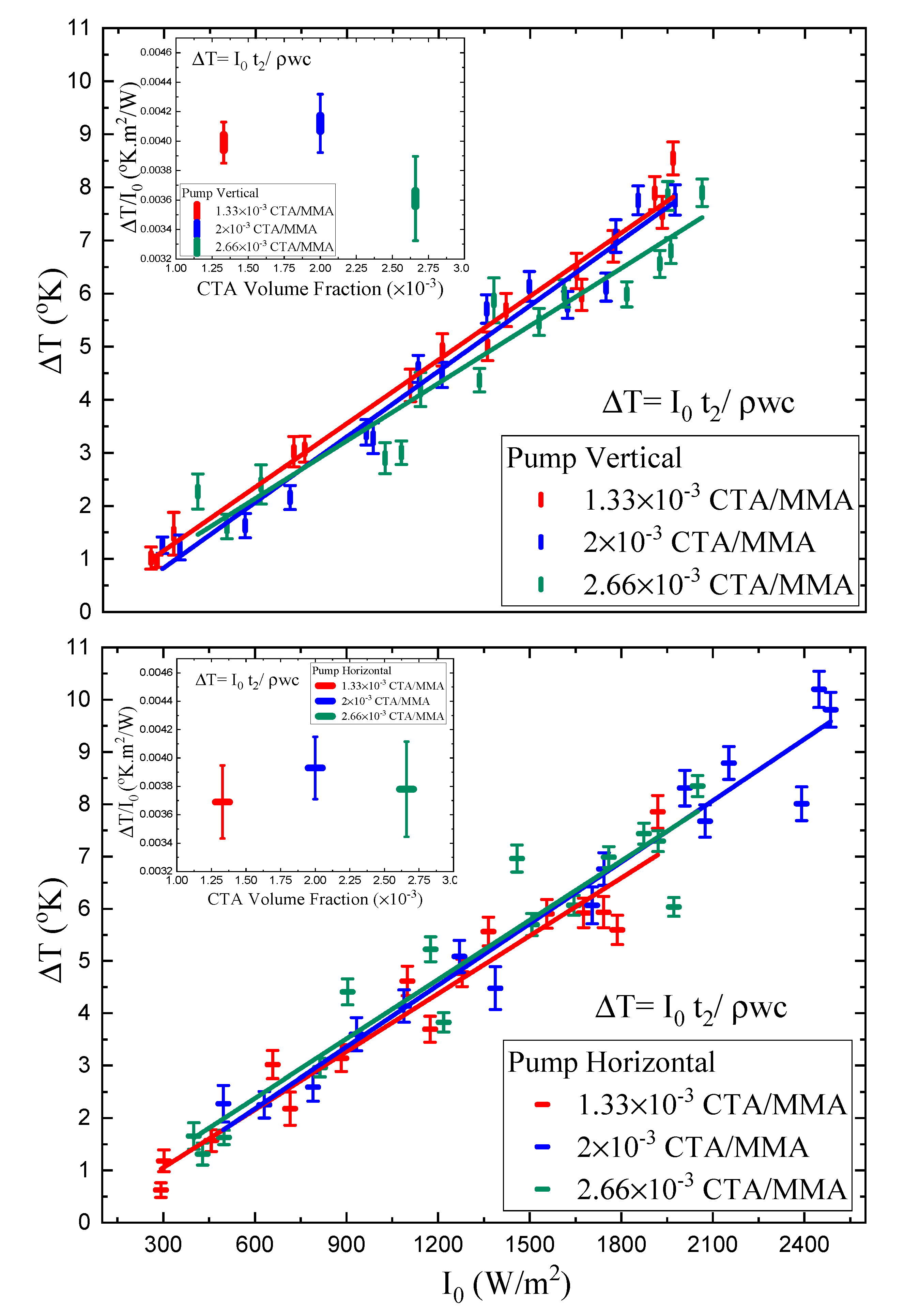
Figure 11 shows a plot of the parameter
, the steady state light-induced stress at infinite time, which is determined from such fits at each pump polarization. Also shown is a fit to the first two terms of the Taylor series expansion given by Equation (
35). This expansion is a good approximation to the intensity-dependent response with a small quadratic contribution.
The results of the linear photomechanical and nonlinear stress response
and
for vertical and horizontal polarization of light and as a function of CTA volume fraction are displayed in
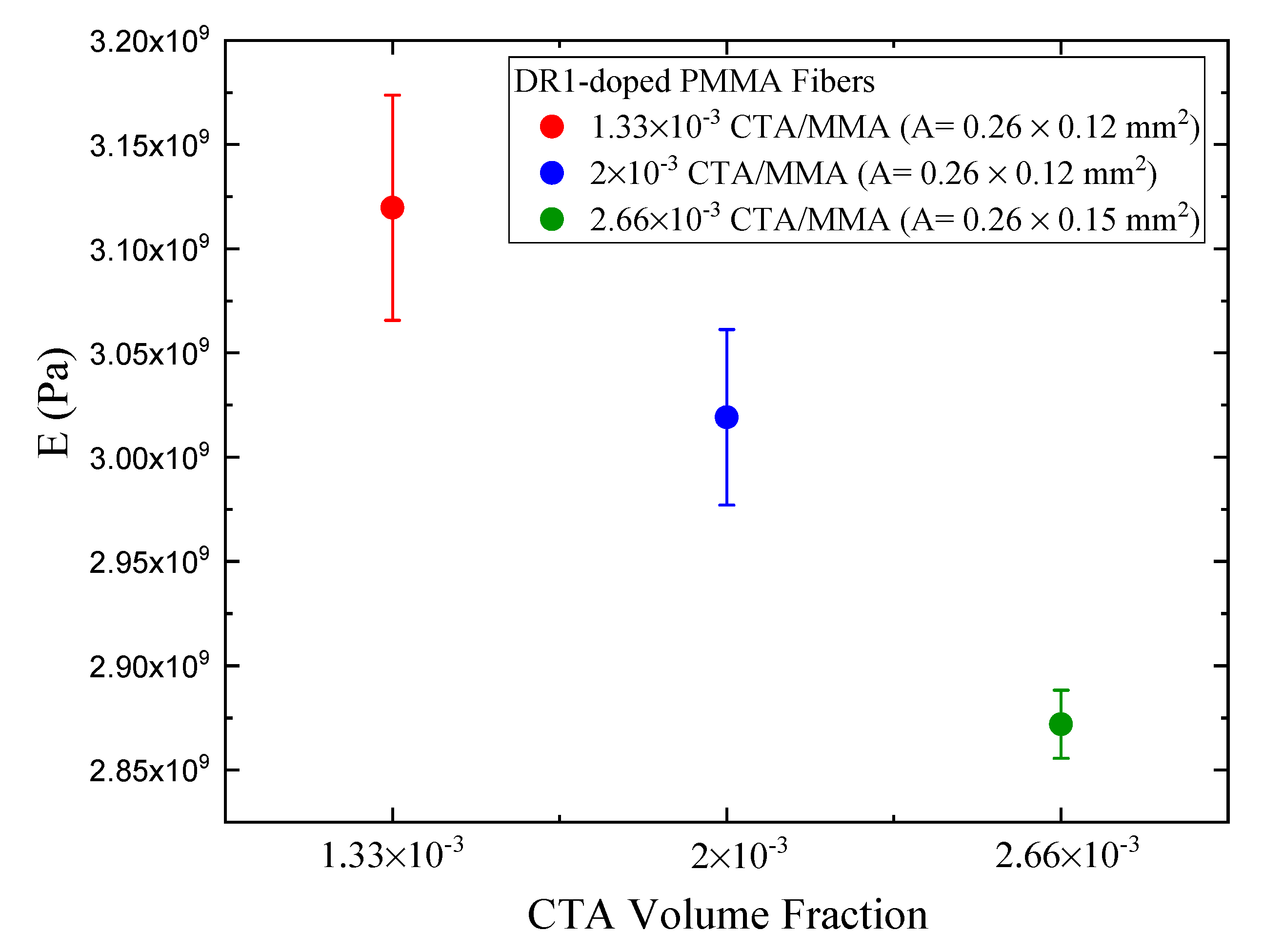
Table 3. The linear photomechanical response is the same within experimental uncertainties for all CTA volume fractions, so is independent of the polymer chain length. Furthermore, the linear response is independent of the polarization within experimental uncertainties, but the smaller values for the vertically polarized pump relative to the horizonal polarization are consistent with a small contribution from molecular reorientation.
The quadratic stress responses for the vertically polarized pump are all the same, within experimental uncertainty, as a function of CTA volume fraction, but the response for a perpendicular pump becomes more negative. This suggest a nonlinearity, due to a mechanism that is not isotropic, as is heating. This increase in the magnitude of the quadratic response correlates with the decrease in Young’s modulus and the decrease in polymer chain length. The chain length and Young’s modulus decreases result in greater chain mobility and, hence, greater mobility of the dopant molecules, allowing them to reorient. However, the negative sign is opposite to what we would expect; with increased molecular reorientation, a horizontal pump would induce an increased stress response along the uniaxial direction.
Figure 12 shows the photomechanical response as a function of intensity calculated from Equation (
36) at the measured intensities using the fit parameters
and
from
Table 3. The intensity-dependent response for pump light polarization along the axis containing the force sensor (which we will call vertical) changes by less than or equal to approximately 10%. The heating model given by Equation (
7) predicts that the change in stress induced by the photothermal heating mechanism is independent of the intensity. The data for vertically polarized pump light, thus, shows that heating is responsible for at least 90% of the observed photomechanical response.
Molecular reorientation is the process by which molecules reorient away from the pump light’s polarization axis, which should lead to an increase in the measured stress as the material’s equilibrium length decreases for a vertical pump polarization. Reorientation thus opposes heating for vertical polarization, which tends to decrease the stress as the sample elongates. As a result, the response is lower than it would be with heating alone. When the light is horizontally polarized, reorientation toward the vertical direction leads to a decrease in the stress as the material’s length increases, thus adding to photothermal heating, which also results in the sample elongating.
For all three polymers of varying polymer chain length,
Figure 12 shows the vertically polarized pump to have a smaller response than the horizonal one in the limit of small intensity, as we would expect for a small reorientational contribution to a response that is dominated by photothermal heating. The difference between the two diagonal tensor components at room temperature is on the other of
to
, suggesting that the reorientational contribution is
to
—a 2%-to-5% contribution.
The intensity dependence is governed by the term, which is more difficult to explain. It is due to both the temperature dependence of photothermal heating—which originates in the temperature dependence of material parameters such as the heat capacity, and molecular reorientation—which depends on the viscoelastic properties of the polymer and the temperature-dependence of the isomer populations. Accounting for these contributions are beyond the scope of our present work but will be the topic of future studies.
Figure 13 shows the turn-on time constants,
—when the pump is on—and
Figure 14 shows the turn-off time constant,
—when the pump is off—as a function of intensity, determined from a fit of the data to the functions in Equations (
33) and (
34). If heating is the only mechanism, then
and
should be the same as the Newton cooling time constant
. The insets show the time constants as a function of CTA volume fraction averaged over all the intensities. The time constant of the polymer made with higher CTA concentrations is marginally higher than those made with lower CTA concentrations.
The Beer–Lambert law [
50]
is used to determine the material absorbance
at a wavelength of
nm by measuring the transmitted intensity
I and the incident intensity
with a photo detector. The fiber thickness through which the light propagates,
w, is measured with a micrometer.
Table 4 shows the absorbance for both incident polarizations. Note that the absorption length is much longer than the fiber thickness, so nonlinearities from depth effects can be ignored [
72,
73], making the assumptions in our heating model appropriate.
The fibers are slightly pre-stressed when first mounted to prevent buckling when they expand in response to light or temperature change. The pre-stress may result in chain alignment and thus affect the photomechanical response. If the chains and molecules were to align along the fiber axes, the vertical absorbance would be greater than the horizontal one. We find horizontal absorbance to be higher, which is inconsistent with the hypothesis that molecular alignment is induced in the fiber drawing process and/or due to prestress.
Table 3 shows the photomechanical response,
and
, of the fiber with 2.66 × 10
CTA volume fraction for pre-stress values of
and
. The response is found to be the same within the large experimental uncertainties for both polarizations, though they are systematically higher for the higher pre-stress values.
The efficiency figure of merit (FOM), which is the material’s efficiency in converting light energy to mechanical work, is defined in Equation (
29) [
49,
50]. We determine the FOM as a function of CTA volume fraction.
Table 3 summarizes the results. Within experimental uncertainties, the FOM is found to be independent of the CTA concentration.
These results emphasize some of the advantages of glassy polymers as host materials. They have a large Young’s modulus, so they are more rigid than other materials, such as liquid crystal elastomers, and also have a larger photomechanical stress response [
49,
50]. Crystals, on the other hand have good photomechanical properties but have lower optical quality and cannot be formed into arbitrary shapes [
74,
75,
76,
77,
78]. These are important qualities for providing a rigid support for the precise positioning of massive objects. Additionally, glassy polymers can be formed into thin films and fibers of high optical quality, making them promising candidates for waveguide devices and smart photomechanically morphing materials [
17].
4.2.1. Photothermal Heating
This section shows how the parameters in the photothermal heating model are determined from the experiment and how the tensor properties of the photomechanical response are used to isolate the heating contribution. These two independent approaches will be used to separate the photothermal response and hole burning/molecular reorientation. The degree of consistency between them will provide an estimate of the reliability of the measurements and their interpretation.
Figure 15 and
Figure 16 show the semi-empirically determined temperature change as a function of intensity using Equation (
4) and the measured time constant of the photomechanical response. The theory has no adjustable parameters, and depends only on known material parameters, such as the specific heat capacity
c and the density
, as well as on the sample’s geometry, characterized by its width,
w. The values of
,
c, and
w that are used to find the temperature increase are listed in
Table 5. The insets in the graphs in
Figure 15 and
Figure 16 show the temperature change per unit of absorbed intensity as a function of CTA volume fraction.
Figure 17 summarizes all the results obtained in this paper by providing plots of the intensity-dependent linear photomechanical response
, defined by Equation (
36) as a function of intensity. Included are plots of the theory of the heating contribution that use the temperature change determined from Equation (
3) in the heating model given by Equation (
7), which draws from the parameters shown in
Table 2. These parameters are determined from fits to the temperature-dependent stress shown in
Figure 9 to the function given by Equation (
6). The response along the two principle axes of the fiber sample are denoted by “|” and “—” for the pump polarization along and perpendicular to the long axis (mounted vertically) for three polymer chain lengths as controlled by the amount of chain transfer agent used during polymerization. Additionally included are plots of the experimentally determined values for the same quantities, which are represented by smooth lines that are determined from fits to the data. The dashed curves are the heating and molecular reorientational contributions determined from the polarization-dependent measurements.
The solid curves, which show the fit functions to the measured values, and the dashed curves, which show the heating contribution determined from the polarization-dependent measurements, fall on top of each other. The polarization-dependent measurement shows that the degree of molecular reorientation is negligible. Aside from the polymer prepared with CTA volume per MMA volume, the pure heating model is consistent with the theoretical results. However, the trend of the heating model’s prediction and the data are opposite to each other, but on average, they agree. This is not surprising, given that the model has no adjustable parameters and relies only on the temperature-dependent stress measurement and time constant measurement, both of which may be biased by systematic errors.
We have found the temperature-dependent stress measurement of the polymer prepared with CTA volume per MMA volume to vary significantly between measurements, perhaps because of the high sensitivity of the material’s properties to temperature variations due to its lower glass transition temperature. This lack of reproducibility for the heating model in this polymer is consistent with the heating theory being off by a factor of two, from the experimentally determined values.
The figure of merit is typically defined by the linear photomechanical response and Young’s modulus
E to be given by
. The intensity-dependent figure of merit is a generalization of the figure of merit for an illuminated sample, providing another degree of freedom that can be controlled. It depends on the intensity-dependent photomechanical response
and the intensity-dependent Young’s modulus
, which are determined using Equation (
32), where
is the slope of the plots in
Figure 15 and
Figure 16 at intensity
, yielding
where
a and
b are the intercept and slope of the graphs in
Figure 15 and
Figure 16. So
Figure 18 shows a plot of the intensity-dependent figure of merit as a function of intensity as determined from fits to the data to get the appropriate parameters. The intensity-dependent figure of merit is observed to decrease as a function of intensity. While the decrease in Young’s modulus with increased intensity works to improve the figure of merit, the decrease in the intensity-dependent photomechanical response wins out, resulting in a decreased figure of merit. As such, the best figure of merit is for a dark sample.
Finally, we determine the temperature-dependent figure of merit due to the heating mechanism for the three polymers to determine if heating the material might improve it. Using the semi-empirical heating model of the photomechanical response given by Equation (
7) and a fit to the measured temperature-dependent Young’s modulus given by Equation (
32), the temperature dependence of the figure of merit is determined to be of the form
Figure 19 shows the temperature dependence of the figure of merit. For all chain lengths and light polarizations, it increases with increased temperature by a factor of about three when the temperature is raised by
. Thus, adjusting the operational temperature might be one avenue for improving the photo-mechanical response of a material.
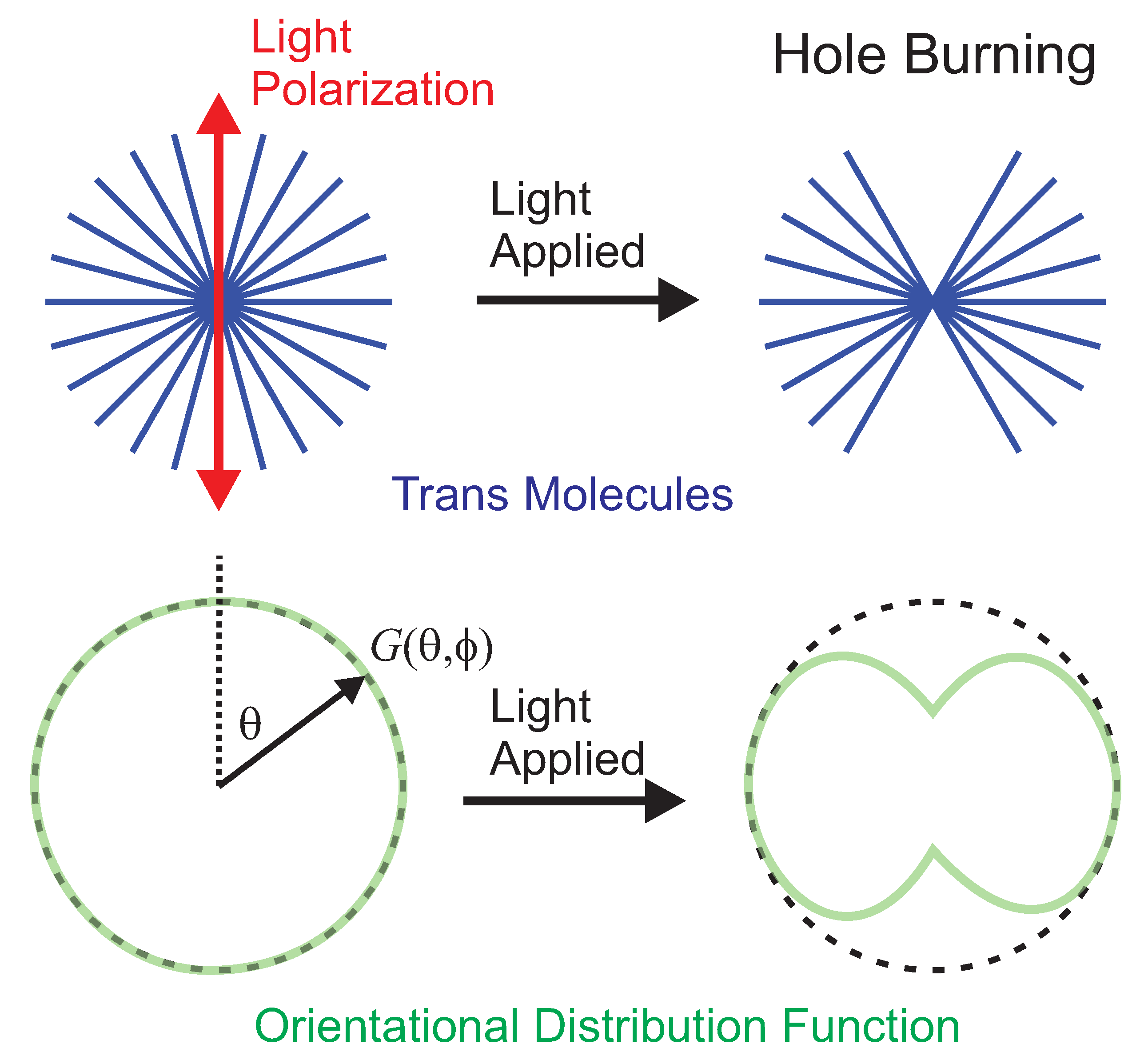
4.2.2. Separating Photothermal Heating and Angular Hole Burning Using the Tensor Response
, the ratio of the orientational response to the heating response for light polarized along the axis between the clamps, is given by Equation (
14), which is expressed in terms of the intensity-dependent photomechanical constants
and
. Equation (
36) relates the intensity-dependent photomechanical constants to the linear and quadratic photomechanical constants. These, in turn, are the fit parameters determined from a plot of the photomechanical stress response as a function of intensity, and determined for the polymers studied here from the data in
Figure 11. The zero-intensity limit,
, is the value we observe in most experiments since the sample is typically not illuminated before the pump light is turned on to actuate a photomechanical response in the material.
Table 6 summarizes the zero-intensity results for the DR1-doped PMMA fibers of varying CTA volume fraction. The fractional contribution to molecular reorientation
is small and consistent with zero within experimental uncertainty, showing that heating is the dominant mechanism.
is positive, indicating that thermal expansion elongates the fiber sample along its long axis. The magnitudes of
and
are also within experimental uncertainty of vanishing, aside from the medium-chain-length sample.
The negative sign of in all measurements, particularly in the medium-chain-length polymer, is marginally statistically significant. The results are consistent with molecular reorientation, which decreases the length of the samples along the polarization axis of the light, and the positive sign of shows an increase in the samples’ length along its long axis when excited by light perpendicular to it. The fact that all of the measured reorientational contributions are consistently of the correct sign suggests that the uncertainties determined from fits to the data might be overestimated.
We find that the effect of heating and molecular reorientation are independent of polymer chain length, since all the photomechanical constants are the same within their experimental uncertainties.
Figure 20 shows a plot of the relative reorientational,
, as a function of background illumination
. Such experiments are not common, but, as with temperature, they provide the experimenter with an additional degree of freedom that can be varied to provide yet another perspective of the photomechanical response. The ratio
is observed to vary significantly over the intensity range measured. The medium-chain-length sample, for example, starts with a ratio of −0.06 and crosses zero at the higher intensity range. The short chain polymer (highest CTA concentration) shows the most dramatic change, with the sign changing at low intensity and the ratio increasing by a factor of almost 4.
This data shows the most dramatic effect as a function of chain length; the average slope becomes more positive as the chain length decreases. Decreased chain length results in greater mobility, which should lead to a larger reorientational contribution, hence a more negative value. A possible explanation of the observation is as follows. For long chain lengths, high background irradiation softens the material, making the molecules more mobile and hence increasing the molecular reorientational contribution. For short chain lengths, the molecules are more mobile when softened by the background light. The light also induces molecular reorientation through light-induced torque on the induced dipole moment. Such effects are the topic of future research.

























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}