1. Introduction
Bentonite is a naturally occurring clay mineral containing mainly montmorillonite (MMT). By forming viscous water suspension, it acts as a bonding, plasticizing, and suspending agent. Its application is diverse and includes the foundry industry, metallurgy, and civil engineering. Bentonite acts as a flocculant in wastewater treatment and as a purifying agent in the food industry. It is also used as a component of insulating materials for large-scale tanks, lakes, and artificial water reservoirs—or even in pyrotechnics to make rocket nozzles and end plugs [
1,
2].
The above-mentioned examples of bentonite applications in various industries constitute only a small part of its wide application field. Nevertheless, the dynamically developing field of science of materials engineering constantly expands its application possibilities. This is related to the growing demand for materials with precisely defined properties, which translates into more and more advanced technologies for the synthesis of new compounds or the modification of existing raw materials [
3,
4].
Due to its specific crystal structure and highly chemically reactive surface, bentonite is one of the most interesting minerals enabling a controlled change of its properties. Computer simulations are currently one of the basic methods supporting the process of designing, manufacturing, and analyzing new materials. They enable both the verification of a theory and the prediction of an outcome in real studies, which is extremely helpful in determining their direction and results in significant time savings [
5,
6]. Modeling methods, such as molecular dynamics (MD) or Monte Carlo (MC) methods, are widely used in studying the dynamic behavior of systems consisting of a large number of atoms or molecules [
7,
8]. The direct information about the spatial distribution of interacting atoms and the paths of their movement in systems affected by given external factors can be reliably provided [
9]. Therefore, the simulations are an extremely useful tool complementing experimental research, and, in special cases, allowing for the description of phenomena or material structures that are impossible to assess using conventional analytical tests.
There is a substantial body of literature describing the use of computer simulation techniques to analyze complex organically modified clay materials [
10,
11,
12,
13]. The interest stems from the difficulty involved in evaluating the conformation of polymers in the clay interlayer space, as well as their interaction being hindered by the occlusion of organic molecules in the mineral structure [
14].
The article complements the recently published results of structural studies of bentonite intercalated with acrylic polymers. The research team carried out an organic modification of montmorillonite in calcium bentonite (SN) with poly (acrylic acid) (PAA) and its sodium salt (PAA/Na). The considered organobentonite is intended to act as a material binding the grains of the mineral matrix in the molding sand, from which a mold with an appropriate cavity shape is prepared. While pouring the mold with liquid metal, the polymer undergoes thermal destruction, forming a specific carbon structure without negative environmental impact. This form, known as lustrous carbon, ensures good quality of the casting surface. Knowledge of the nature of the arising interactions based on the results of computer simulations is already useful at the material verification stage, and further, in terms of recognizing the course of component bonding and their thermal degradation. The simulations were carried out on a simplified model system that maintains the physicochemical properties of the components of actual organobentonites. The obtained inorganic–organic composites were considered as prospective, environmentally friendly binders in foundry technology, which is a response to the harmfulness of the carbon and organic additives, which are introduced to molding sands in order to ensure good quality of the casting surface [
15]. The collective analysis of the research results (FTIR, XRD, BET measurements) used to consider the mechanism of interaction in the mineral/polymer system indicates surface adsorption combined with intercalation of the PAA monolayer into the mineral interlayer spaces. Based on the X-ray diffraction (XRD) analysis, it was found that the influence of PAA/Na on the layered structure of aluminosilicate is destructive, which may adversely affect the binding properties of its composites. This phenomenon was not found in the case of composites containing PAA; hence, it was concluded that it could act as a binding agent in the technology of synthetic molding sand despite the polymer adsorption on mineral surfaces. The risk of losing the bentonite binding capacity is reduced due to the good bonding properties of the pol (acrylic acid) itself.
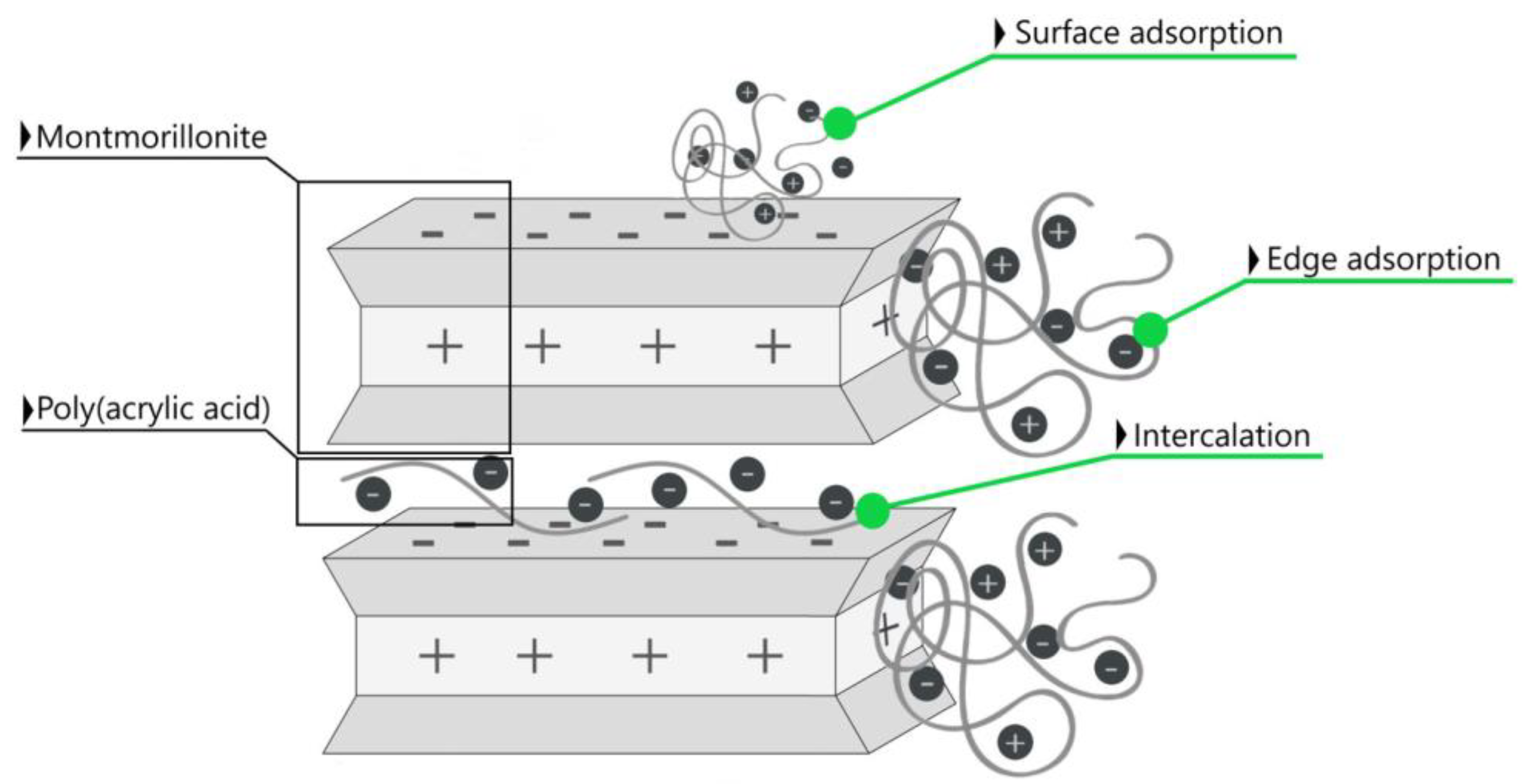
Considering the anionic nature of poly (acrylic acid) chains in the reaction medium, their electrostatic interaction with the positively charged edges of the mineral sheets seems to be the most likely. The adsorption mechanism may also result from the ion exchange between the OH− groups on the surface of mineral particles and the anionic part of the polymer (
Figure 1).
Computer simulations were carried out using classical molecular dynamics (MD) on a simplified montmorillonite/mer system in the form of acrylic acid (AA) or acrylate ion (NaA) to confirm the promoting effect of the carboxyl group on the polymer adsorption to the mineral surface. The simplification of the system was necessary, but it did not adversely affect the consideration of interactions between characteristic groups in either montmorillonite or poly (acrylic acid); therefore, the simulation results can be related to the results obtained using the analytical techniques.
2. Methods of Simulation
To investigate the interaction between the montmorillonite layer and acrylic acid or acrylate anions, molecular dynamics simulations were performed. The composition of the montmorillonite layer was given by the formula Na
0.75[Si
7.75Al
0.25][Al
3.5Mg
0.5]O
20(OH)
4. This is a simplified composition of Na-montmorillonite (Wyoming) [
16]. There is little experimental work that accurately describes the structure of montmorillonite. The initial model of the mineral layer was built on the base structural data reported by D. Gournis et al. [
17], which is similar to Na-montmorillonite.
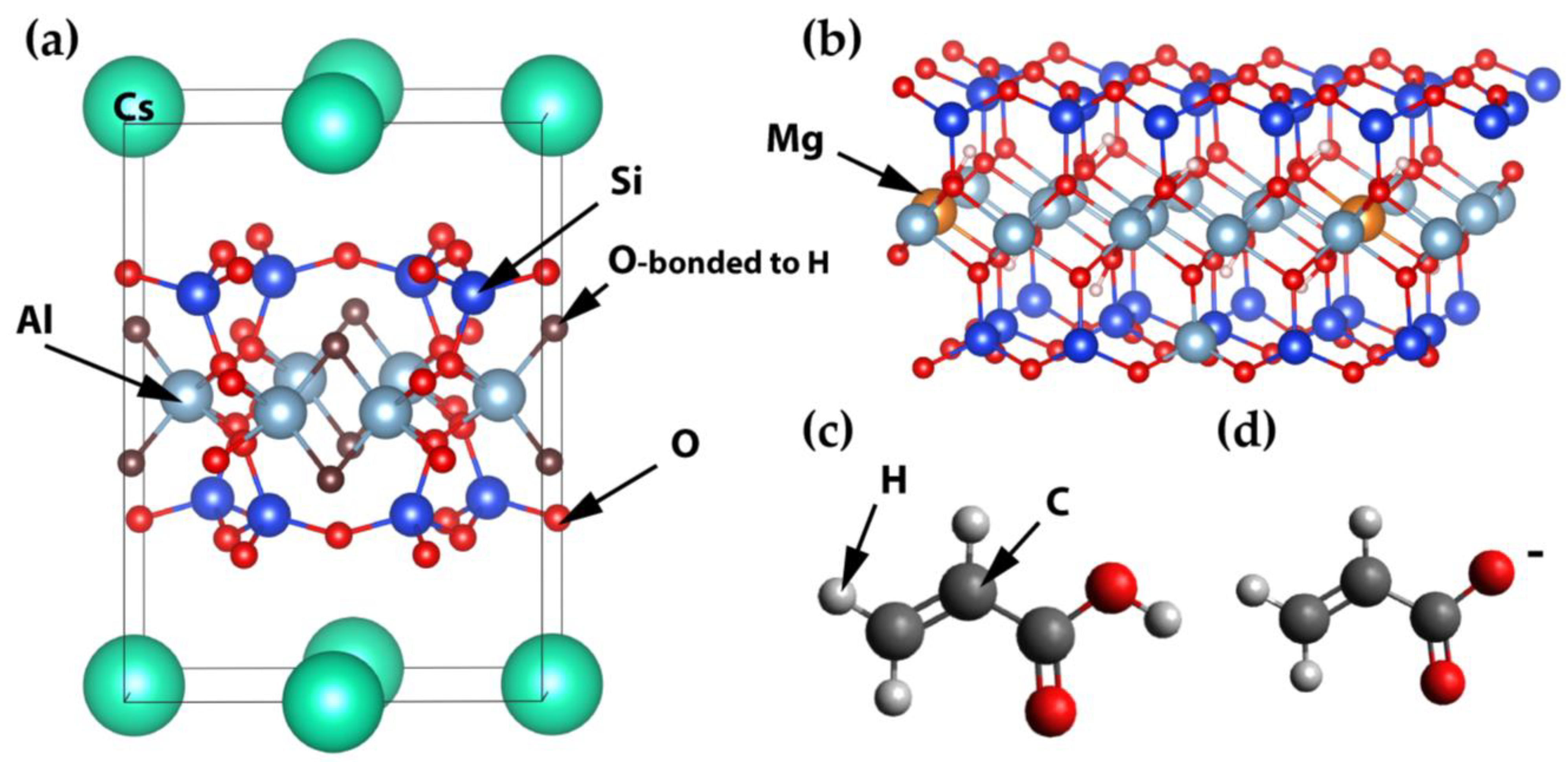
Figure 2a shows the unit cell of montmorillonite from [
17] of composition Cs
0.62[Al
3.01Fe(III)
0.41Fe(II)
0.04Mg
0.54][Si
7.8Al
0.2]O
20(OH)
4. The octahedral positions of Al can be substituted by Mg and Fe, and tetrahedral Si positions can be substituted by Al. Cesium positions in this unit cell are only partially filled by Cs (occupancy equal to 0.08). This material has a C/2m space group, and their unit call parameters are a = 5.181, b = 8.945, c = 12.34 Å, α = γ = 90, β = 99.62°.
The supercell of the montmorillonite layer was created by the multiplication of the unit cells in x and y directions by 16 × 8. The Cs has been replaced with the appropriate amount of Na. Part of the Al positions was substituted with Mg and part of the Si was substituted with Al to obtain Na
0.75[Si
7.75Al
0.25][Al
3.5Mg
0.5]O
20(OH)
4 stoichiometry. The initial layer dimensions were 82.90 × 71.56 Å. Water molecules were placed around the montmorillonite layer so that the initial model had 12,896 atoms. The initial simulation box height was about 30 Å. The CVFF-interface force field [
19] was used to describe the interaction between atoms in the system. This force field is successfully used for modeling clay minerals-organics interfaces [
19,
20,
21,
22]. Initial models of acrylic acid, acrylate anions, and water molecules were built based on structural data from [
18,
23]. Classical molecular dynamics simulations were performed using LAMMPS software [
24]. The periodic boundary conditions were used to remove the surface effect of the simulation box. The simulations were performed in terms of constant pressure equal to 1 bar and temperature equal to 300 K. At first, systems were relaxed during 70 ps with the time step equal to 0.005 fs. Such a long time was necessary not only to relax the montmorillonite layer but also to homogenize and relax the water around the montmorillonite. Then, the acrylic acid or sodium acrylate molecules were added to the system randomly at a distance greater than 10 Å from the montmorillonite layer. One molecule was added every 2.5 ps. In total, 40 molecules in each simulation were added into the system. For the next 63.00 ps, data of the molecules’ localization was collected. The systems were visualized using Ovito software [
25]. Carbon quantity contour maps (located in acrylic acid or acrylate anions) on the xy-plane parallel to the montmorillonite layer were created using python library matplotlib [
26]. The size of the bins in which the carbon atoms were counted to create contour maps was 4 × 4 Å.
3. Results and Discussion
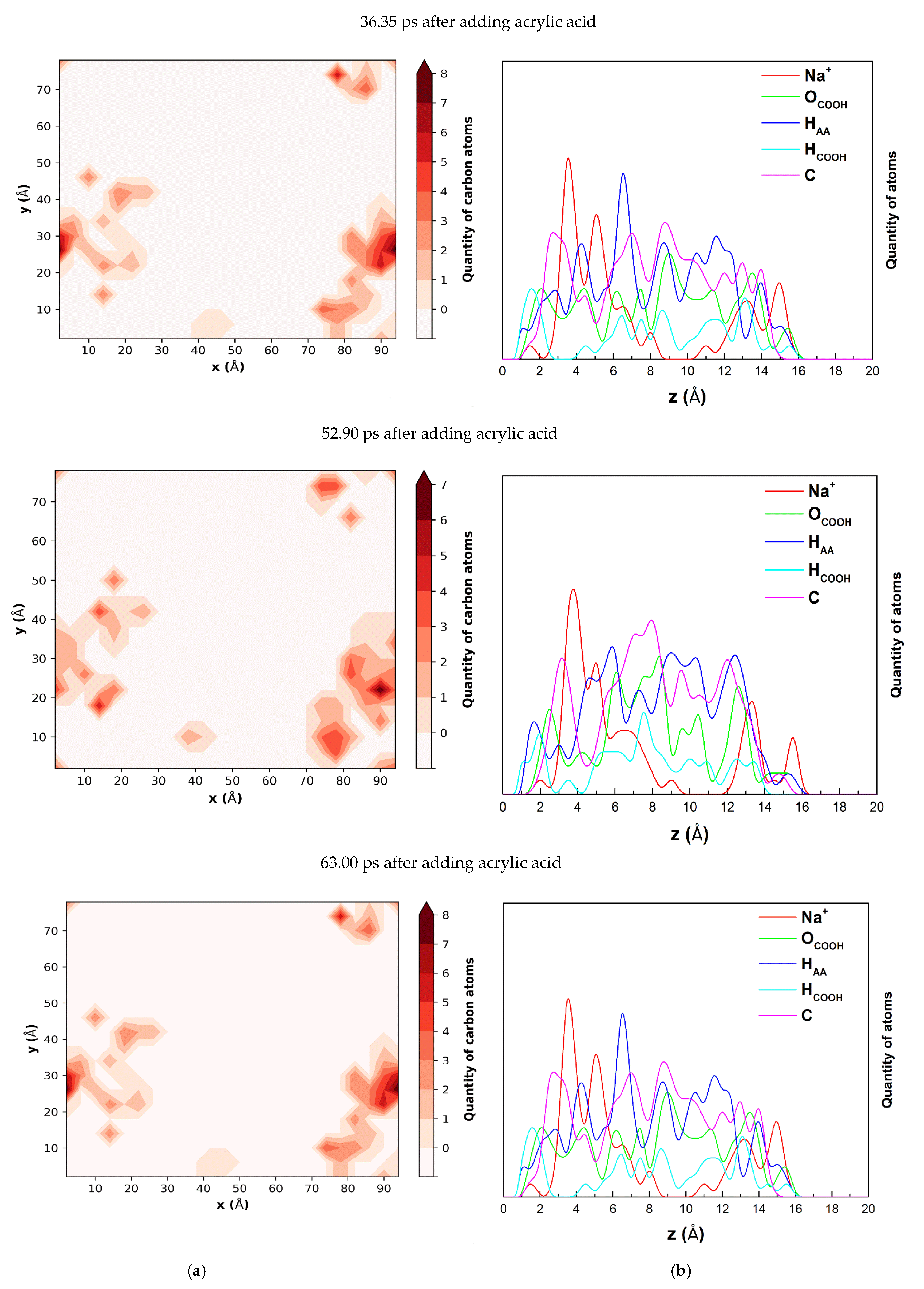
To show the position and arrangement of acrylic acid and sodium acrylate in relation to the montmorillonite layer, the carbon quantity contour maps and graphs of the number of atoms belonging to acrylic acid, sodium acrylate, and Na
+ as a function of distance from the montmorillonite layer surface were made. The carbon quantity contour maps and quantity of atoms belonging to acrylic acid as a function of distance from the montmorillonite layer in 36.35, 52.90, and 63.00 ps after adding acrylic acid are shown in
Figure 3. The contour map for 36.35 ps clearly shows the grouping of carbon atoms that make up the acrylic acid molecules (
Figure 3a). Similar groupings are visible for 52.90 and 63.00 ps. The atom quantity graphs as a function of distance from the surface of the montmorillonite layer (
Figure 3b) show that the atoms that make up acrylic acid and Na
+ are grouped at distances up to 16.0(3) Å. Nearest to the surface of the layer are hydrogens atoms from the COOH group (H
COOH) at 1.2(3) Å and hydrogens bonded to carbon (H
AA) a bit further at 1.5(3). These atoms are much closer than the positively charged Na
+ compensating for the negative charge of the layer, which is the most frequent at about 3.5 Å. The oxygen molecules from the COOH group (O
COOH) are located a little further from the surface at about 2 Å. This shows that the molecules closest to the surface are directed to the surface with H
COOH and H
AA. Both contour maps and graphs of the number of atoms in the dependence of distance from the montmorillonite surface show little changes over time in the position of the atoms groupings and the arrangement of the atoms within the group. These changes are caused by the random thermal movement of the water molecules.
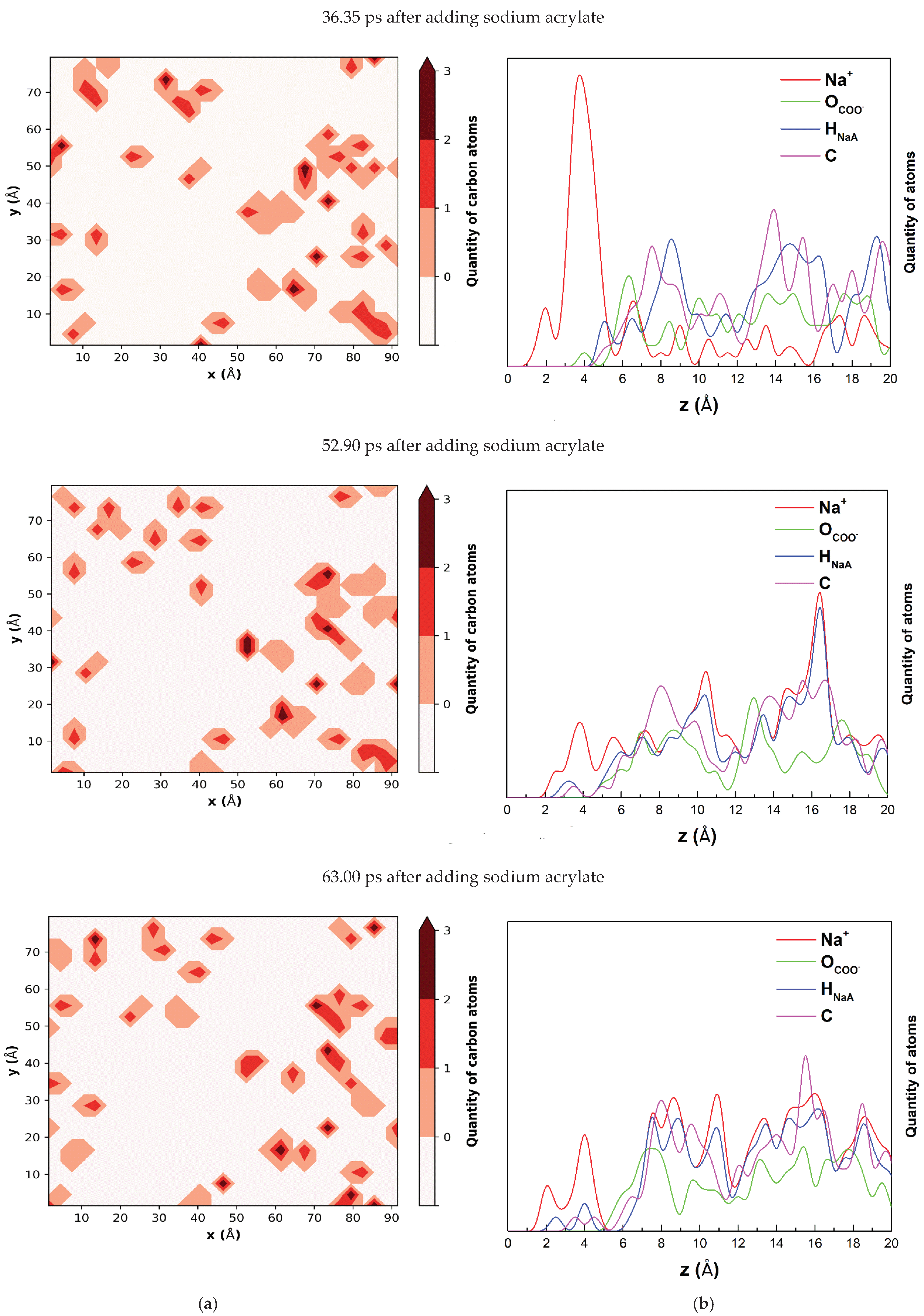
The simulations showed that acrylate anions do not tend to group. Carbon quantity contour maps (
Figure 4a) show the scattered groupings of carbon atoms that correspond to single acrylate anions. There is a maximum of 3 carbon atoms. Na
+ cations are nearest to the montmorillonite surface (
Figure 4b). Only at above 4 Å from the surface do the atoms building acrylate anions begin to appear.
Figure 3b shows neither sodium nor the atoms building acrylate ions grouped close to the surface. Both Na
+ and acrylate anions are present far from the surface, above 20 Å in the bulk of the water, as well as a short distance from the surface. The presence of sodium cations far from the surface is due to the presentence of CH
2=CHCOO
− balancing the charge of Na
+.
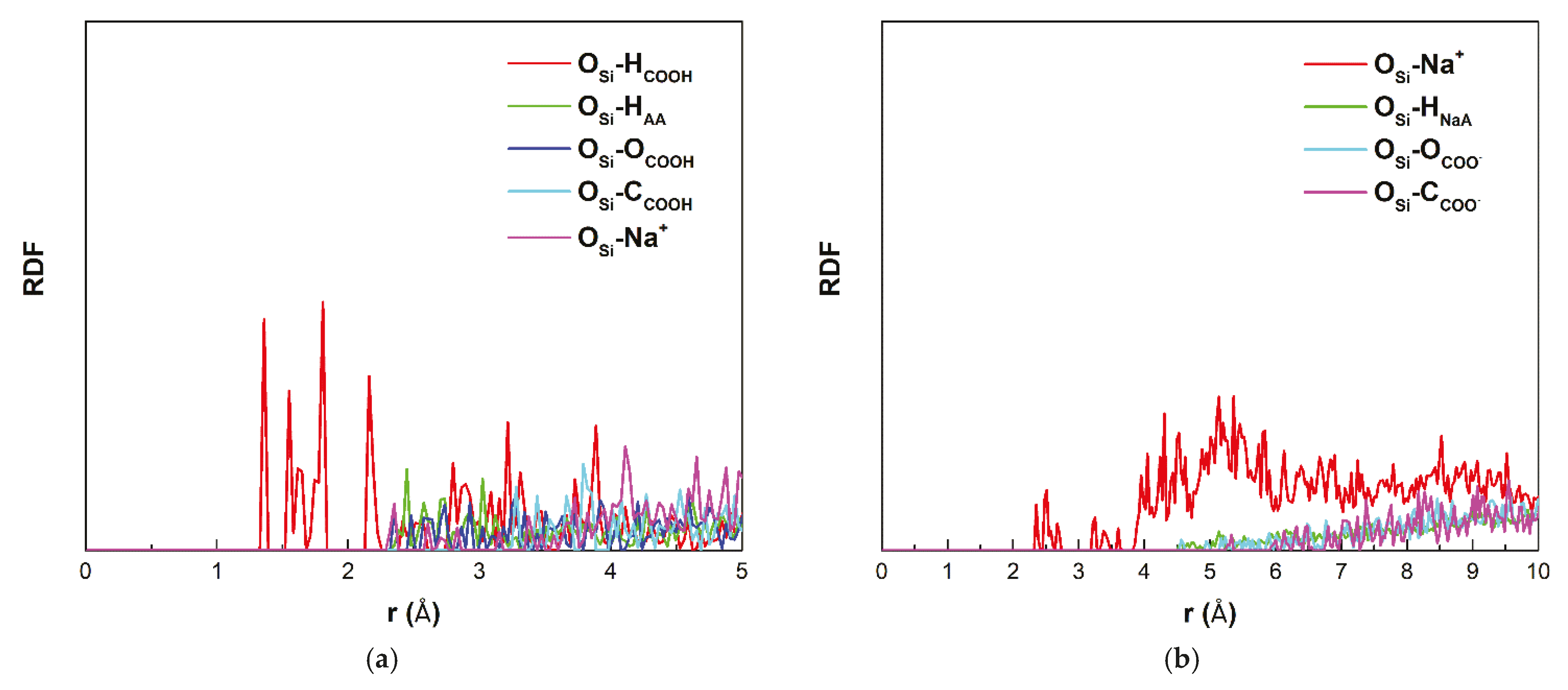
To describe the surroundings of oxygen atoms bonded to Si (O
Si) on the surface of the montmorillonite layer, radial distribution functions (RDF) of O
Si–I atom pairs were prepared.
Figure 5 shows the RDF functions of O
Si–H
COOH, O
Si–H
AA, O
Si–O
COOH, O
Si–C
COOH, and O
Si–Na
+ atoms averaged over time, ranging from 62 to 63 ps, after adding acrylic acid (
Figure 5a) and O
Si–Na
+, O
Si–H
NaA, O
Si–O
COO–, and O
Si–C
COO– after adding sodium acrylate (
Figure 5b). The sharp peaks of the RDF function for O
Si-H
COOH occur for distances from 1.40 Å to 2.16 Å (mean 1.75 Å). This distance range corresponds to the distances between the O
Si and neighboring H
COOH atoms. The RDF functions for other atoms belonging to acrylic acid molecule and Na
+ do not have such distinct peaks, and the RDF function for these molecules is greater than zero for distances above 2.34 Å. The lack of distinct peaks is due to the random arrangement of atoms around O
Si above 2.34 Å. In the case of acrylate anions, the closest to O
Si is Na
+ (
Figure 5b). The first peak of the RDF function for the O
si–Na
+ atoms pair is for a distance of 2.35 Å. The atoms that make up the acrylic anions are located above 4.5 Å. This shows that there is no direct interaction between acrylic anion molecules and O
si. The difference between the distances of the H
COOH–surface (1.2(3) Å) and O
Si–H
COOH (mean 1.75 Å) exists because H
COOH is not located directly above O
Si but is slightly shifted toward the center of the [Si
6O
18]
−12 ring.
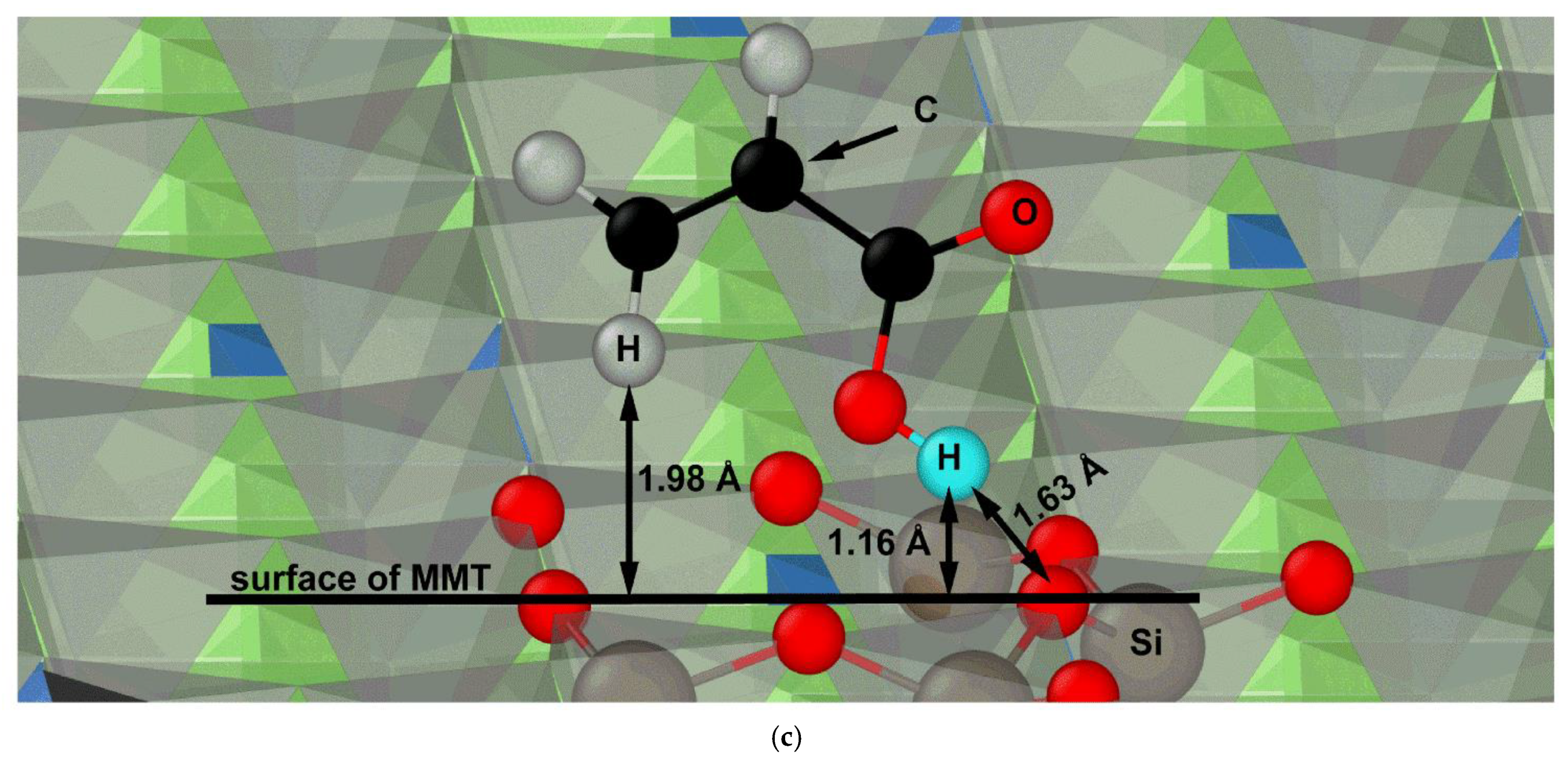
Figure 5c shows exemplary acrylic acid molecules in the neighborhood of O
Si.
The simulations suggest that the –COOH group plays the most important role in the deposition of organic acid molecules on the montmorillonite surface. A greater attraction of the polymer to the numerous carboxyl groups in the molecule stabilizing the system and making it resistant to thermal movement of water can be expected.
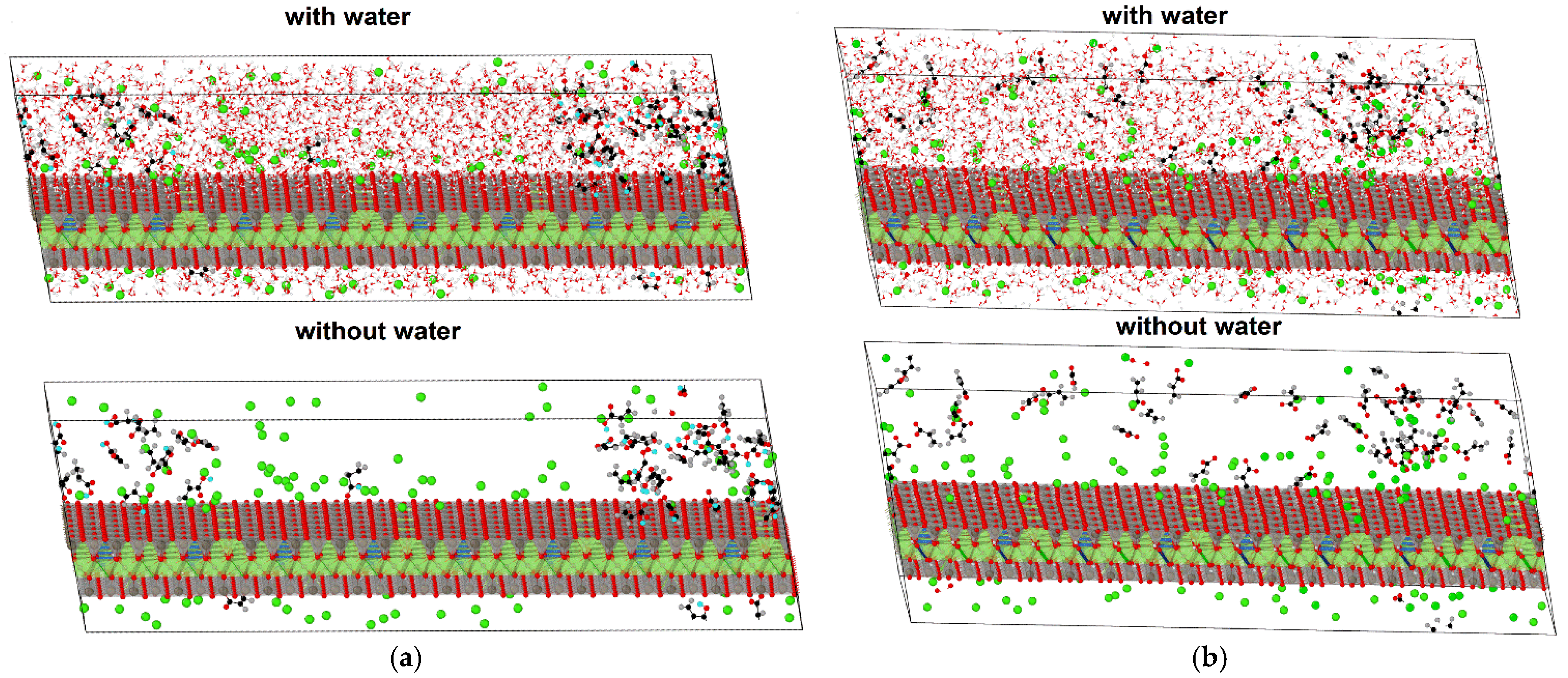
Figure 6 presents the montmorillonite/organic compound systems in which acrylic acid molecules and dispersed acrylate ions are visible after a simulation time of 63.00 ps.
The accumulation of acid molecules in larger groups at the mineral surface is evident (
Figure 6a), while acrylate ions are randomly distributed in the simulated space and are reluctant to interact with the surface of the mineral (
Figure 6b). This may be related to the electrostatic repulsion between the ions and the negatively charged clay surface.
4. Conclusions
Molecular dynamics simulation is a useful tool in assessing the mutual behavior of organic and inorganic compounds within clay-polymer systems. It provides insight into both their interlayer structure and the possible reactivity of their components. The performed investigations aimed at evaluating the influence of functional groups of organic molecules next to the non-rigid clay sheets using large-scale MD methods showed that the –COOH group plays the most important role in the adsorption mechanism of acrylic compounds. This study’s outcome matches well with the experimental results presented in the article [
11], which indicates its relevance in the production of a new group of complex materials. While verifying the composite components, it is important not only to have physicochemical knowledge about the materials but also to predict their interactions in a given environment. Computer simulations of complex systems—even those simplified but reflecting the physicochemical essence of the real state—make it possible to discern the intermolecular bonding interactions. This is of key importance in research on composites containing layered minerals. In the present case, the main goal was to learn whether it is possible to obtain the polymer-intercalated form of montmorillonite, or a mineral coated with polymer particles via their surface adsorption. The experimental results in this matter turned out to be fully consistent with the carried-out simulation.
The simulation results clearly show that the appropriate assumption of the model system, even with its significant simplification, provides important information about the phenomena and interactions that may occur between the components during the preparation of organobentonites. The results of the simulation of the model system may be a determining element in making a decision about the legitimacy of its production, and further, the advisability of conducting research in the context of the assumed application. The obtained knowledge is useful in terms of recognizing the course of binding of components in the molding sands and their thermal degradation during the casting process. Nevertheless, in the next stage of works, the validity of the simulation on systems directly representing the organobentonites analyzed in the mentioned article will be reconsidered.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






