
Photochemistry of 1-Phenyl-4-Allyl-Tetrazol-5-One: A Theoretical Study Contribution towards Mechanism Elucidation




Abstract
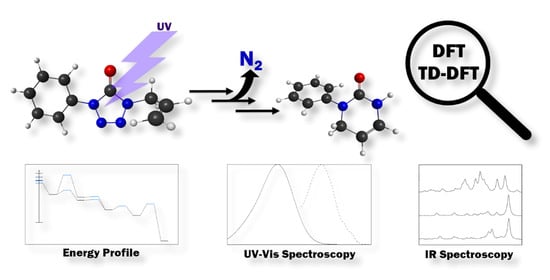
1. Introduction
2. Methods
3. Results and Discussion
3.1. ATZ Singlet Electronic States
3.2. Triplet Electronic States
3.3. Pyrimidinone
3.4. Future Experimental Validation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Myznikov, L.V.; Hrabalek, A.; Koldobskii, G.I. Drugs in the tetrazole series. Review. Chem. Heterocycl. Compd. 2007, 43, 1–9. [Google Scholar] [CrossRef]
- Zhao, H.; Qu, Z.R.; Ye, H.Y.; Xiong, R.G. In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties. Chem. Soc. Rev. 2008, 37, 84–100. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Kurpiewska, K.; Kalinowska-Tłuscik, J.; Herdtweck, E.; Dömling, A. α-Amino Acid-Isosteric α-Amino Tetrazoles. Chem. Eur. J. 2016, 22, 3009–3018. [Google Scholar] [CrossRef] [PubMed]
- Rostom, S.A.F.; Ashour, H.M.A.; El Razik, H.A.A.; Abd El Fattah, H.; El-Din, N.N. Azole antimicrobial pharmacophore-based tetrazoles: Synthesis and biological evaluation as potential antimicrobial and anticonvulsant agents. Bioorganic Med. Chem. 2009, 17, 2410–2422. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, V.H.; Mohite, P.B. Synthesis, characterization and evaluation of anticancer activity of some tetrazole derivatives. J. Optoelectron. Biomed. Mater. 2010, 2, 249–259. [Google Scholar]
- Khanage, S.G.; Raju, A.; Mohite, P.B.; Pandhare, R.B. Analgesic activity of some 1,2,4-triazole heterocycles clubbed with pyrazole, tetrazole, isoxazole and pyrimidine. Adv. Pharm. Bull. 2013, 3, 13–18. [Google Scholar]
- Trindade, N.R.; Lopes, P.R.; Naves, L.M.; Fajemiroye, J.O.; Alves, P.H.; Amaral, N.O.; Lião, L.M.; Rebelo, A.C.S.; Castro, C.H.; Braga, V.A.; et al. The Newly Synthesized Pyrazole Derivative 5-(1-(3 Fluorophenyl)-1H-Pyrazol-4-yl)-2H-Tetrazole Reduces Blood Pressure of Spontaneously Hypertensive Rats via NO-cGMO Pathway. Front. Physiol. 2018, 9, 1073. [Google Scholar] [CrossRef]
- Ghule, V.D.; Radhakrishnan, S.; Jadhav, P.M. Computational studies on tetrazole derivatives as potential high energy materials. Struct. Chem. 2011, 22, 775–782. [Google Scholar] [CrossRef]
- Shi, Y. Computational design of tetrazolone-based high-energy density energetic materials: Property prediction and decomposition mechanism. J. Phys. Org. Chem. 2018, 31, e3733. [Google Scholar] [CrossRef]
- Fischer, D.; Klapötke, T.M.; Stierstorfer, J. 1, 5-Di (nitramino) Tetrazole: High Sensitivity and Superior Explosive Performance. Angew. Chem. Int. Ed. 2015, 54, 10299–10302. [Google Scholar] [CrossRef]
- Klapötke, T.M.; Karaghiosoff, K.; Mayer, P.; Penger, A.; Welch, J.M. Synthesis and Characterization of 1,4-Dimethyl-5-Aminotetrazolium 5-Nitrotetrazolate. Propellants Explos. Pyrotech. Int. J. Deal. Sci. Technol. Asp. Energ. Mater. 2006, 31, 188–195. [Google Scholar] [CrossRef]
- Ismael, A.; Serpa, C.; Cristiano, M.L.S. Photochemistry of 1-allyl-4-aryltetrazolones in solution; Structural effects on photoproduct selectivity. Photochem. Photobiol. Sci. 2013, 12, 272–283. [Google Scholar] [CrossRef]
- Hyatt, J.A.; Swenton, J.S. A facile synthesis of 9H-pyrimido [4,5-b] indole via photolysis of 4-azido-5-phenylpyrimidine. J. Heterocycl. Chem. 1972, 9, 409–410. [Google Scholar] [CrossRef]
- Frija, L.M.T.; Ismael, A.; Cristiano, M.L.S. Photochemical transformations of tetrazole derivatives: Applications in organic synthesis. Molecules 2010, 15, 3757–3774. [Google Scholar] [CrossRef]
- Quast, H.; Fuss, A.; Nüdling, W. Photoextrusion of Molecular Nitrogen from Annulated 5-Alkylidene-4, 5-dihydro-1H-tetrazoles: Annulated Iminoaziridines and the First Triplet Diazatrimethylenemethane. Eur. J. Org. Chem. 1998, 28, 317–327. [Google Scholar] [CrossRef]
- Quast, H.; Nahr, U. Photochemische Stickstoff-Eliminierung aus 1, 4-Dihydro-1-phenyl-5H-tetrazol-5-onen und-thionen. Benzimidazolone und Carbodiimide. Chem. Berichte 1985, 118, 526–540. [Google Scholar] [CrossRef]
- Frija, L.M.T.; Khmelinskii, I.V.; Serpa, C.; Reva, I.D.; Fausto, R.; Cristiano, M.L.S. Photochemistry of 5-allyloxy-tetrazoles: Steady-state and laser flash photolysis study. Org. Biomol. Chem. 2008, 6, 1046–1055. [Google Scholar] [CrossRef]
- Frija, L.M.; Khmelinskii, I.V.; Cristiano, M.L.S. Mechanistic investigations into the photochemistry of 4-allyl-tetrazolones in solution: A new approach to the synthesis of 3,4-dihydro-pyrimidinones. J. Org. Chem. 2006, 71, 3583–3591. [Google Scholar] [CrossRef]
- Frija, L.M.; Reva, I.D.; Gómez-Zavaglia, A.; Cristiano, M.L.; Fausto, R. UV-induced photochemistry of matrix-isolated 1-phenyl-4-allyl-tetrazolone. Photochem. Photobiol. Sci. 2007, 6, 1170–1176. [Google Scholar] [CrossRef]
- Dunkin, I.R.; Shields, C.J.; Quast, H. The photochemistry of 1,4-dihtdro-5h-tetrazole derivatives isolated in low-temperature matrices. Tetrahedron 1989, 45, 259–268. [Google Scholar] [CrossRef]
- Quast, H.; Bieber, L. Synthese und Photolyse von 1, 4-Dialkyl-1, 4-dihydro-5H-tetrazol-5-onen und-thionen: Neue Wege zu Diaziridinonen und Carbodiimiden1. Chem. Berichte 1981, 114, 3253–3272. [Google Scholar] [CrossRef]
- Gómez-Zavaglia, A.; Reva, I.D.; Frija, L.; Cristiano, M.L.; Fausto, R. Molecular structure, vibrational spectra and photochemistry of 2-methyl-2H-tetrazol-5-amine in solid argon. J. Phys. Chem. A 2005, 109, 7967–7976. [Google Scholar] [CrossRef]
- Gómez-Zavaglia, A.; Reva, I.D.; Frija, L.; Cristiano, M.L.; Fausto, R. Photochemistry of 1-phenyl-tetrazolone isolated in solid argon. J. Photochem. Photobiol. A Chem. 2006, 179, 243–255. [Google Scholar] [CrossRef]
- Ismael, A.; Cristiano, M.L.; Fausto, R.; Gómez-Zavaglia, A. Tautomer selective photochemistry in 1-(tetrazol-5-yl)ethanol. J. Phys. Chem. A 2010, 114, 13076–13085. [Google Scholar] [CrossRef]
- Alawode, O.E.; Robinson, C.; Rayat, S. Clean photodecomposition of 1-methyl-4-phenyl-1 H-tetrazole-5 (4 H)-thiones to carbodiimides proceeds via a biradical. J. Org. Chem. 2011, 76, 216–222. [Google Scholar] [CrossRef]
- Frija, L.M.; Khmelinskii, I.V.; Cristiano, M.L.S. Novel efficient synthesis of 3,4-dihydro-6-substituted-3-phenylpyrimidin-2(1H)-ones. Tetrahedron Lett. 2005, 46, 6757–6760. [Google Scholar] [CrossRef]
- Hirata, S.; Head-Gordon, M. Time-dependent density functional theory within the Tamm-Dancoff approximation. Chem. Phys. Lett. 1999, 314, 291–299. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other function. Theor. Chem. Accounts 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward reliable density functional methods without adjustable parameters: The PBE0 model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of AB initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Li, H.; Jensen, J.H. Improving the efficiency and convergence of geometry optimization with the polarizable continuum model: New energy gradients and molecular surface tessellation. J. Comput. Chem. 2004, 25, 1449–1462. [Google Scholar] [CrossRef]
- Barca, G.M.; Bertoni, C.; Carrington, L.; Datta, D.; De Silva, N.; Deustua, J.E.; Fedorov, D.G.; Gour, J.R.; Gunina, A.O.; Guidez, E.; et al. Recent developments in the general atomic and molecular electronic structure system. J. Chem. Phys. 2020, 152, 154102. [Google Scholar] [CrossRef]
- Cramer, C.J.; Truhlar, D.G. Implicit Solvation Models: Equilibria, Structure, Spectra, and Dynamics. Chem. Rev. 1999, 99, 2161–2200. [Google Scholar] [CrossRef]
- Laurent, A.D.; Jacquemin, D. TD-DFT benchmarks: A review. Int. J. Quantum Chem. 2013, 113, 2019–2039. [Google Scholar] [CrossRef]
- Coskun, D.; Jerome, S.V.; Friesner, R.A. Evaluation of the Performance of the B3LYP, PBE0, and M06 DFT Functionals, and DBLOC-Corrected Versions, in the Calculation of Redox Potentials and Spin Splittings for Transition Metal Containing Systems. J. Chem. Theory Comput. 2016, 12, 1121–1128. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 10089. [Google Scholar] [CrossRef]
- Tawada, Y.; Tsuneda, T.; Yanagisawa, S.; Yanai, T.; Hirao, K. A long-range-corrected time-dependent density functional theory. J. Chem. Phys. 2004, 120, 8425–8433. [Google Scholar] [CrossRef]
- Chai, J.D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Serpa, C.; Arnaut, L.G.; Formosinho, S.J.; Naqvi, K.R. Calculation of triplet-triplet energy transfer rates from emission and absorption spectra. The quenching of hemicarcerated triplet biacetyl by aromatic hydrocarbons. Photochem. Photobiol. Sci. 2003, 2, 616–623. [Google Scholar] [CrossRef]
- Mckinlay, R.G.; Zurek, J.M.; Paterson, M.J. Vibronic Coupling in Inorganic Systems. Photochemistry, Conical Intersections, and the Jahn-Teller and Pseudo-Jahn-Teller Effects; Academic Press: Cambridge, MA, USA, 2010; Volume 62, pp. 351–390. [Google Scholar]
- Rehmat, N.; Kurganskii, I.V.; Mahmood, Z.; Guan, Q.L.; Zhao, J.; Xing, Y.H.; Gurzadyan, G.G.; Fedin, M.V. Spin–Orbit Charge-Transfer Intersystem Crossing in Anthracene–Perylenebisimide Compact Electron Donor–Acceptor Dyads and Triads and Photochemical Dianion Formation. Chem. Eur. J. 2021. [Google Scholar] [CrossRef]
- Cammi, R.; Corni, S.; Mennucci, B.; Tomasi, J. Electronic excitation energies of molecules in solution: State specific and linear response methods for nonequilibrium continuum solvation models. J. Chem. Phys. 2005, 122, 104513. [Google Scholar] [CrossRef]
- Jacquemin, D.; Perpète, E.A.; Scuseria, G.E.; Ciofini, I.; Adamo, C. TD-DFT performance for the visible absorption spectra of organic dyes: Conventional versus long-range hybrids. J. Chem. Theory Comput. 2008, 4, 123–135. [Google Scholar] [CrossRef]
- Fang, C.; Oruganti, B.; Durbeej, B. How method-dependent are calculated differences between vertical, adiabatic, and 0-0 excitation energies? J. Phys. Chem. A 2014, 118, 4157–4171. [Google Scholar] [CrossRef]
- Gräfenstein, J.; Kraka, E.; Filatov, M.; Cremer, D. Can Unrestricted Density-Functional Theory Describe Open Shell Singlet Biradicals? Int. J. Mol. Sci. 2002, 3, 360–394. [Google Scholar] [CrossRef]
- Coe, J.D.; Levine, B.G.; Martínez, T.J. Ab initio molecular dynamics of excited-state intramolecular proton transfer using multireference perturbation theory. J. Phys. Chem. A 2007, 111, 11302–11310. [Google Scholar] [CrossRef]
- Raucci, U.; Perrella, F.; Donati, G.; Zoppi, M.; Petrone, A.; Rega, N. Ab-initio molecular dynamics and hybrid explicit-implicit solvation model for aqueous and nonaqueous solvents: GFP chromophore in water and methanol solution as case study. J. Comput. Chem. 2020, 41, 2228–2239. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B3LYP | M06-HF | PBE0 | ||||
|---|---|---|---|---|---|---|
| Property | ||||||
| 1.354 | 1.362 | 1.338 | 1.337 | 1.343 | 1.351 | |
| 1.369 | 1.364 | 1.347 | 1.355 | 1.355 | 1.353 | |
| 1.272 | 1.460 | 1.250 | 1.254 | 1.266 | 1.440 | |
| 1.224 | 1.210 | 1.217 | 1.217 | 1.219 | 1.205 | |
| 1.385 | 1.396 | 1.368 | 1.365 | 1.378 | 1.390 | |
| 1.397 | 1.419 | 1.379 | 1.394 | 1.388 | 1.410 | |
| 2.147 | 2.209 | 2.120 | 2.131 | 2.130 | 2.193 | |
| 1.101 | 1.101 | 1.097 | 1.097 | 1.101 | 1.100 | |
| 108.7 | 103.3 | 108.9 | 109.1 | 108.7 | 103.2 | |
| 111.3 | 110.6 | 111.0 | 111.3 | 111.4 | 110.2 | |
| 110.2 | 109.9 | 110.2 | 109.5 | 110.5 | 109.6 | |
| 108.7 | 103.9 | 108.9 | 109.0 | 108.7 | 103.8 | |
| −123.2 | 0.0 | 0.0 | −1.7 | −4.1 | 0.0 | |
| 0.1 | 8.0 | 1.1 | 0.2 | 1.5 | 9.3 | |
| −13.7 | −7.9 | −9.4 | −10.8 | −13.4 | −7.7 | |
| 0.2 | 18.2 | −13.0 | −6.8 | −13.7 | 19.2 | |
| 179.8 | −178.6 | 165.9 | 172.5 | 165.5 | −179.5 | |
| 171.8 | −170.7 | 179.0 | 179.5 | 179.1 | −170.8 | |
| 8.6 | −29.6 | 0.4 | 0.6 | 0.0 | −31.7 | |
| −680.9182942 | −680.806160 | −681.161206 | −681.007779 | −680.550204 | −680.43570 | |
| 507.83 | 499.73 | 522.35 | 508.83 | 513.99 | 506.16 | |
| 1772 | 1796 | 1810 | 1806 | 1824 | 1849 | |
| 1647 | 1640 | 1702 | 1694 | 1681 | 1673 | |
| 1524 | 1515 | 1565 | 1458 | 1544 | 1532 | |
| State | f | ||||
|---|---|---|---|---|---|
| B3LYP | |||||
| 4.828 | 1.7950 | 0.0207 | −0.1817 | 0.3850 | |
| 5.021 | −0.5511 | −0.0515 | 0.0439 | 0.0379 | |
| 5.266 | 0.0730 | −0.0003 | 0.2090 | 0.0063 | |
| 5.438 | −0.1353 | 0.0251 | −0.0600 | 0.0030 | |
| 5.670 | 0.6462 | −0.2856 | 0.0912 | 0.0705 | |
| M06-HF | |||||
| 5.677 | 0.2548 | 0.0990 | −0.0488 | 0.0107 | |
| 6.003 | −0.0488 | 0.0136 | −0.3475 | 0.0181 | |
| 6.085 | 2.0851 | 0.1603 | −0.2364 | 0.6603 | |
| 6.544 | −0.2037 | −0.0297 | 0.1090 | 0.0087 | |
| 7.082 | −1.0043 | −0.1280 | 0.0216 | 0.1779 | |
| PBE0 | |||||
| 5.042 | −1.7551 | −0.0594 | 0.1793 | 0.3849 | |
| 5.203 | −0.8622 | −0.0361 | 0.0711 | 0.0956 | |
| 5.499 | −0.1097 | −0.0062 | −0.2154 | 0.0079 | |
| 5.749 | 0.1360 | −0.0439 | 0.0636 | 0.0034 | |
| 5.962 | -0.5593 | 0.1908 | −0.1240 | 0.0533 | |
| M06-HF | PBE0 | B3LYP | ||||
|---|---|---|---|---|---|---|
| 398.9 | 334.7 | 282.4 | ||||
| TS1 | 458.9 | 60.0 | 396.2 | 61.5 | 354.5 | 72.1 |
| M0 | 318.4 | 269.9 | 219.3 | |||
| TS2 | 340.6 | 22.3 | 283.3 | 13.4 | 223.2 | 3.8 |
| M1 | 201.1 | 171.7 | 11.5 | |||
| TS3 | 254.0 | 52.8 | 228.0 | 56.2 | 77.0 | 65.5 |
| M2 | 117.0 | 103.3 | −17.3 | |||
| TS4 | 287.9 | 170.9 | 279.2 | 175.9 | 152.9 | 170.2 |
| P() | 149.5 | 141.7 | 119.4 | |||
| P() | −247.6 | −201.0 | −308.7 | |||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duque-Prata, A.; Serpa, C.; Caridade, P.J.S.B. Photochemistry of 1-Phenyl-4-Allyl-Tetrazol-5-One: A Theoretical Study Contribution towards Mechanism Elucidation. Appl. Sci. 2021, 11, 4045. https://doi.org/10.3390/app11094045
Duque-Prata A, Serpa C, Caridade PJSB. Photochemistry of 1-Phenyl-4-Allyl-Tetrazol-5-One: A Theoretical Study Contribution towards Mechanism Elucidation. Applied Sciences. 2021; 11(9):4045. https://doi.org/10.3390/app11094045
Chicago/Turabian StyleDuque-Prata, Amilcar, Carlos Serpa, and Pedro J. S. B. Caridade. 2021. "Photochemistry of 1-Phenyl-4-Allyl-Tetrazol-5-One: A Theoretical Study Contribution towards Mechanism Elucidation" Applied Sciences 11, no. 9: 4045. https://doi.org/10.3390/app11094045
APA StyleDuque-Prata, A., Serpa, C., & Caridade, P. J. S. B. (2021). Photochemistry of 1-Phenyl-4-Allyl-Tetrazol-5-One: A Theoretical Study Contribution towards Mechanism Elucidation. Applied Sciences, 11(9), 4045. https://doi.org/10.3390/app11094045