Abstract
DNA nanotechnology has reported a wide range of structurally tunable scaffolds with precise control over their size, shape and mechanical properties. One promising application of these nanodevices is as probes for protein function or determination of protein structure. In this perspective we cover several recent examples in this field, including determining the effect of ligand spacing and multivalency on cell activation, applying forces at the nanoscale, and helping to solve protein structure by cryo-EM. We also highlight some future directions in the chemistry necessary for integrating proteins with DNA nanoscaffolds, as well as opportunities for computational modeling of hybrid protein-DNA nanomaterials.
1. Introduction
The ability to probe protein structure and function at the single-molecule level has been a key motivation of biology, chemistry and physics for decades. Innovations like super-resolution microscopy, optical tweezers, atomic force microscopy (AFM) probing, and Förster resonance energy transfer (FRET) studies have opened new windows into the biological world by enabling analysis of single protein molecules with unprecedented precision. Structural biology, ranging from X-ray crystallography and nuclear magnetic resonance (NMR) to the recent revolution in cryo-electron microscopy (cryo-EM), has also played an indispensable role in determining the three-dimensional conformation of proteins, and thereby elucidating their function at the molecular level. In the past 15 years or so, DNA mechanical nanodevices have emerged as powerful new alternative tools for probing proteins with single-molecule resolution. The DNA origami technique [1,2], by which a long DNA scaffold strand is folded into a well-defined, addressable, and mechanically robust nanoobject, has in particular been a boon for designing structures on a similar length scale as proteins, which can immobilize them or apply forces to them in interesting ways to elucidate their function. In this perspective, we outline recent advances in the use of rigid DNA origami objects and lattices to probe the structure of proteins, or to interrogate their function at the single-molecule level. We focus on three key properties that make DNA nanoscaffolds particularly well-suited to this task (Figure 1): (1) their rigidity, which in turn allows for protein attachment with a controlled orientation for structural biology studies; (2) their ability to space multiple proteins with nanoscale control, or to confine them within a defined volume; and (3) their ability to impart forces at the nanoscale, and thereby probe the biophysics of attached proteins. We then close with a brief discussion of two areas for future investigation for improving DNA nanomechanical devices: (1) rigid, multipoint attachment of proteins on a DNA scaffold and (2) computational modeling of hybrid protein-DNA nanomaterials. Our goal is to highlight some key recent advances in these areas and to focus on how the mechanical nature of DNA scaffolds can facilitate new insights into protein science, not to provide an exhaustive overview of the rich intersection of protein science and DNA nanotechnology. For a more complete treatment of hybrid protein-DNA nanomaterials beyond the scope of this perspective, we refer the interested reader to several key reviews [3,4,5,6].
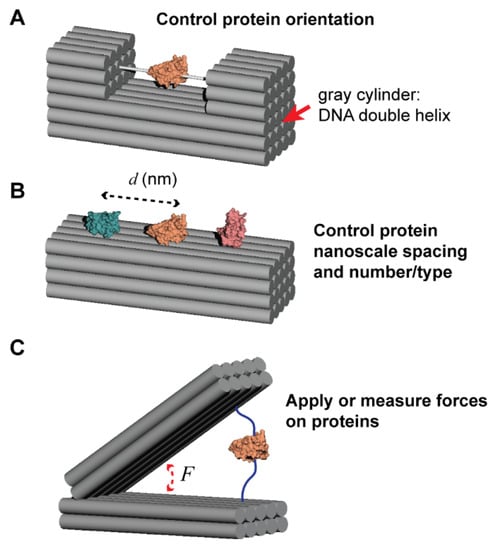
Figure 1.
Scope of this Perspective. (A) Controlling protein orientation by attachment to rigid DNA nanostructures that enables visualization by another technique (e.g., cryo-EM). (B) Controlling protein spacing (at the nanometer scale) and the number or type of protein attached. (C) Using a DNA nanostructure to either apply a force to, or measure the force exerted by, an attached protein.
2. Structural Characterization of Proteins on DNA Nanoscaffolds
The original goal of DNA nanotechnology, as conceived by Ned Seeman in 1982 [7], was to generate a three-dimensional crystal from the self-assembly of individual DNA strands linked by immobile Holliday junctions. Such a crystal could, in turn, be used to scaffold proteins in a repeating lattice in space and solve their structure using X-ray crystallography without having to crystalize the proteins (Figure 2A) [8]. Key to this endeavor is the rigid attachment of the proteins to a mechanically robust, 3D DNA latticework, since even slight variations in their orientation would result in poor diffraction and preclude high-resolution characterization. The first rationally designed, self-assembled DNA crystal was reported by Seeman, Mao and coworkers in 2009, based on a tensegrity triangle motif—a nanoscale analogue of a mechanically rigid macroscopic object—comprised of three DNA strands (Figure 2B) [9]. This crystal was characterized to 4–14 Å resolution (depending on the design), with the largest cavities surpassing 1100 nm3 in volume. In 2016 and 2017, Yan, Seeman and coworkers reported a second DNA crystal design, once again employing three strands but relying on four stacked duplexes linked by Holliday junctions as a key motif (Figure 2C,D) [10,11]. These motifs diffracted to ~3 Å, and a subsequent report that optimized the Holliday junction sequence yielded crystals that diffracted to 2.6 Å, which allowed for visualization of individual purine-pyrimidine base stacks (Figure 2E,F) [12]. Critically, this latter design allowed for crystal cavities with ~50% larger edges (albeit at a reduced resolution) and volumes of 1250 nm3, paving the way for incorporation of larger proteins.
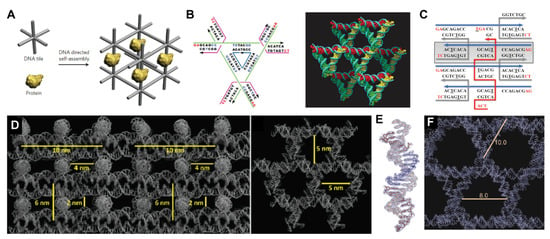
Figure 2.
Self-assembled DNA crystal lattices. (A) Original concept of DNA nanotechnology as conceived by Seeman: using a self-assembled DNA lattice to scaffold proteins in well-defined orientations in 3D space [7].Reprinted with permission from ref. [8], Copyright 2011 Springer Nature. (B) Design (left) and 3D crystal structure (right) of Seeman and Mao’s original tensegrity triangle crystal design. Reprinted with permission from ref. [9], Copyright 2009 Springer Nature. (C) Crystal design with a weaving strand (red) linking four duplexes via Holliday junctions. Reprinted with permission from ref. [11], Copyright 2016 American Chemical Society. (D) Two views of the 3D crystal structure using the weaving strand with stacked duplexes design. Reprinted with permission from ref. [11], Copyright 2017 American Chemical Society. (E) The asymmetric unit of a crystal design with 2.7 Å resolution, showing individual base stacks. Reprinted with permission from ref. [12], Copyright 2020 John Wiley and Sons. (F) This design enabled expanded cavities in the crystal, with void spaces approaching 10 nm in diameter.
The above examples demonstrated that mechanically rigid 3D lattices of DNA duplexes could form crystals with defined void spaces that could host proteins. A key next step to this work will be to demonstrate attachment of proteins, either by using DNA binding proteins, or through chemical conjugation (see Section 4), in a rigid and predictable fashion, in order to solve their structure using the crystal with a known structure as a host. In 2006 Paukstelis demonstrated a different DNA crystal design could serve as a “molecular sieve”, allowing the incorporation of a small protein (GFP, 28 kDa) but not a larger one (MBP-RFP, 280 kDa) [13]. This report was followed in 2014 with a demonstration that enzymes immobilized inside crystals were still catalytically active [14]. Although these reports did not attempt to immobilize the proteins for structural solution, the fact that they could permeate the 3D volume of the crystal and retain their structure and function (as evidenced by the GFP fluorescence and enzyme activity) bode well for achieving structural solution in the future.
In recent years, rigid DNA nanostructures have also been applied to protein structural determination using cryoelectron microscopy (cryo-EM). Although cryo-EM methods are constantly improving, it is still difficult to characterize the structure of small proteins (<100 kDa) due to the lack of image contrast with the background. To circumvent this limitation, proteins can be complexed with larger species like antibodies to increase their effective size and make them easier to discern in the image. Inspired by these studies, several groups have explored using DNA nanostructures, which are readily visualized by cryo-EM due to their large size (~tens of nanometers) and high contrast relative to the background, as “markers” to find the protein. Furthermore, because DNA origami nanostructures can be highly anisotropic, they can serve as “nanoscale goniometers” [15,16] that define the absolute orientation of the attached molecules, further facilitating class-averaging and solution of protein structure. Unlike DNA crystals, which, for the time being, are restricted to rather small proteins (~30–50 kDa) that can fit in their cavities, DNA origami nanostructures can be designed with much larger void spaces for hosting proteins. Indeed, work by Turberfield and coworkers demonstrated that simple 2D DNA lattices could host proteins like RuvA [17] or G-protein coupled receptors (GPCRs) [18] in their cavities to generate repeating arrays that could be visualized by cryo-EM. Although these approaches gave only low resolution (20–30 Å) reconstructions of the protein, they demonstrated the potential for a DNA scaffold as a “sample holder” to image proteins, while preventing aggregation or undesired surface adhesion effects.
Cryo-EM had been used to characterize the 3D structure of DNA cages and to demonstrate their three-dimensionality for many years [19,20], but not until 2012 was the structure of a compact, rigid and block-like DNA nanostructure reported for the goal of protein structural characterization [21]. Dietz, Scheres, and coworkers designed a block-like “pointer” object with densely packed helices on a square lattice, and solved its structure to a maximum resolution of 9.7 Å (for the more rigid core), and 14 Å at the more flexible outer periphery (Figure 3A). This resolution, nonetheless, allowed clear discernment of the DNA strand routing in the object, including the crossover points, stacked Holliday junctions or deviations from the designed lattice geometry due to backbone repulsion. In 2016, these same groups applied this technique to solving the structure of a DNA-binding protein, the transcription factor p53 (Figure 3B) [15]. The researchers designed a DNA origami cage to specifically sit perpendicular to, and vertically span the vitreous ice sheet containing the sample. The center of the structure was spanned by a DNA duplex bearing the binding site for the p53 protein. Changing the relative location of this binding site (relative to the helical axis of the duplex, Figure 3C) resulted in a different viewing angle of the protein. In this way, the DNA origami served as a nanoscale goniometer, enabling different perspectives of the attached protein. The authors used the origami-enforced images to solve the structure of the protein to ~15 Å, and, thereby, elucidate new details about the symmetry of the oligomer. The ultimate resolution was limited, however, by the flexibility of the DNA origami construct, and specifically the single duplex spanning the cavity. Two years later, Mao and coworkers reported the reconstitution of the membrane protein α-hemolysin in a hydrophobic DNA origami cavity, and probed its structure by cryo-EM [22]. They were able to observe the structure of the protein at a resolution of 30 Å, partly due to the absence of interactions that pinned it down in one conformation in the lipid cavity.
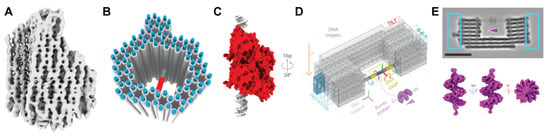
Figure 3.
DNA nanostructure analysis by cryo-electron microscopy. (A) Structure of an asymmetric DNA origami “pointer” object at 9–14 Å resolution [21]. (B) DNA origami “frame” with a spanning helix for binding the transcription factor p. 53 [15]. (C) The orientation of the protein on the spanning duplex can be tuned by the relative positioning (and thus rotation) of the binding site on the helix. (D) Design of a 3D origami with a spanning helix, and several “bits” to determine the orientation, for binding the protein BurrH. Reprinted with permission from ref. [16], Copyright 2020 Springer Nature. (E) Cryo-EM image of the structure (top) with encoding bits boxed in blue; 3D reconstruction of BurrH (bottom).
In 2020, Douglas and coworkers extended the nanogoniometer approach to solve the structure of BurrH (another DNA-binding protein) to a resolution of 6.5 Å (Figure 3D,E) [16]. The DNA origami served as a barcoded, asymmetric object that could directly report the relative conformation of the protein, which was again attached to DNA duplex spanning a cavity in the nanostructure. The nanostructure “barcodes” on the origami specified the rotation angle of the protein on the bound duplex, and the rotational tilt of the duplex between two orientations. Compared with Dietz and Scheres’s work, Douglas and coworkers were able to obtain higher resolution by increasing the yield of origami structures bearing proteins (resulting in more particles per image) and tuning the origami aspect ratio to ensure proper orientation upon adhesion to the transmission electron microscopy (TEM) grid. Three key innovations will likely enable researchers to push the resolution limit of DNA-scaffolded proteins down to <3 Å, which would enable atomic resolution. The first is chemical conjugation to the scaffold in a rigid fashion, as outlined in Section 4 below. Interestingly, Douglas and coworkers proposed that highly rigid attachment of their protein was not strictly necessary and could even be detrimental, but chemical conjugation could also enable analysis of non-DNA binding proteins. The second innovation is increasing the resolution of the DNA scaffold itself to the atomic level. A third advancement would be to use computational simulation with atomic resolution to better fit cryo-EM maps, as also discussed in Section 4.
In 2020, the Dietz and Scheres groups reported that by taking into account structural fluctuations in DNA origami, and interpreting the cryo-EM maps with molecular dynamics simulations, they could further improve the resolution of 3D origami nanostructures to ~4 Å [23]. In this fashion, they could distinguish the fine features of the objects, including major and minor grooves, single-strand breaks and crossover positions. Importantly, this technique demonstrated the inherent flexibility and undesired or unintended deformations in DNA nanostructures, and allowed the authors to correct them through sequence design or covalent crosslinking. In this fashion, it should be possible to design a rigid and well-defined object with atomically-defined locations for protein attachment. By combining this advanced method with better chemical immobilization of arbitrary proteins on a DNA scaffold, as well as methods for covalently locking flexible points in the DNA structure itself [24], the potential for structural solution of individual proteins becomes ever more tantalizing. We also highlight that while atomic resolution may be a worthwhile ultimate goal for these structures, even determining protein structure to 3–6 Å could be useful for many studies, especially in conjunction with mechanical force application as in Section 3 below. For example, the unfolding of a multidomain protein whose structure is already known could probably be probed sufficiently even at nonatomic resolutions.
3. DNA Nanoscaffolds for Interrogating Ligand Spacing, Valency, and Confinement
A second area where DNA nanomechanical devices have played an important role in biology is in probing the required distance and multivalency of proteins for effectively binding to cell receptors or triggering a biological effect. DNA origami nanostructures are particularly effective at this role for several reasons. First, they are rigid objects (especially if multilayer, block-like nanostructures are used), so thermal fluctuations do not change the distance between attached ligands much [25]. Second, they typically span dimensions of tens to hundreds of nanometers, a size scale well matched to many biological receptor complexes on cells. Third, it is relatively straightforward to change the distance between ligands, or the number of ligands, by extending staple strands at arbitrary points on the structure (with a resolution of ~5–10 nm). Fourth, the shape of a DNA nanostructure can be tuned to match the application at hand, e.g., a linear rod to serve as a molecular caliper [26], a wireframe polyhedron to mimic a virus [27], or a ring to recapitulate the nuclear pore [28,29].
Since the inception of DNA origami [1], one of the most attractive applications for these structures was as a “molecular breadboard” to attach other species, like nanoparticles or proteins, with nanoscale control. Indeed, a vast and rich literature exists on the attachment of proteins to DNA nanoscaffolds, with particular interest in enzymatic cascades as molecular assembly lines. One potential limitation of the original, two-dimensional designs reported by Rothemund, and used by many others since, is that they are quite flexible. With the advent of three-dimensional origami comprised of several layers of stacked helices linked by multiple crossovers [2], much more rigid objects can be obtained, which do not deform significantly and can thus enforce specific distances between bound ligands. Here we discuss several seminal examples of biological studies that would not be possible, or be much more difficult to carry out, without the unique properties of origami. Due to space limitations, we will not discuss the extensive work on enzymatic cascades, and instead direct the interested reader to several excellent reviews on this topic [30,31,32].
In 2014, the Hogberg and Teixera labs used a 3D origami scaffold as a “nanocaliper” to position the ephrin-A5 protein ligand by attaching it to a single-stranded (ss) DNA handle complementary to extensions of staple strands from the nanostructure (Figure 4A) [26]. In this way, the authors were able to control not only the number of ligands, but also the distance separating them (either 40 or 100 nm). This technique demonstrated that not only were two ligands necessary for receptor activation (compared with a monovalent system), but the sample with 100 nm spacing worked just as well as the one with 40 nm distance between proteins. Furthermore, using eight ligands spaced 14 nm apart did not yield any further improvements in bioactivity compared with the two ligands. A key to this approach’s success was the rigidity of the nanocaliper, which enforced the desired distances and allowed for a systematic study of both spacing and absolute number of receptors that would not be possible with other approaches (like antibody clustering of the receptors). In a follow-up study, Hogberg and coworkers were able to use a similar nanocaliper to immobilize small molecule antigens with controlled distances to probe the spatial tolerance of antibody binding [33]. Once again, spacing the molecular species on a rigid and tunable scaffold allowed for a precise investigation of IgG bivalent binding not possible with traditional methods like surface plasmon resonance. The ability of DNA origami to control the nanoscale spacing and number of ligands can also be applied to study the proteins that must oligomerize to function, like the potassium channel Kir3 [34], or caspase 9 [35]. In the caspase 9 example, the addressable pegboard also allowed the authors to determine that clustering of the proteins enhanced activity; even though dimerization alone did activate the enzymes, spatially tunable clusters of three or four enzymes worked even better, but only if the proteins were also within a given distance.
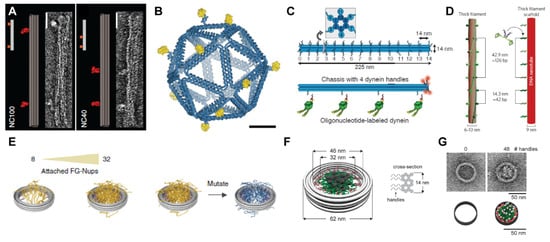
Figure 4.
Controlling protein spacing, valence, and confinement with DNA objects. (A) DNA “nanocaliper” for spacing proteins at distances of 100 nm (left) and 40 nm (right). Reprinted with permission from ref. [26], Copyright 2014 Springer Nature. Insets show negative-stain TEM images of the calipers. (B) Icosahedral DNA cage for presenting the eOD-GT8 antigen (in yellow) to create synthetic vaccines. Reprinted with permission from ref. [27], Copyright 2020 Springer Nature. (C,D) DNA origami templates for controlling the spacing and stoichiometry of molecular motors like dynein. Reprinted with permission from ref. [37], Copyright 2012 The American Association for the Advancement of Science. (C) or myosin Reprinted with permission from ref. [38], Copyright 2015 Springer Nature. (D). (E,F) DNA origami rings for attaching FG-Nup proteins to mimic the nuclear pore complex. Reprinted with permission from ref. [29], Copyright 2018 Springer Nature. Reprinted with permission from ref. [28], Copyright 2018 American Chemical Society. (G) Negative stain TEM images (top) and cartoons (bottom) of rings with zero or 48 handles for FG-Nup proteins. Reprinted with permission from ref. [28], Copyright 2018 American Chemical Society.
Although the above example used linear templates to space proteins or ligands, a key advantage of DNA nanotechnology is the high degree of programmability in shape that it allows. In 2020, Irvine and Bathe and coworkers described a polyhedral DNA origami scaffold for presenting B-cell ligands in order to create a vaccine (Figure 4B) [27]. This approach allowed the researchers to investigate the effect of protein number and spacing, as well as compare the spherical particle to a linear scaffold (akin to the Hogberg caliper). The authors found that increasing the ligand spacing resulted in more potent B-cell receptor activation, and the rigid segregation of ligands on the origami scaffolds, enhanced activity compared with more flexible ssDNA or polyethylene glycol (PEG) linkers. As with the nanocaliper, the programmability of DNA nanostructures enabled a direct, apples-to-apples comparison of various ligand presentations on a readily tunable scaffold, which would have been impossible with other approaches. Very recently, DNA origami scaffolds were also used to study the clustering of T-cell receptors (TCRs) by attaching binding antibody fragments to the DNA scaffold [36]. Furthermore, the origami structures were modified with cholesterol and embedded in a supported lipid bilayer, facilitating the motion and clustering of the TCRs in a highly biomimetic fashion, allowing the authors to determine that T cell activation required at least two TCRs within a distance of 20 nm. The DNA origami thus enabled both nanoscale control of ligand spacing, but also served as a nanoscale “adapter” to mimic the biological arrangement by embedding the proteins in a lipid membrane.
Another powerful application of DNA nanostructure templates in biology is to probe the cooperative actions of multiple proteins working in tandem. In 2012, Reck-Peterson and colleagues used a DNA origami 12-helix bundle to position multiple copies of the motor proteins dynein or kinesin-1 (Figure 4C) [37]. The rigid origami nanostructure thus served to place these proteins with controlled valence and spacing to probe their cooperativity, and found that increasing the number of motors did not increase the velocity of the origami on a microtubule-modified surface. However, the spatial addressability of the handles enabled a “tug of war” arrangement between the two types of proteins, which are opposite-polarity molecular motors. Furthermore, introducing photocleavable linkers in the DNA handles for one type of motor allowed their triggered “release” from the scaffold with light, resulting in the other protein dominating. In 2015, the Sivaramakrishnan lab used a DNA origami nanotube to mimic a myosin fiber, and used DNA handles to position myosin heads with the same pitch (14.3 nm) as in the natural filament (Figure 4D) [38]. Once again, the authors were able to probe the synergistic effects of multiple myosin heads on nanotube motion on an actin-modified surface, and found that neither the number of myosin heads, nor their density, affected the gliding speed. In this way, the researchers validated a key biochemical mechanism for these molecular motors. In both of these examples, the DNA origami nanostructures provide an unparalleled tool for positioning these motor proteins with not just controlled spacing and valency but, as in the Reck-Peterson paper, precise stoichiometry between multiple types of proteins. The rigidity of these multihelical objects is critical to prevent spatial fluctuations that would perturb the desired distances or allow for undesired interactions between the proteins.
A third distinct area of biological exploration enabled by DNA nanostructures is probing the function of proteins in confined nanoscale volumes. Two sets of researchers in 2018, one led by the Dietz and Dekker groups [29], and one by the Lin and Lusk labs [28], used a rigid DNA origami ring to mimic the nuclear pore complex. The goal of both teams was to understand the collective assembly and pore blocking function of proteins called FG-nups, by confining them to a cylindrical volume ~40 nm in diameter (Figure 4E–G). The use of DNA origami also allowed for a tunable incorporation of proteins (from 32–48), and independent modulation of their orientation (i.e., pointing “in” vs. “out” of the ring, with only the former being mimetic of the nuclear pore). It is hard to envision another system that can control both the nanoscale area/volume available to proteins with high precision, but also the number of proteins attached to that volume, and the direction in which those proteins are oriented relative to it. The DNA origami rings balance flexibility (to create a circular structure) with rigidity (to prevent its deformation), while allowing for a stoichiometrically-defined number of proteins via the DNA handles used. Using a similar ring-like structure that encircled a liposome and could position soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) proteins, the Shih and Rothman groups were able to determine that only one to two SNAREs were sufficient for membrane fusion [39]. This work was particularly notable because the DNA nanostructure controlled both the number of the proteins, but also their proximity to a liposome, helping recreate a complex biological arrangement that would not have been possible without the rigidity and shape control intrinsic to the origami, as well as the multiple chemical functionalities that the structure can present.
4. Measuring or Applying Forces with DNA Nanodevices
The third application of DNA nanostructures that we discuss is in measuring or applying forces on proteins at the single-molecule level. Although other methods, like optical tweezers or attachment to an AFM tip, exist for pulling on proteins with forces in the piconewton (pN) regime, these require specialized equipment and are limited to analyzing one protein at a time. DNA nanodevices have the advantage that they are relatively inexpensive and allow analysis of many proteins in parallel in a single sample. Furthermore, the biophysical properties of DNA are fairly well understood, or can be readily probed using computational simulations, as we discuss in Section 4, and readily tunable with only minor changes to the DNA sequences that comprise the origami nanoobject. We also note that simple DNA duplexes are extremely promising as biophysical sensors, e.g., to probe the forces applied by cell integrins to the extracellular matrix [40,41], but since these do not involve DNA nanostructures we will not cover them here.
The structural programmability of DNA origami structures led several groups to probe their use as nanoscale force-generating devices akin to calipers or mechanical jacks. An added advantage to these systems is that DNA duplexes could be used as “cranks” to apply a given force. Alternatively, purely nanoscale effects absent from the macroscale world (like electrostatic repulsion or entropic springs) can be employed as well. Funke and Dietz reported a hinged DNA origami nanostructure where the distance between the rigid arms could be tuned with several DNA “adjuster” duplexes, with adjustments tunable down to a single base pair (Figure 5A) [42]. Remarkably, this device could achieve displacement steps of only 0.04 nanometers, slightly less than the Bohr radius, due to the lever-arm effect. Tinnefield, Liedl, and coworkers demonstrated a different nanomechanical device based on the entropic forces that oppose extension of a single strand of DNA [43]. By reducing the number of nucleotides in these strands, the force could be readily tuned. As an alternative trigger for applying forces at the nanoscale, Stephanopoulos and coworkers described a DNA tweezer with a hairpin loop that could be triggered to open with a displacement strand. By incorporating the displacement strand into the nanostructure, but photocaging it, the authors were able to generate tens of piconewtons of force with a brief pulse of UV light that removed the cages and allowed the strand to open the tweezer (Figure 5B) [44]. The Castro lab reported a host of nanoscale analogues to macroscale mechanical devices, like crank-sliders, pistons, Bennett linkages and a waterbomb base [45,46], which can readily be extended to biophysical studies on proteins. Fygenson and Schulman described DNA “nunchuck” devices, comprised of two long DNA nanotubes linked by a flexible hinge that can mechanically magnify the angle of the hinge and measure single-molecule forces [47,48]. All the above demonstrations relied intimately on both the rigidity of the DNA helices comprising them and the ability to use DNA hybridization or coiling to apply precise forces.
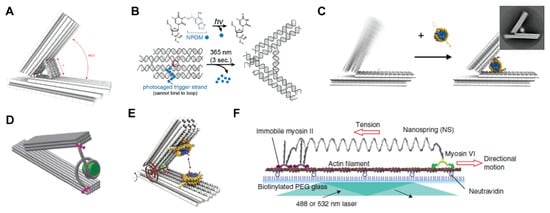
Figure 5.
Applying forces using DNA nanodevices. (A) DNA nanocaliper with “adjuster” helices for Bohr radius resolution. Reprinted with permission from ref. [42], Copyright 2015 Springer Nature. (B) DNA nanotweezer with a photocaged trigger strand, allowing for rapid opening with a short pulse of UV light. Reprinted with permission from ref. [44], Copyright 2018 John Wiley and Sons. (C,D) Application of hinged DNA nanostructures to measure the biophysical properties of nucleosomes binding to DNA. Reprinted with permission from ref. [49,50], Copyright 2016 American Chemical Society. (E) DNA nanostructure for measuring the forces between two nucleosomes. Reprinted with permission from ref. [51], Copyright 2016 American Association for the Advancement of Science. (F) DNA origami nanospring for measuring the stall force of myosin motors. Reprinted with permission from ref. [52], Copyright 2016 Springer Nature.
In two reports from 2016, these caliper- and jack-like structures were applied to measure the forces between double stranded DNA and nucleosomes, the protein complexes that wrap the genomic material in the nucleus. Both works, one by the Poirier and Castro groups [49] and the other by the Dietz lab [50], used a hinge-like structure to position double stranded DNA between the arms of the device and allow the nucleosomes to bind (Figure 4C,D). By measuring the angle between the arms of the caliper, which could be closed by the nucleosome binding to and winding the DNA, various molecular details of these protein complexes could be determined such as the effect of salt on binding strength, the number and orientation of neighboring nucleosomes, the length of DNA or transcription factor binding on the nucleosome. In a related study that same year, the Dietz group used their caliper device to measure the forces between two different nucleosomes (as opposed to between nucleosomes and DNA), which is key for understanding the higher-order organization of chromatin into structures in the nucleus (Figure 4E) [51]. Both the relative force applied on the nucleosomes, as well as their distance, orientation and chemical modification (e.g., acetylation) could be probed to yield an energy landscape as a function of internucleosome distance. These examples all demonstrate the great potential in DNA nanodevices for studying DNA-binding proteins with single-molecule resolution, with an emphasis on controlling their displacement and energy landscape, or other molecular properties of the proteins and their substrates. As we discuss in Section 4 below, using site-specific bioconjugation methods would extend these studies to arbitrary proteins, and enhancing the rigidity of attachment would enable greater control over protein orientation for measuring protein-protein interaction energies.
An alternative design for applying and measuring forces on proteins was reported by Iwaki and coworkers in 2016, and relied on a DNA nanostructure “spring” to tug on the motor protein myosin VI (Figure 4F) [52]. Unlike the caliper-like designs mentioned above, this spring consisted of a two-helix DNA bundle with negative superhelical strain (following design rules for curved and twisted origami) [53] to give a structure similar to a macroscopic spring. The authors first characterized the force-extension curve of this spring using an optical trap and demonstrated that the DNA nanostructure had a roughly 10-fold lower spring constant than double-stranded (ds) DNA. As a result, their design matched the stall force of the myosin motors (~2 pN) at a much shorter length than dsDNA, which in turn allowed for shorter run lengths (~700–800 nm). The nanostructure could thus precisely “match” the intrinsic biophysical properties of the motor, whereas simpler force-applying devices like dsDNA were unsuited to this particular protein. Using this device, the authors were able to demonstrate that at higher forces the mechanism of myosin VI motion switched (from hand-over-hand to inchworm motion), as well as set up a tug of war between myosin VI and myosin V by attaching them to opposite ends of the spring. The programmable mechanical properties of DNA nanostructures played a key role in the ultimate application, and although optical tweezers were used to calibrate the device it could be used thereafter to probe protein function without a complicated experimental setup. The application of DNA origami to single molecule biophysics also presents challenges in the correct interpretation of the measured force-extension properties, as one needs to correctly interpret measured values of a statistical ensemble of forces and extensions [54], as well as take into account the mechanical properties and extensions of double-stranded and single-stranded segments under tension in the DNA origami constructs that are used to apply force [55].
5. Future Enabling Technologies for Protein-DNA Nanodevices
In the previous three sections we outlined key applications for DNA nanodevices. There is no doubt that future researchers will continue to explore all three areas, with exciting advances in structural determination, basic biological studies of protein distribution or the nanoscale forces that govern protein activity. In addition, the mechanical and programmable nature of DNA nanostructures will undoubtedly play a role in many additional areas that we cannot even conceive of at the moment. Here, however, we wish to cover two enabling technologies that will enhance DNA nanodevice functionality for probing proteins and allow their use in new areas as well: (1) rigid, multipoint attachment of proteins on a DNA nanoscaffold and (2) computational modeling of hybrid protein-DNA nanostructures. The ultimate goal of both technologies is to create a highly integrated system of both molecules, where the mechanical properties of the DNA scaffold can best communicate with, or influence, those of the protein.
5.1. Rigid Attachment of Proteins to DNA Scaffolds
It is still challenging to link proteins to DNA scaffolds through either covalent or supramolecular means, especially if rigid and multipoint attachment is desired. Here we highlight a few key aspects of this challenge as it pertains to the three applications covered in this perspective. First, most chemical linkages between proteins and DNA are accomplished through heterobifunctional linkers (Figure 6A), where a surface residue on a protein (e.g., cysteine) is linked to an oligonucleotide bearing a unique reactive group (e.g., an amine) at the 5′ or 3′ end. Most such linkers incorporate a flexible connector, such as an aliphatic chain or oligoethylene glycol, between the two reactive ends of the linker. In addition, commercially obtained, functionalized DNA often has an aliphatic linker (ranging from 3–12 carbons) between the 3′ or 5′ end and the terminal moiety (Figure 6B). Although this additional flexibility and length (on the order of 2–3 nm for a fully extended linker) facilitate the reaction yield between the protein and DNA, they preclude a rigid attachment of the protein to the DNA scaffold, and reduce the spatial accuracy of protein placement. Another common strategy for attaching a protein of interest (POI) to a DNA scaffolds is by fusing it to a protein that ligates itself to a specific functional group on the DNA oligonucleotide. These self-ligating proteins, such as the Halo, SNAP, or Clip tags, are highly efficient and site-specific, but introduce extra bulk to the POI and are often linked to it through flexible amino acid linkers [56].
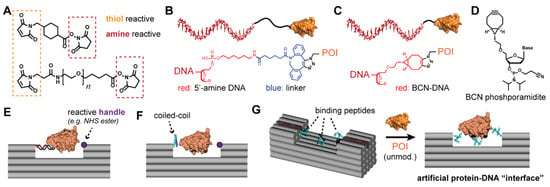
Figure 6.
Chemical conjugation strategies. (A) Examples of two commercially available linkers for attaching thiols (e.g., a cysteine on a protein) to amines (e.g., a modified oligonucleotide). (B) Example of a protein-DNA conjugate from strain-promoted azide-alkyne cycloaddition (SPAAC) between an azide-containing protein and DNA modified with dibenzocyclooctyne (DBCO)-N-hydroxysuccinimide (NHS) ester [57]. Both the DNA amine modification (red) and the DBCO-NHS (blue) introduce flexible alkyl linkers between the oligonucleotide and the protein of interest (POI). (C) Using a bicyclononyne (BCN) directly attached to the 5′ end of DNA for SPAAC reduces the linker length by more than half compared with DBCO-NHS [58]. (D) Structure of a BCN phosphoramidite for introduction of the cyclooctyne at the 5′ end of DNA via solid-phase synthesis. (E) Attaching a POI to a DNA nanostructure with a first handle, and positioning a second reactive handle (purple) on the DNA structure in close proximity to the protein surface. (F) Using a coiled-coil, with one peptide attached to the DNA structure and the other genetically fused to the protein, avoids initial protein-DNA conjugation. (G) Positioning three binding peptides on a DNA nanostructure may allow for immobilization of an unmodified POI simply through multivalent binding interactions.
Thus, a key opportunity for chemists and bioengineers is to design conjugation strategies that enable short, rigid linkers between the DNA and protein, as well as multisite attachment to more effective “pin” the protein on a DNA nanoscaffold. We have described the range of protein-DNA bioconjugation strategies in extensive detail elsewhere, [6] but here we highlight two approaches that will facilitate these goals: (1) custom synthesis of modified DNA, with use of efficient bioconjugation reactions, and (2) proximity-enhanced reactions for modifying proteins in a second location driven by hybridization of a protein-DNA conjugate to a DNA scaffold. To the first point, the main reason many protein-DNA studies use long and flexible bifunctional linkers is because they are commercially available and can target common residues on a protein surface (e.g., lysine), or uniquely reactive amino acids that can readily introduced through genetic methods (e.g., cysteine). However, solid-phase DNA synthesis can incorporate arbitrarily complex (and arbitrarily many) unique chemical moieties by introducing appropriately-functionalized phosphoramidites into the solid-phase synthesis protocol. It is not trivial, however, to make modified phosphoramidites, so this task will require collaboration between synthetic organic chemists and DNA nanotechnologists and engineers. As one illustration, we contrast two approaches that our lab has pursued [57,58] to click chemistry with a protein or peptide modified with a noncanonical azide residue: (1) using a commercially available N-hydroxysuccinimide (NHS) ester linker with amine-DNA (Figure 6B), which is ~2 nm in length, and (2) using a strand with a 5′ custom-synthesized phosphoramidite (Figure 6C,D), which is around half that size. However, a range of additional, efficient and high-yielding bioconjugation reactions have been used to attach proteins to DNA in recent years (for example, the trans-cyclooctene/tetrazine reaction) [59], and can, in turn, be incorporated into phosphoramidites in order to control the linker length, rigidity and point of attachment (e.g., off the DNA backbone or base, instead of the termini).
The second key advantage that we propose for protein-DNA nanomechanical devices is multisite tethering of proteins on a DNA scaffold to enforce the spatial relationship between them. Although it is possible to attach two or more unique DNA strands to a protein, this task is quite challenging because it requires two selective and orthogonal bioconjugation reactions, and attachment of the second handle is often made sterically more challenging due to the first strand. As an alternative, we suggest that the first strand (introduced using site-specific chemistry) can be used to anchor the protein on a DNA nanostructure bearing a strand with a second reactive handle. The proximity of this handle to the protein surface can then drive the second reaction due enhanced local concentration. A seminal report by Gothelf and coworkers demonstrated that one ssDNA handle attached to a protein (via the His6-Ni(NTA) interaction) could spatially direct the bioconjugation of a complementary strand bearing a reactive NHS ester moiety [60]. In a follow-up work, Gothelf and coworkers demonstrated this approach for attaching IgG antibodies to a cavity in a two-dimensional origami [61]. We envision widespread use of this concept in the future to attach arbitrary proteins to a nanostructure of interest, as in Figure 6E. The chemistry for this second reaction could also be site-specific (with proximity simply serving to increase the yield), or nonselective, like the lysine-targeting acylation described by Gothelf.
In this fashion, proteins could be rigidly attached in a fixed orientation on 3D DNA crystals, or DNA origami nanogoniometers for cryo-EM experiments. Indeed, modifying the second site of attachment of the protein on the scaffold (e.g., by moving around a reactive amino acid through mutagenesis) could provide another way to direct protein orientation for optimal “viewing” in the cryo-EM image. This approach would also be powerful for biophysical studies of proteins, for example by probing the forces required to unfold a protein by tugging on different parts of its surface, or to determine allosteric effects. The ability to tether any protein to a DNA nanoscaffold will circumvent the restriction to DNA-binding proteins, like transcription factors or histones, used in many of the examples discussed in Section 2 and Section 4. Finally, the ability to control protein orientation on a DNA scaffold will facilitate more precise biological studies. In native biological settings, protein surfaces are tightly controlled, and orientation is critical to ligand-receptor binding. By more accurately recapitulating these orientations, studies using these systems would provide greater insight. For example, using flexible linkers and single-site attachment precludes tight spatial control, which may obscure biological effects that rely on subnanometer accessibility. Finally, by more effectively tethering proteins to DNA scaffolds, it is possible to envision using the protein to apply a force on the DNA nanostructure, e.g., by incorporating motor proteins, photoswitchable domains, or proteins that undergo conformational changes upon ligand binding, to create nanomachines that are actuated by the proteins.
The strategy for attaching a given protein to a DNA scaffold is likely to vary depending on the target with respect to its size, charge, amenability to mutagenesis, shape and accessibility of the surface to the solvent, among other considerations. And while dual-site modification with DNA handles might provide one avenue to rigid attachment, we foresee alternate approaches that altogether avoid prefunctionalization of the protein with even a single DNA handle. Fusing a coiled-coil peptide to the protein of interest, for example, could bind it to a DNA nanostructure bearing a complementary coil, [58] and enable a second site-specific conjugation reaction (Figure 6F). It may also be possible to directly attach proteins to a DNA scaffold by attaching several short peptides to the oligonucleotides to create an interface similar to that between proteins, where multiple weak interactions mediate a tight interaction (Figure 6G). In this fashion, the DNA nanostructure becomes an addressable material for spatial control of multiple peptides, the way that many proteins (e.g., antibodies) control the presentation of key loops for binding a target. This concept was indeed demonstrated by Sacca and coworkers using DNA origami presenting multiple peptides for binding to a spherical protein cage, [62] and future studies that move beyond multivalent targets and position several different peptides would expand the utility of the method. In order to rationalize design of such hybrid structures, however, researchers will likely require new computational methods that can simulate both DNA and protein to the requisite accuracy, so we discuss this area next.
5.2. Computational Simulation of Protein-DNA Nanostructures
The ability to simulate hybrid protein-DNA nanostructures to atomic accuracy would enable great advances in the design of these systems and facilitate future applications in the areas discussed in Section 2, Section 3 and Section 4. Such capability would also enable the design of hybrid nanostructures comprised of both building blocks, [6] as well as help interpret experimental data (e.g., protein deformation using DNA nanodevices or interpretation of cryo-EM maps for proteins scaffolded on origami). Protein design software, such as Rosetta [63], has enabled remarkable advances in the design of complex protein nanostructures and devices. A similar software package for protein-DNA nanotechnology, however, is lacking. Below we discuss DNA nanostructure modeling and outline the challenges, and some recent advances, in merging these methods with protein simulation.
Since the invention of DNA origami, various computational design tools have been used to obtain increasingly complex nanostructures. The most commonly used tools include Cadnano [64] for 2D and 3D origami designs, and Athena [65] for 3D wireframe designs. However, these design tools have been, for the most part, limited to constructs comprised of DNA only. Currently, only the ADENITA [66] and oxView tools [67] (Figure 7) allow for representing a protein along with a designed DNA nanostructure. The inclusion of proteins (typically based on a known structure from the PDB database) in the 3D representation of the design tool is crucial for determining the optimal placement of linkers for rigid positioning of the proteins, which in turn is also critical for spacing proteins appropriately or determining the forces exerted upon them by a DNA nanodevice. The modeling tools that can incorporate proteins in this way have, however, been introduced only recently and are currently still missing an accurate representation for different types of linkers. Besides using computer-aided design, molecular simulation has also become an important part of the DNA structure characterization pipeline [68]. Simulations can be used to optimize different designs in silico, explain and rationalize behavior observed in experiments and probe nanostructures at a level of detail not accessible experimentally.
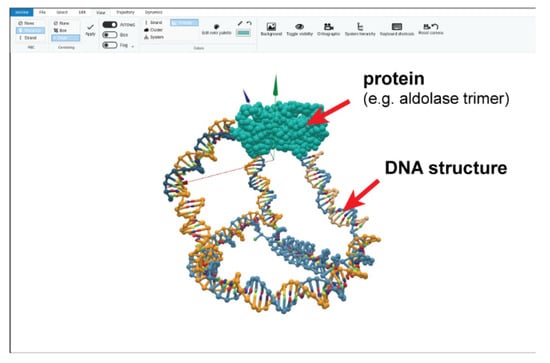
Figure 7.
A DNA-protein nanocage as represented in the ANM-oxDNA model using the oxView editing tool. Reprinted with permission from ref. [67], Copyright 2020 Oxford University Press.
The most commonly used models to simulate molecules in a biological context are atomic-resolution models, where each particle corresponds to a single atom. Programs such as AMBER, GROMACS, [69] or NAMD [70] can be used to study the molecules and their interactions, relying on parallel computations on multiple CPUs and/or GPUs. The parametrization of interactions between atoms is called a force-field. Two of the most popular ones for protein simulation are based on CHARMM and AMBER-based force fields [71]. Recently, there has been significant effort to also develop force-fields that better represent DNA [72] and RNA [73]. However, probing the interactions between proteins and nucleic acids remains a challenging topic in terms of parametrization of the force-field in order to reproduce interaction free-energies more accurately or predict protein-DNA interactions.
Besides the difficulties mentioned above, fully atomistic models are also limited in the timescales that can be studied. With the current latest hardware, up to tens of microseconds can be simulated, requiring up to several months of simulation on multicore systems. DNA nanostructures like origami are typically made up of tens of thousands of nucleotides, and include events on timescales that can reach several hours, making the systems extremely challenging targets for simulation. So far, fully atomistic studies of DNA nanostructures have been generally limited to conformational sampling up to fractions of a microsecond in length [74]. There are ongoing efforts to further improve the force fields to capture both the properties of nucleic acids as well as proteins, and to quantitatively reproduce free-energies of their interactions [75].
An alternative to atomistic simulation is to use coarse-grained models, which represent groups of atoms as a single particle, with interactions between them parameterized to reproduce a particular set of properties of the system: e.g., thermodynamics, structural motifs or mechanical properties. The trade-off in reduced accuracy is balanced by the models’ ability to study larger systems (up to hundreds of thousands of nucleotides for the model oxDNA) and longer timescales, including processes that happen on the order of seconds. Examples of models that can represent both proteins and nucleic acids include the MARTINI [76] 3SPN.2 DNA model with the incorporated AWSEM protein model, [77] and the recently introduced ANM-oxDNA model [78] (Figure 7). These coarse-grained models differ in terms of the detail of their representation as well as the set of properties their parameters are fitted to reproduce. ANM-oxDNA, which uses the established oxDNA model [79,80,81] for DNA nanotechnology and combines it with an anisotropic network model (ANM) of proteins [82], has been used to simulate an experimentally realized DNA-protein hybrid nanocage reported by the Stephanopoulos lab [57]. The ANM model represents the protein as a string of rigid beads connected by a parametrized spring potential. The ANM-oxDNA further parametrizes the linkers connecting the proteins and DNA by a potential function that reproduces the behavior of a linker in a fully atomistic simulation. The use of other coarse-grained models that support nucleic acids as well as proteins has so far been limited to biomolecular systems, and it would be interesting to benchmark them against DNA-protein structures.
Most current modeling approaches for DNA-protein systems have focused on biological problems, such as the estimation of DNA-protein interaction energies, docking or the effect of proteins binding to DNA [75]. As a result, these methods rarely address the same behavior relevant to DNA-protein hybrid nanosystems, like the mechanical effect of pulling a protein attached to a nanostructure, the protein-DNA nanostructure response to change in temperature, and the accurate representation of linkers are necessary to realistically capture the experimental behavior of hybrid nanosystems. New experimental datasets would greatly help in benchmarking existing models, as well as developing new ones with fine-tuned parameters to better represent the protein-hybrid nanostructures. Examples of data that would be useful to the modeling community include cryo-EM imaging of protein-DNA hybrid constructs, studies of assembly yield as a function of temperature, and imaging nanostructures where the protein and DNA are subject to mechanical stresses. Not only can such experiments provide important data on parametrization of the properties of the linkers, but can also help uncover properties of proteins that can further improve model parameters, also benefiting their accuracy for biomolecular simulations.
As was the case with the emergence of DNA nanotechnology, we can expect that further examples of successful experimental realization of DNA nanostructures interacting with proteins will lead to development of more accurate models, which will, in turn, enable design of more complex nanodevices. New modeling and design tools can help to interpret data produced from interactions between protein and DNA, be used for more accurate reconstruction of nanostructures with bound proteins via cryo-EM and also probe designs for optimal placements of protein interacting sites in the nanostructures.
Author Contributions
N.S. primarily wrote Section 1, Section 2, Section 3 and Section 4 and Section 5.1. P.Š. primarily wrote Section 5.2. Both authors edited the final manuscript. The authors gratefully acknowledge Minghui Liu for help preparing Figure 1 and Figure 6. All authors have read and agreed to the published version of the manuscript.
Funding
The authors acknowledge funding from the National Science Foundation (DMR-BMAT CAREER award 1753387 to N.S.; OAC award 1931487 to P.Š. and N.S.).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rothemund, P.W.K. Folding DNA to create nanoscale shapes and patterns. Nat. Cell Biol. 2006, 440, 297–302. [Google Scholar] [CrossRef]
- Douglas, S.M.; Dietz, H.; Liedl, T.; Högberg, B.; Graf, F.; Shih, W.M. Self-assembly of DNA into nanoscale three-dimensional shapes. Nat. Cell Biol. 2009, 459, 414–418. [Google Scholar] [CrossRef] [PubMed]
- Sacca, B.; Niemeyer, C.M. Functionalization of DNA nanostructures with proteins. Chem. Soc. Rev. 2011, 40, 5910–5921. [Google Scholar] [CrossRef] [PubMed]
- Madsen, M.; Gothelf, K.V. Chemistries for DNA Nanotechnology. Chem. Rev. 2019, 119, 6384–6458. [Google Scholar] [CrossRef]
- Wilner, O.I.; Willner, I. Functionalized DNA Nanostructures. Chem. Rev. 2012, 112, 2528–2556. [Google Scholar] [CrossRef]
- Stephanopoulos, N. Hybrid Nanostructures from the Self-Assembly of Proteins and DNA. Chem 2020, 6, 364–405. [Google Scholar] [CrossRef]
- Seeman, N.C. Nucleic acid junctions and lattices. J. Theor. Biol. 1982, 99, 237–247. [Google Scholar] [CrossRef]
- Pinheiro, A.V.; Han, D.; Shih, W.M.; Yan, H. Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 2011, 6, 763–772. [Google Scholar] [CrossRef]
- Zheng, J.; Birktoft, J.J.; Chen, Y.; Wang, T.; Sha, R.; Constantinou, P.E.; Ginell, S.L.; Mao, C.; Seeman, N.C. From molecular to macroscopic via the rational design of a self-assembled 3D DNA crystal. Nat. Cell Biol. 2009, 461, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.R.; Zhang, F.; MacCulloch, T.; Fahmi, N.; Stephanopoulos, N.; Liu, Y.; Seeman, N.C.; Yan, H. Tuning the Cavity Size and Chirality of Self-Assembling 3D DNA Crystals. J. Am. Chem. Soc. 2017, 139, 11254–11260. [Google Scholar] [CrossRef]
- Simmons, C.R.; Zhang, F.; Birktoft, J.J.; Qi, X.; Han, D.; Liu, Y.; Sha, R.; Abdallah, H.O.; Hernandez, C.; Ohayon, Y.P.; et al. Construction and Structure Determination of a Three-Dimensional DNA Crystal. J. Am. Chem. Soc. 2016, 138, 10047–10054. [Google Scholar] [CrossRef] [PubMed]
- Simmons, C.R.; MacCulloch, T.; Zhang, F.; Liu, Y.; Stephanopoulos, N.; Yan, H. A Self-Assembled Rhombohedral DNA Crystal Scaffold with Tunable Cavity Sizes and High-Resolution Structural Detail. Angew. Chem. Int. Ed. 2020, 59, 18619–18626. [Google Scholar] [CrossRef]
- Paukstelis, P.J. Three-Dimensional DNA Crystals as Molecular Sieves. J. Am. Chem. Soc. 2006, 128, 6794–6795. [Google Scholar] [CrossRef]
- Geng, C.; Paukstelis, P.J. DNA Crystals as Vehicles for Biocatalysis. J. Am. Chem. Soc. 2014, 136, 7817–7820. [Google Scholar] [CrossRef]
- Martin, T.G.; Bharat, T.A.M.; Joerger, A.C.; Bai, X.-C.; Praetorius, F.; Fersht, A.R.; Dietz, H.; Scheres, S.H.W. Design of a molecular support for cryo-EM structure determination. Proc. Natl. Acad. Sci. USA 2016, 113, E7456–E7463. [Google Scholar] [CrossRef]
- Aksel, T.; Yu, Z.; Cheng, Y.; Douglas, S.M. Molecular goniometers for single-particle cryo-electron microscopy of DNA-binding proteins. Nat. Biotechnol. 2020, 1–9. [Google Scholar] [CrossRef]
- Malo, J.; Mitchell, J.C.; Harris, J.R.; Wille, H.; Sherratt, D.J.; Turberfield, A.J.; Vénien-Bryan, C. Engineering a 2D Protein-DNA Crystal. Angew. Chem. Int. Ed. 2005, 44, 3057–3061. [Google Scholar] [CrossRef] [PubMed]
- Selmi, D.N.; Adamson, R.J.; Attrill, H.; Goddard, A.D.; Gilbert, R.J.C.; Watts, A.; Turberfield, A.J. DNA-Templated Protein Arrays for Single-Molecule Imaging. Nano Lett. 2011, 11, 657–660. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Ye, T.; Su, M.; Zhang, C.; Ribbe, A.E.; Jiang, W.; Mao, C. Hierarchical self-assembly of DNA into symmetric supramolecular polyhedra. Nat. Cell Biol. 2008, 452, 198–201. [Google Scholar] [CrossRef]
- Shih, W.M.; Quispe, J.D.; Joyce, G.F. A 1.7-kilobase single-stranded DNA that folds into a nanoscale octahedron. Nat. Cell Biol. 2004, 427, 618–621. [Google Scholar] [CrossRef]
- Bai, X.-C.; Martin, T.G.; Scheres, S.H.W.; Dietz, H. Cryo-EM structure of a 3D DNA-origami object. Proc. Natl. Acad. Sci. USA 2012, 109, 20012–20017. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.C.; Chen, S.B.; Zhang, S.J.; Sodroski, J.; Yang, Z.Q.; Liu, D.S.; Mao, Y.D. Folding DNA into a Lipid-Conjugated Nanobarrel for Controlled Reconstitution of Membrane Proteins. Angew. Chem. Int. Ed. 2018, 57, 2094–2098. [Google Scholar] [CrossRef]
- Kube, M.; Kohler, F.; Feigl, E.; Nagel-Yüksel, B.; Willner, E.M.; Funke, J.J.; Gerling, T.; Stömmer, P.; Honemann, M.N.; Martin, T.G.; et al. Revealing the structures of megadalton-scale DNA complexes with nucleotide resolution. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Gerling, T.; Kube, M.; Kick, B.; Dietz, H. Sequence-programmable covalent bonding of designed DNA assemblies. Sci. Adv. 2018, 4, eaau1157. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-N.; Kilchherr, F.; Dietz, H.; Bathe, M. Quantitative prediction of 3D solution shape and flexibility of nucleic acid nanostructures. Nucleic Acids Res. 2011, 40, 2862–2868. [Google Scholar] [CrossRef]
- Shaw, A.; Lundin, V.; Petrova, E.; Fördős, F.; Benson, E.; Al-Amin, A.; Herland, A.; Blokzijl, A.; Högberg, B.; Teixeira, A. Spatial control of membrane receptor function using ligand nanocalipers. Nat. Methods 2014, 11, 841–846. [Google Scholar] [CrossRef] [PubMed]
- Veneziano, R.; Moyer, T.J.; Stone, M.B.; Wamhoff, E.-C.; Read, B.J.; Mukherjee, S.; Shepherd, T.R.; Das, J.; Schief, W.R.; Irvine, D.J.; et al. Role of nanoscale antigen organization on B-cell activation probed using DNA origami. Nat. Nanotechnol. 2020, 15, 716–723. [Google Scholar] [CrossRef]
- Fisher, P.D.E.; Shen, Q.; Akpinar, B.; Davis, L.K.; Chung, K.K.H.; Baddeley, D.; Šarić, A.; Melia, T.J.; Hoogenboom, B.W.; Lin, C.; et al. A Programmable DNA Origami Platform for Organizing Intrinsically Disordered Nucleoporins within Nanopore Confinement. ACS Nano 2018, 12, 1508–1518. [Google Scholar] [CrossRef] [PubMed]
- Ketterer, P.; Ananth, A.N.; Trip, D.S.L.; Mishra, A.; Bertosin, E.; Ganji, M.; Van Der Torre, J.; Onck, P.; Dietz, H.; Dekker, C. DNA origami scaffold for studying intrinsically disordered proteins of the nuclear pore complex. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Teller, C.; Willner, I. Organizing protein–DNA hybrids as nanostructures with programmed functionalities. Trends Biotechnol. 2010, 28, 619–628. [Google Scholar] [CrossRef]
- Rajendran, A.; Nakata, E.; Nakano, S.; Morii, T. Nucleic-Acid-Templated Enzyme Cascades. ChemBioChem 2017, 18, 696–716. [Google Scholar] [CrossRef]
- Linko, V.; Nummelin, S.; Aarnos, L.; Tapio, K.; Toppari, J.J.; Kostiainen, M.A. DNA-Based Enzyme Reactors and Systems. Nanomaterials 2016, 6, 139. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.; Hoffecker, I.T.; Smyrlaki, I.; Rosa, J.; Grevys, A.; Bratlie, D.; Sandlie, I.; Michaelsen, T.E.; Andersen, J.T.; Högberg, B. Binding to nanopatterned antigens is dominated by the spatial tolerance of antibodies. Nat. Nanotechnol. 2019, 14, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Kurokawa, T.; Kiyonaka, S.; Nakata, E.; Endo, M.; Koyama, S.; Mori, E.; Tran, N.H.; Dinh, H.; Suzuki, Y.; Hidaka, K.; et al. DNA Origami Scaffolds as Templates for Functional Tetrameric Kir3 K+ Channels. Angew. Chem. Int. Ed. 2018, 57, 2586–2591. [Google Scholar] [CrossRef] [PubMed]
- Rosier, B.J.H.M.; Markvoort, A.J.; Audenis, B.G.; Roodhuizen, J.A.L.; Hamer, A.D.; Brunsveld, L.; De Greef, T.F.A. Proximity-induced caspase-9 activation on a DNA origami-based synthetic apoptosome. Nat. Catal. 2020, 3, 295–306. [Google Scholar] [CrossRef]
- Hellmeier, J.; Platzer, R.; Eklund, A.S.; Schlichthaerle, T.; Karner, A.; Motsch, V.; Schneider, M.C.; Kurz, E.; Bamieh, V.; Brameshuber, M.; et al. DNA origami demonstrate the unique stimulatory power of single pMHCs as T cell antigens. Proc. Natl. Acad. Sci. USA 2021, 118, 2016857118. [Google Scholar] [CrossRef] [PubMed]
- Derr, N.D.; Goodman, B.S.; Jungmann, R.; Leschziner, A.E.; Shih, W.M.; Reck-Peterson, S.L. Tug-of-War in Motor Protein Ensembles Revealed with a Programmable DNA Origami Scaffold. Science 2012, 338, 662–665. [Google Scholar] [CrossRef]
- Hariadi, R.F.; Sommese, R.F.; Adhikari, A.S.; Taylor, R.; Sutton, S.; Spudich, J.; Sivaramakrishnan, S. Mechanical coordination in motor ensembles revealed using engineered artificial myosin filaments. Nat. Nanotechnol. 2015, 10, 696–700. [Google Scholar] [CrossRef]
- Xu, W.; Nathwani, B.; Lin, C.; Wang, J.; Karatekin, E.; Pincet, F.; Shih, W.; Rothman, J.E. A Programmable DNA Origami Platform to Organize SNAREs for Membrane Fusion. J. Am. Chem. Soc. 2016, 138, 4439–4447. [Google Scholar] [CrossRef]
- Wang, X.; Ha, T. Defining Single Molecular Forces Required to Activate Integrin and Notch Signaling. Science 2013, 340, 991–994. [Google Scholar] [CrossRef]
- Zhang, Y.; Ge, C.; Zhu, C.; Salaita, K. DNA-based digital tension probes reveal integrin forces during early cell adhesion. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef]
- Funke, J.J.; Dietz, H. Placing molecules with Bohr radius resolution using DNA origami. Nat. Nanotechnol. 2015, 11, 47–52. [Google Scholar] [CrossRef]
- Nickels, P.C.; Wünsch, B.; Holzmeister, P.; Bae, W.; Kneer, L.M.; Grohmann, D.; Tinnefeld, P.; Liedl, T. Molecular force spectroscopy with a DNA origami–based nanoscopic force clamp. Science 2016, 354, 305–307. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Jiang, S.; Loza, O.; Fahmi, N.E.; Šulc, P.; Stephanopoulos, N. Rapid Photoactuation of a DNA Nanostructure using an Internal Photocaged Trigger Strand. Angew. Chem. Int. Ed. 2018, 57, 9341–9345. [Google Scholar] [CrossRef] [PubMed]
- Marras, A.E.; Zhou, L.; Su, H.-J.; Castro, C.E. Programmable motion of DNA origami mechanisms. Proc. Natl. Acad. Sci. USA 2015, 112, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Marras, A.E.; Huang, C.-M.; Castro, C.E.; Su, H.-J. Paper Origami-Inspired Design and Actuation of DNA Nanomachines with Complex Motions. Small 2018, 14, e1802580. [Google Scholar] [CrossRef]
- Cai, X.; Arias, D.S.; Velazquez, L.R.; Vexler, S.; Bevier, A.L.; Fygenson, D.K. DNA Nunchucks: Nanoinstrumentation for Single-Molecule Measurement of Stiffness and Bending. Nano Lett. 2019, 20, 1388–1395. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, A.M.; Velazquez, L.; Chisenhall, A.; Schiffels, D.; Schulman, R.; Fygenson, D.K. Self-Assembly of Precisely Defined DNA Nanotube Superstructures Using DNA Origami Seeds. Nanoscale 2016, 9, 522–526. [Google Scholar] [CrossRef]
- Le, J.V.; Luo, Y.; Darcy, M.A.; Lucas, C.R.; Goodwin, M.F.; Poirier, M.G.; Castro, C.E. Probing Nucleosome Stability with a DNA Origami Nanocaliper. ACS Nano 2016, 10, 7073–7084. [Google Scholar] [CrossRef]
- Funke, J.J.; Ketterer, P.; Lieleg, C.; Korber, P.; Dietz, H. Exploring Nucleosome Unwrapping Using DNA Origami. Nano Lett. 2016, 16, 7891–7898. [Google Scholar] [CrossRef]
- Funke, J.J.; Ketterer, P.; Lieleg, C.; Schunter, S.; Korber, P.; Dietz, H. Uncovering the forces between nucleosomes using DNA origami. Sci. Adv. 2016, 2, e1600974. [Google Scholar] [CrossRef]
- Iwaki, M.; Wickham, S.F.; Ikezaki, K.; Yanagida, T.; Shih, W.M. A programmable DNA origami nanospring that reveals force-induced adjacent binding of myosin VI heads. Nat. Commun. 2016, 7, 13715. [Google Scholar] [CrossRef]
- Dietz, H.; Douglas, S.M.; Shih, W.M. Folding DNA into Twisted and Curved Nanoscale Shapes. Science 2009, 325, 725–730. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.; Benetatos, P. Inequivalence of fixed-force and fixed-extension statistical ensembles for a flexible polymer tethered to a planar substrate. Soft Matter 2018, 14, 6857–6866. [Google Scholar] [CrossRef]
- Engel, M.C.; Romano, F.; Louis, A.A.; Doye, J.P.K. Measuring Internal Forces in Single-Stranded DNA: Application to a DNA Force Clamp. J. Chem. Theory Comput. 2020, 16, 7764–7775. [Google Scholar] [CrossRef] [PubMed]
- Kolberg, K.; Puettmann, C.; Pardo, A.; Fitting, J.; Barth, S. SNAP-Tag Technology: A General Introduction. Curr. Pharm. Des. 2013, 19, 5406–5413. [Google Scholar] [CrossRef]
- Xu, Y.; Jiang, S.; Simmons, C.R.; Narayanan, R.P.; Zhang, F.; Aziz, A.-M.; Yan, H.; Stephanopoulos, N. Tunable Nanoscale Cages from Self-Assembling DNA and Protein Building Blocks. ACS Nano 2019, 13, 3545–3554. [Google Scholar] [CrossRef] [PubMed]
- Buchberger, A.; Simmons, C.R.; Fahmi, N.E.; Freeman, R.; Stephanopoulos, N. Hierarchical Assembly of Nucleic Acid/Coiled-Coil Peptide Nanostructures. J. Am. Chem. Soc. 2020, 142, 1406–1416. [Google Scholar] [CrossRef]
- Van Buggenum, J.A.G.L.; Gerlach, J.P.; Eising, S.; Schoonen, L.; Van Eijl, R.A.P.M.; Tanis, S.E.J.; Hogeweg, M.; Hubner, N.C.; Van Hest, J.J.; Bonger, K.M.; et al. A covalent and cleavable antibody-DNA conjugation strategy for sensitive protein detection via immuno-PCR. Sci. Rep. 2016, 6, 22675. [Google Scholar] [CrossRef]
- Rosen, C.B.; Kodal, A.L.B.; Nielsen, J.S.; Schaffert, D.H.; Scavenius, C.; Okholm, A.H.; Voigt, N.V.; Enghild, J.J.; Kjems, J.; Tørring, T.; et al. Template-directed covalent conjugation of DNA to native antibodies, transferrin and other metal-binding proteins. Nat. Chem. 2014, 6, 804–809. [Google Scholar] [CrossRef]
- Ouyang, X.; De Stefano, M.; Krissanaprasit, A.; Kodal, A.L.B.; Rosen, C.B.; Liu, T.; Helmig, S.; Fan, C.; Gothelf, K.V. Docking of Antibodies into the Cavities of DNA Origami Structures. Angew. Chem. Int. Ed. 2017, 56, 14423–14427. [Google Scholar] [CrossRef]
- Sprengel, A.; Lill, P.; Stegemann, P.; Bravo-Rodriguez, K.; Schöneweiß, E.-C.; Merdanovic, M.; Gudnason, D.; Aznauryan, M.; Gamrad, L.; Barcikowski, S.; et al. Tailored protein encapsulation into a DNA host using geometrically organized supramolecular interactions. Nat. Commun. 2017, 8, 14472. [Google Scholar] [CrossRef]
- Das, R.; Baker, D. Macromolecular Modeling with Rosetta. Annu. Rev. Biochem. 2008, 77, 363–382. [Google Scholar] [CrossRef] [PubMed]
- Douglas, S.M.; Marblestone, A.H.; Teerapittayanon, S.; Vazquez, A.; Church, G.M.; Shih, W.M. Rapid prototyping of 3D DNA-origami shapes with caDNAno. Nucleic Acids Res. 2009, 37, 5001–5006. [Google Scholar] [CrossRef]
- Jun, H.; Wang, X.; Bricker, W.P.; Jackson, S.; Bathe, M. Rapid Prototyping of Wireframe Scaffolded DNA Origami using ATHENA. bioRxiv 2020. [Google Scholar] [CrossRef]
- de Llano, E.; Miao, H.; Ahmadi, Y.; Wilson, A.J.; Beeby, M.; Viola, I.; Barisic, I. Adenita: Interactive 3D modelling and visualization of DNA nanostructures. Nucleic Acids Res. 2020, 48, 8269–8275. [Google Scholar] [CrossRef]
- Poppleton, E.; Bohlin, J.; Matthies, M.; Sharma, S.; Zhang, F.; Šulc, P. Design, optimization and analysis of large DNA and RNA nanostructures through interactive visualization, editing and molecular simulation. Nucleic Acids Res. 2020, 48, e72. [Google Scholar] [CrossRef] [PubMed]
- Doye, J.P.K.; Ouldridge, T.E.; Louis, A.A.; Romano, F.; Šulc, P.; Matek, C.; Snodin, B.E.K.; Rovigatti, L.; Schreck, J.S.; Harrison, R.M.; et al. Coarse-graining DNA for simulations of DNA nanotechnology. Phys. Chem. Chem. Phys. 2013, 15, 20395–20414. [Google Scholar] [CrossRef] [PubMed]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Ivani, I.; Dans, P.D.; Noy, A.; Pérez, A.; Faustino, I.; Hospital, A.; Walther, J.; Andrio, P.; Goñi, R.; Balanceanu, A.; et al. Parmbsc1: A refined force field for DNA simulations. Nat. Methods 2016, 13, 55–58. [Google Scholar] [CrossRef]
- Kührová, P.; Mlýnský, V.; Zgarbová, M.; Krepl, M.; Bussi, G.; Best, R.B.; Otyepka, M.; Šponer, J.; Banáš, P. Improving the Performance of the Amber RNA Force Field by Tuning the Hydrogen-Bonding Interactions. J. Chem. Theory Comput. 2019, 15, 3288–3305. [Google Scholar] [CrossRef]
- Maffeo, C.; Yoo, J.; Aksimentiev, A. De novoreconstruction of DNA origami structures through atomistic molecular dynamics simulation. Nucleic Acids Res. 2016, 44, 3013–3019. [Google Scholar] [CrossRef]
- Yoo, J.; Winogradoff, D.; Aksimentiev, A. Molecular dynamics simulations of DNA-DNA and DNA-protein interactions. Curr. Opin. Struct. Biol. 2020, 64, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Honorato, R.V.; Roel-Touris, J.; Bonvin, A.M.J.J. MARTINI-Based Protein-DNA Coarse-Grained HADDOCKing. Front. Mol. Biosci. 2019, 6, 6. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Bueno, C.; Schafer, N.P.; Moller, J.; Jin, S.; Chen, X.; Chen, M.; Gu, X.; Davtyan, A.; de Pablo, J.J.; et al. OpenAWSEM with Open3SPN2: A fast, flexible, and accessible framework for large-scale coarse-grained biomolecular simulations. PLoS Comput. Biol. 2021, 17, e1008308. [Google Scholar] [CrossRef] [PubMed]
- Procyk, J.; Poppleton, E.; Šulc, P. Coarse-grained nucleic acid–protein model for hybrid nanotechnology. Soft Matter 2020. [Google Scholar] [CrossRef]
- Snodin, B.E.K.; Randisi, F.; Mosayebi, M.; Šulc, P.; Schreck, J.S.; Romano, F.; Ouldridge, T.E.; Tsukanov, R.; Nir, E.; Louis, A.A.; et al. Introducing improved structural properties and salt dependence into a coarse-grained model of DNA. J. Chem. Phys. 2015, 142, 234901. [Google Scholar] [CrossRef]
- Šulc, P.; Romano, F.; Ouldridge, T.E.; Rovigatti, L.; Doye, J.P.K.; Louis, A.A. Sequence-dependent thermodynamics of a coarse-grained DNA model. J. Chem. Phys. 2012, 137, 135101. [Google Scholar] [CrossRef]
- Ouldridge, T.E.; Louis, A.A.; Doye, J.P.K. Structural, mechanical, and thermodynamic properties of a coarse-grained DNA model. J. Chem. Phys. 2011, 134, 085101. [Google Scholar] [CrossRef] [PubMed]
- Xia, K.; Opron, K.; Wei, G.-W. Multiscale Gaussian network model (mGNM) and multiscale anisotropic network model (mANM). J. Chem. Phys. 2015, 143, 204106. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).