
The Influence of Transitional Metal Dopants on Reducing Chlorine Evolution during the Electrolysis of Raw Seawater

 , ,
, , 
Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Electrode Fabrication and Structure
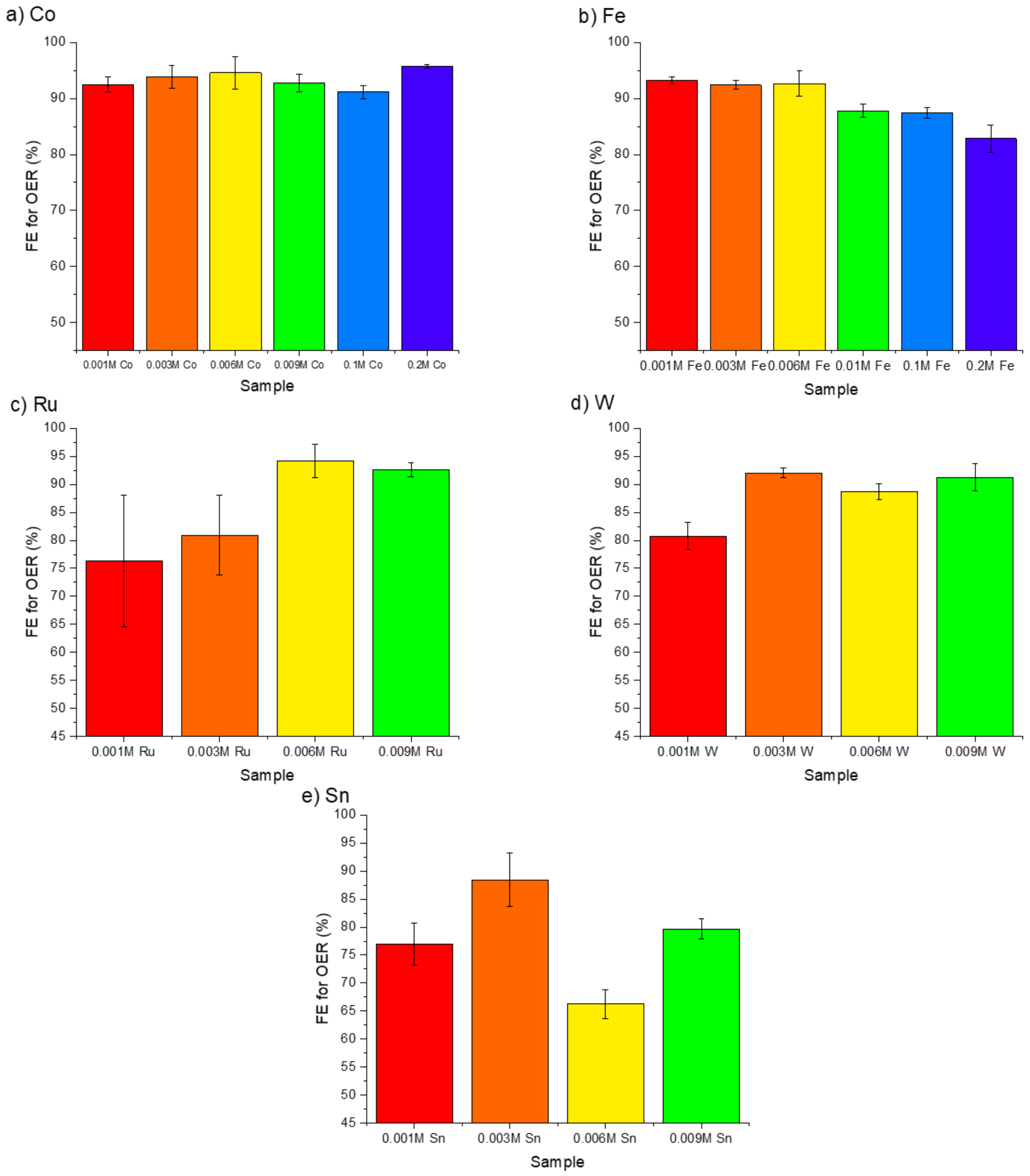
3.2. OER Efficiency in Saline and Seawater Environments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tong, W.; Forster, M.; Dionigi, F.; Dresp, S.; Sadeghi Erami, R.; Strasser, P.; Cowan, A.J.; Farràs, P. Electrolysis of low-grade and saline surface water. Nat. Energy 2020, 5, 367–377. [Google Scholar] [CrossRef]
- Webber, M.E. The water intensity of the transitional hydrogen economy. Environ. Res. Lett. 2007, 2, 034007. [Google Scholar] [CrossRef] [Green Version]
- Dresp, S.R.; Dionigi, F.; Klingenhof, M.; Strasser, P. Direct electrolytic splitting of seawater: Opportunities and challenges. ACS Energy Lett. 2019, 4, 933–942. [Google Scholar] [CrossRef]
- Hansen, H.A.; Man, I.A.; Studt, F.; Abild-Pedersen, F.; Bligaard, T.; Rossmeisl, J. Electrochemical chlorine evolution at rutile oxide (110) surfaces. Phys. Chem. Chem. Phys. 2010, 12, 283–290. [Google Scholar] [CrossRef]
- El-Moneim, A.A.; Bhattarai, J.; Kato, Z.; Izumiya, K.; Kumagai, N.; Hashimoto, K. Mn-Mo-Sn oxide anodes for oxygen evolution in seawater electrolysis for hydrogen production. ECS Trans. 2010, 25, 127. [Google Scholar] [CrossRef]
- El-Moneim, A.A.; Kumagai, N.; Asami, K.; Hashimoto, K. Nanocrystalline manganese-molybdenum-tungsten oxide anodes for oxygen evolution in acidic seawater electrolysis. Mater. Trans. 2005, 46, 309–316. [Google Scholar] [CrossRef] [Green Version]
- Vos, J.; Koper, M. Measurement of competition between oxygen evolution and chlorine evolution using rotating ring-disk electrode voltammetry. J. Electroanal. Chem. 2018, 819, 260–268. [Google Scholar] [CrossRef]
- Vos, J.G.; Wezendonk, T.A.; Jeremiasse, A.W.; Koper, M.T. MnOx/IrOx as selective oxygen evolution electrocatalyst in acidic chloride solution. J. Am. Chem. Soc. 2018, 140, 10270–10281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amikam, G.; Nativ, P.; Gendel, Y. Chlorine-free alkaline seawater electrolysis for hydrogen production. Int. J. Hydrogen Energy 2018, 43, 6504–6514. [Google Scholar] [CrossRef]
- Kuang, Y.; Kenney, M.J.; Meng, Y.; Hung, W.-H.; Liu, Y.; Huang, J.E.; Prasanna, R.; Li, P.; Li, Y.; Wang, L. Solar-driven, highly sustained splitting of seawater into hydrogen and oxygen fuels. Proc. Natl. Acad. Sci. USA 2019, 116, 6624–6629. [Google Scholar] [CrossRef] [Green Version]
- Juodkazytė, J.; Šebeka, B.; Savickaja, I.; Petrulevičienė, M.; Butkutė, S.; Jasulaitienė, V.; Selskis, A.; Ramanauskas, R. Electrolytic splitting of saline water: Durable nickel oxide anode for selective oxygen evolution. Int. J. Hydrogen Energy 2019, 44, 5929–5939. [Google Scholar] [CrossRef]
- Dresp, S.R.; Dionigi, F.; Klingenhof, M.; Merzdorf, T.; Schmies, H.; Drnec, J.; Poulain, A.; Strasser, P. Molecular Understanding of the Impact of Saline Contaminants and Alkaline pH on NiFe Layered Double Hydroxide Oxygen Evolution Catalysts. ACS Catal. 2021, 11, 6800–6809. [Google Scholar] [CrossRef]
- Karlsson, R.K.; Cornell, A. Selectivity between oxygen and chlorine evolution in the chlor-alkali and chlorate processes. Chem. Rev. 2016, 116, 2982–3028. [Google Scholar] [CrossRef]
- Exner, K.S.; Over, H. Beyond the rate-determining step in the oxygen evolution reaction over a single-crystalline IrO2 (110) model electrode: Kinetic scaling relations. ACS Catal. 2019, 9, 6755–6765. [Google Scholar] [CrossRef]
- Exner, K.S. Controlling stability and selectivity in the competing chlorine and oxygen evolution reaction over transition metal oxide electrodes. ChemElectroChem 2019, 6, 3401–3409. [Google Scholar] [CrossRef]
- Zheng, J. Seawater splitting for high-efficiency hydrogen evolution by alloyed PtNix electrocatalysts. Appl. Surf. Sci. 2017, 413, 360–365. [Google Scholar] [CrossRef]
- Zheng, J. Pt-free NiCo electrocatalysts for oxygen evolution by seawater splitting. Electrochim. Acta 2017, 247, 381–391. [Google Scholar] [CrossRef]
- Cheng, F.; Feng, X.; Chen, X.; Lin, W.; Rong, J.; Yang, W. Synergistic action of Co-Fe layered double hydroxide electrocatalyst and multiple ions of sea salt for efficient seawater oxidation at near-neutral pH. Electrochim. Acta 2017, 251, 336–343. [Google Scholar] [CrossRef]
- Hsu, G.-S.W.; Lu, Y.-F.; Hsu, S.-Y. Effects of electrolysis time and electric potential on chlorine generation of electrolyzed deep ocean water. J. Food Drug Anal. 2017, 25, 759–765. [Google Scholar] [CrossRef] [PubMed]
- Fujimura, K.; Izumiya, K.; Kawashima, A.; Akiyama, E.; Habazaki, H.; Kumagai, N.; Hashimoto, K. Anodically deposited manganese-molybdenum oxide anodes with high selectivity for evolving oxygen in electrolysis of seawater. J. Appl. Electrochem. 1999, 29, 769–775. [Google Scholar] [CrossRef]
- Fujimura, K.; Matsui, T.; Izumiya, K.; Kumagai, N.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Asami, K.; Hashimoto, K. Oxygen evolution on manganese–molybdenum oxide anodes in seawater electrolysis. Mater. Sci. Eng. A 1999, 267, 254–259. [Google Scholar] [CrossRef]
- El-Moneim, A. Mn–Mo–W-oxide anodes for oxygen evolution during seawater electrolysis for hydrogen production: Effect of repeated anodic deposition. Int. J. Hydrogen Energy 2011, 36, 13398–13406. [Google Scholar] [CrossRef]
- Dionigi, F.; Reier, T.; Pawolek, Z.; Gliech, M.; Strasser, P. Design criteria, operating conditions, and nickel-iron hydroxide catalyst materials for selective seawater electrolysis. ChemSusChem 2016, 9, 962–972. [Google Scholar] [CrossRef]
- Vesborg, P.C.; Jaramillo, T.F. Addressing the terawatt challenge: Scalability in the supply of chemical elements for renewable energy. Rsc Adv. 2012, 2, 7933–7947. [Google Scholar] [CrossRef] [Green Version]
- Kato, Z.; Sato, M.; Sasaki, Y.; Izumiya, K.; Kumagai, N.; Hashimoto, K. Electrochemical characterization of degradation of oxygen evolution anode for seawater electrolysis. Electrochim. Acta 2014, 116, 152–157. [Google Scholar] [CrossRef]
- Fujimura, K.; Matsui, T.; Habazaki, H.; Kawashima, A.; Kumagai, N.; Hashimoto, K. The durability of manganese–molybdenum oxide anodes for oxygen evolution in seawater electrolysis. Electrochim. Acta 2000, 45, 2297–2303. [Google Scholar] [CrossRef]
- Tian, L.; Li, Z.; Xu, X.; Zhang, C. Advances in noble metal (Ru, Rh, and Ir) doping for boosting water splitting electrocatalysis. J. Mater. Chem. A 2021, 9, 13459–13470. [Google Scholar] [CrossRef]
- Gwag, E.H.; Moon, S.Y.; Mondal, I.; Park, J.Y. Influence of carbon doping concentration on photoelectrochemical activity of TiO 2 nanotube arrays under water oxidation. Catal. Sci. Technol. 2019, 9, 688–694. [Google Scholar] [CrossRef]
- Moon, S.Y.; Gwag, E.H.; Park, J.Y. Hydrogen Generation on Metal/Mesoporous Oxides: The Effects of Hierarchical Structure, Doping, and Co-catalysts. Energy Technol. 2018, 6, 459–469. [Google Scholar] [CrossRef]
- Holt-Hindle, P.; Nigro, S.; Asmussen, M.; Chen, A. Amperometric glucose sensor based on platinum–iridium nanomaterials. Electrochem. Commun. 2008, 10, 1438–1441. [Google Scholar] [CrossRef]
- Subow, N. Oceanographical Tables; USSR. Oceanogr. Inst. Hydro-Meteorol. Com Mosc.: Moscow, Russia, 1931; Volume 208. [Google Scholar]
- Kim, K.; Winograd, N. X-ray photoelectron spectroscopic studies of ruthenium-oxygen surfaces. J. Catal. 1974, 35, 66–72. [Google Scholar] [CrossRef]
- Yu, J.; He, Q.; Yang, G.; Zhou, W.; Shao, Z.; Ni, M. Recent Advances and Prospective in Ruthenium-Based Materials for Electrochemical Water Splitting. ACS Catal. 2019, 9, 9973–10011. [Google Scholar] [CrossRef]
- Exner, K.S. Beyond thermodynamic-based material-screening concepts: Kinetic scaling relations exemplified by the chlorine evolution reaction over transition-metal oxides. Electrochim. Acta 2020, 334, 135555. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis in the anodic evolution of oxygen and chlorine. Electrochim. Acta 1984, 29, 1503–1512. [Google Scholar] [CrossRef]
- Izumiya, K.; Akiyama, E.; Habazaki, H.; Kawashima, A.; Asami, K.; Hashimoto, K.; Kumagai, N. Surface activation of manganese oxide electrode for oxygen evolution from seawater. J. Appl. Electrochem. 1997, 27, 1362–1368. [Google Scholar] [CrossRef]
- Abe, H.; Murakami, A.; Tsunekawa, S.; Okada, T.; Wakabayashi, T.; Yoshida, M.; Nakayama, M. Selective Catalyst for Oxygen Evolution in Neutral Brine Electrolysis: An Oxygen-Deficient Manganese Oxide Film. ACS Catal. 2021, 11, 6390–6397. [Google Scholar] [CrossRef]
- Exner, K.S. Design criteria for the competing chlorine and oxygen evolution reactions: Avoid the OCl adsorbate to enhance chlorine selectivity. Phys. Chem. Chem. Phys. 2020, 22, 22451–22458. [Google Scholar] [CrossRef]
- Keane, T.P.; Nocera, D.G. Selective Production of Oxygen from Seawater by Oxidic Metallate Catalysts. ACS Omega 2019, 4, 12860–12864. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Davis, J.T.; Harvey, A.D.; Esposito, D.V. Framework for evaluating the performance limits of membraneless electrolyzers. Energy Environ. Sci. 2020, 13, 3663–3678. [Google Scholar] [CrossRef]
- Hadikhani, P.; Hashemi, S.M.H.; Schenk, S.A.; Psaltis, D. A membrane-less electrolyzer with porous walls for high throughput and pure hydrogen production. Sustain. Energy Fuels 2021, 5, 2419–2432. [Google Scholar] [CrossRef]
- Abbaspour, A.; Esmaeilbeig, A.R.; Jarrahpour, A.A.; Khajeh, B.; Kia, R. Aluminium(III)-selective electrode based on a newly synthesized tetradentate Schiff base. Talanta 2002, 58, 397–403. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spectrum | O (at%) | Mn (at%) | Mo (at%) | Ru (at%) |
|---|---|---|---|---|
| 1 | 58.23 | 41.49 | 0.28 | 0.00 |
| 2 | 58.17 | 41.29 | 0.40 | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adiga, P.; Doi, N.; Wong, C.; Santosa, D.M.; Kuo, L.-J.; Gill, G.A.; Silverstein, J.A.; Avalos, N.M.; Crum, J.V.; Engelhard, M.H.; et al. The Influence of Transitional Metal Dopants on Reducing Chlorine Evolution during the Electrolysis of Raw Seawater. Appl. Sci. 2021, 11, 11911. https://doi.org/10.3390/app112411911
Adiga P, Doi N, Wong C, Santosa DM, Kuo L-J, Gill GA, Silverstein JA, Avalos NM, Crum JV, Engelhard MH, et al. The Influence of Transitional Metal Dopants on Reducing Chlorine Evolution during the Electrolysis of Raw Seawater. Applied Sciences. 2021; 11(24):11911. https://doi.org/10.3390/app112411911
Chicago/Turabian StyleAdiga, Prajwal, Nathan Doi, Cindy Wong, Daniel M. Santosa, Li-Jung Kuo, Gary A. Gill, Joshua A. Silverstein, Nancy M. Avalos, Jarrod V. Crum, Mark H. Engelhard, and et al. 2021. "The Influence of Transitional Metal Dopants on Reducing Chlorine Evolution during the Electrolysis of Raw Seawater" Applied Sciences 11, no. 24: 11911. https://doi.org/10.3390/app112411911
APA StyleAdiga, P., Doi, N., Wong, C., Santosa, D. M., Kuo, L.-J., Gill, G. A., Silverstein, J. A., Avalos, N. M., Crum, J. V., Engelhard, M. H., Stoerzinger, K. A., & Asmussen, R. M. (2021). The Influence of Transitional Metal Dopants on Reducing Chlorine Evolution during the Electrolysis of Raw Seawater. Applied Sciences, 11(24), 11911. https://doi.org/10.3390/app112411911