
Calcium and Strontium Coordination Polymers as Controlled Delivery Systems of the Anti-Osteoporosis Drug Risedronate and the Augmenting Effect of Solubilizers


 ,
,  and
and
Abstract
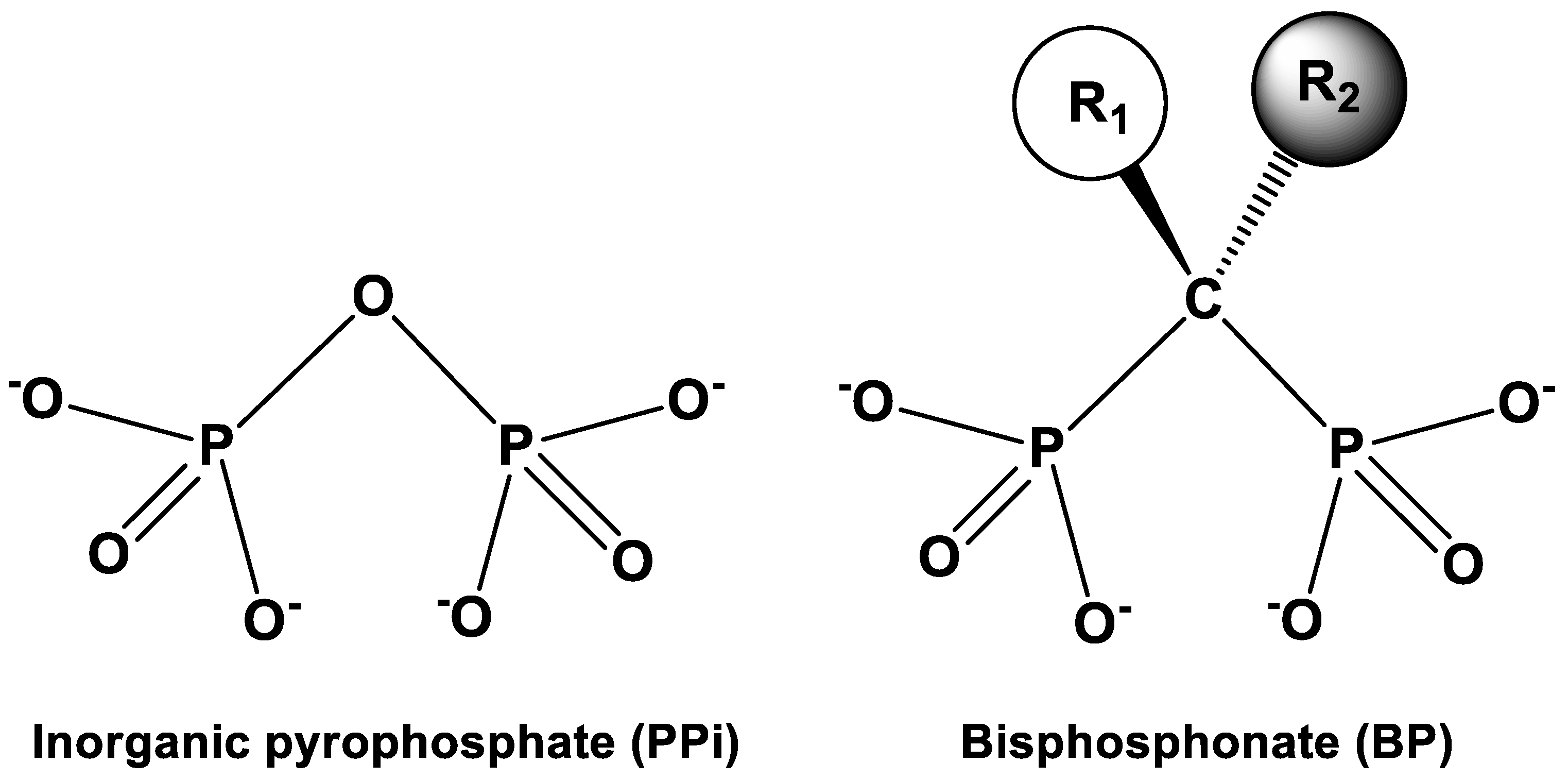
:
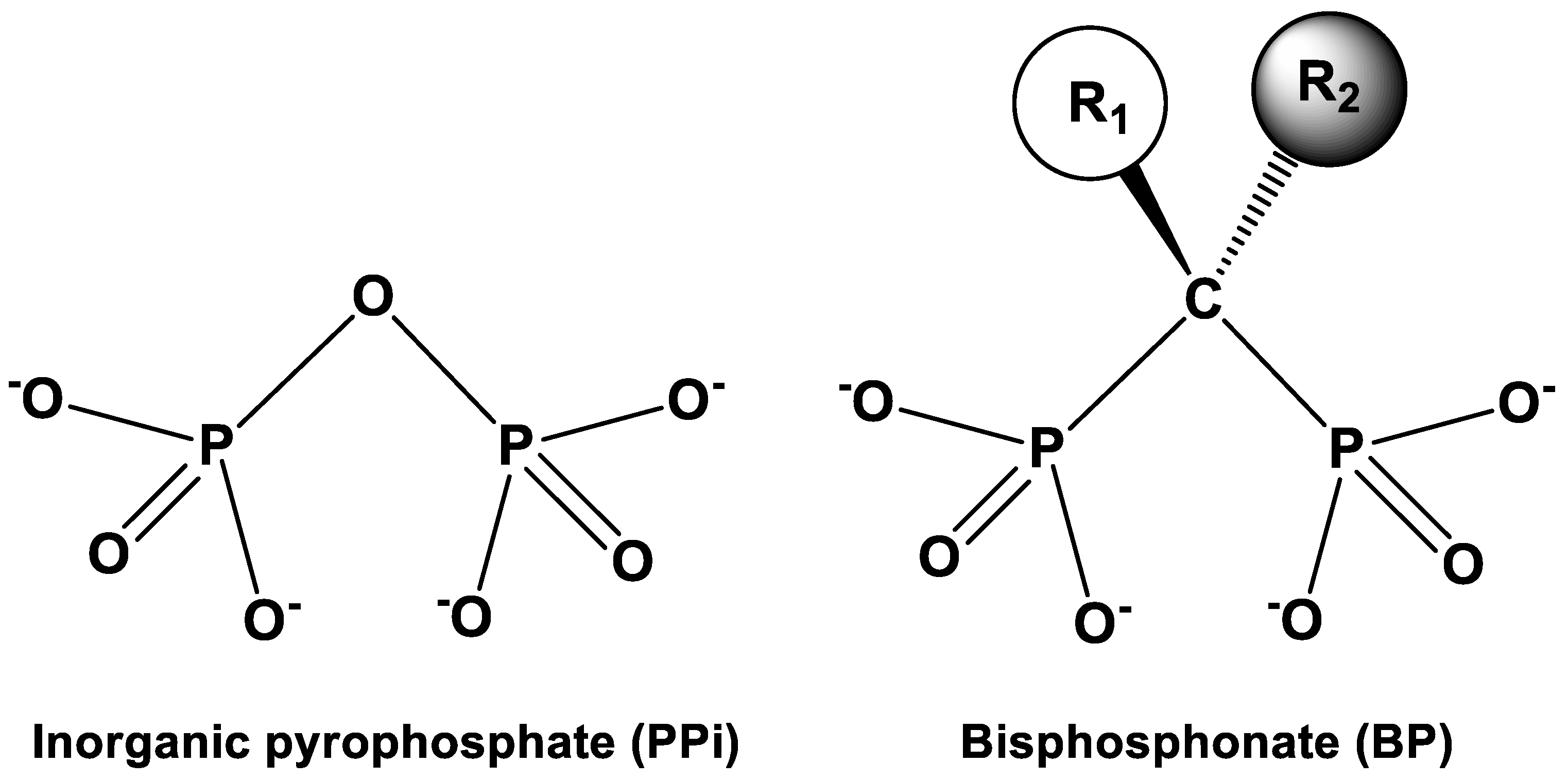
1. Introduction
2. Materials and Methods
2.1. Instrumentation
2.2. Materials
2.3. Synthetic Protocols
2.4. Preparation of Tablets for Drug Release
2.5. Quantification of Drug Controlled Release
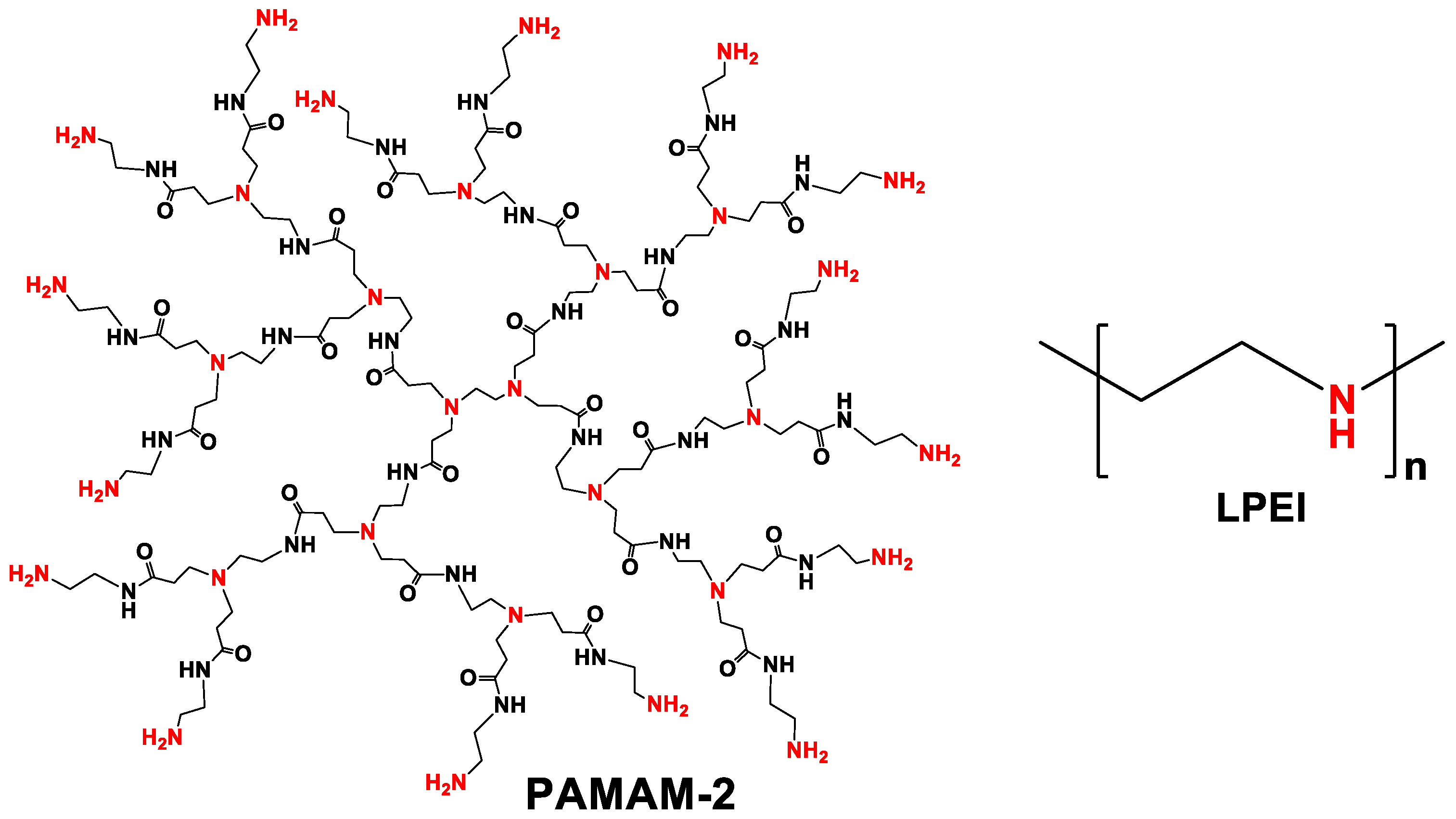
2.6. Quantification of Drug Controlled Release in the Presence of PAMAM-2 Dendrimer
2.7. Quantification of Drug Controlled Release in the Presence of LPEI
3. Results
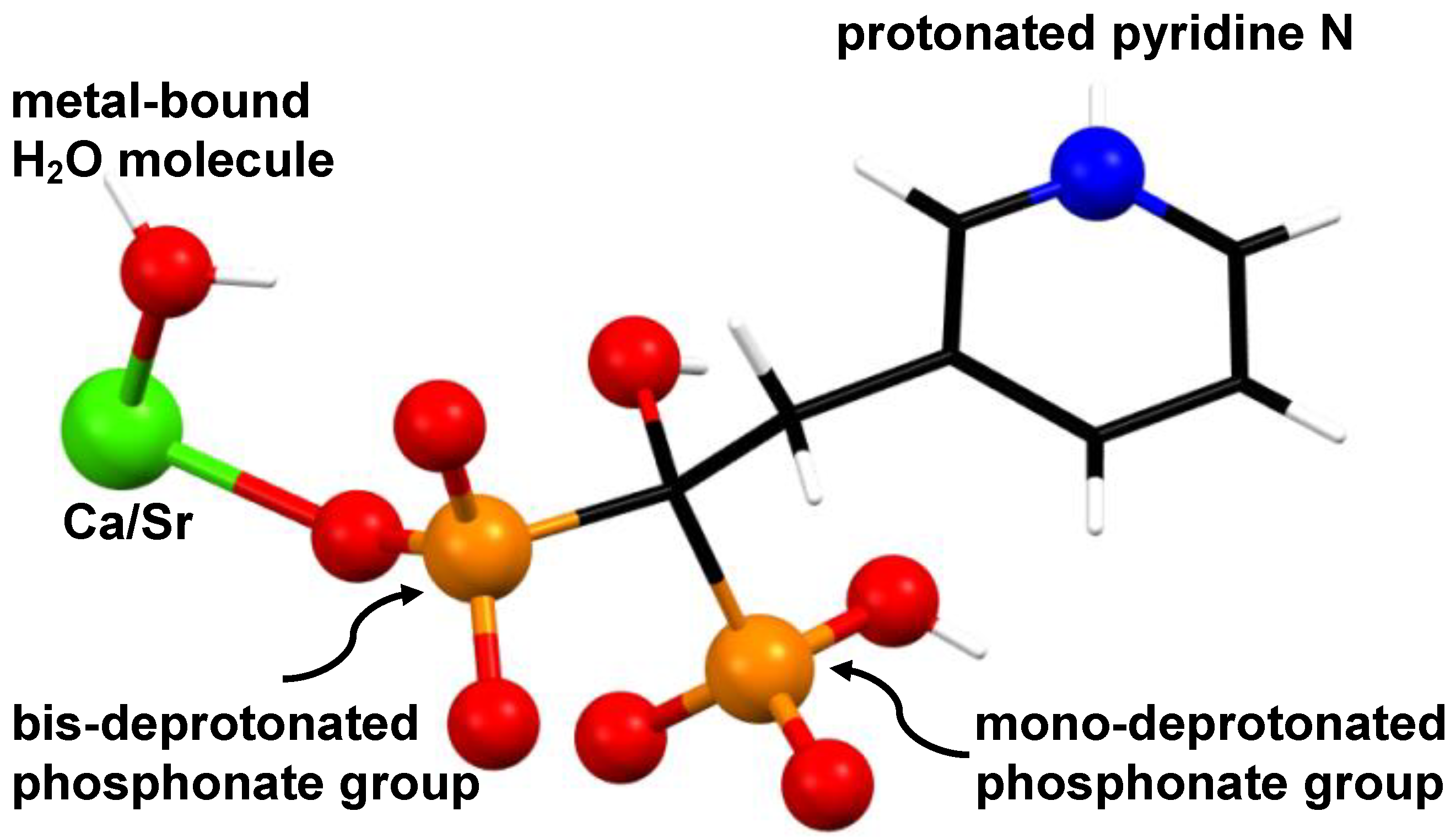
3.1. Synthesis and Characterization of (Ca/Sr)–RIS Compounds
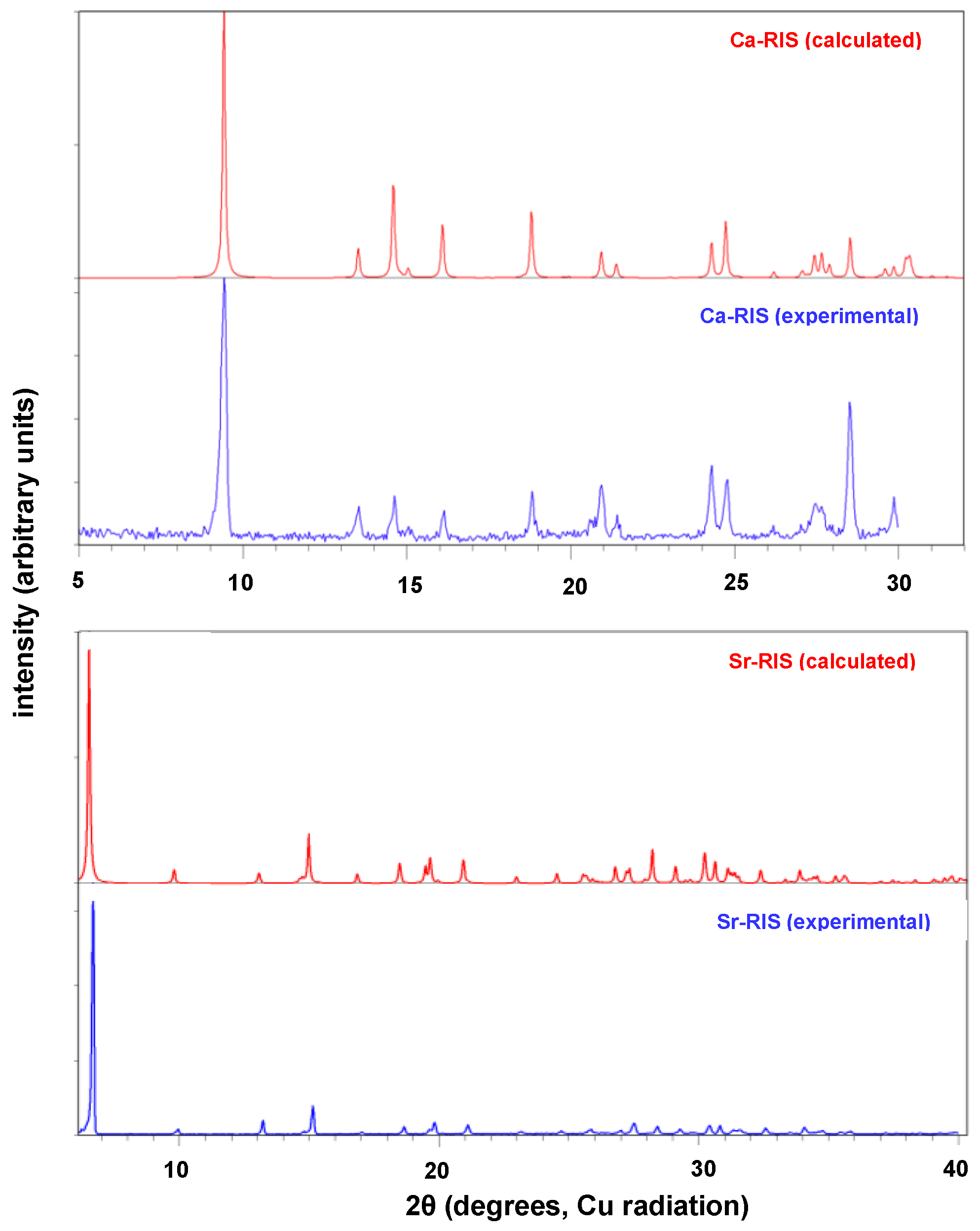
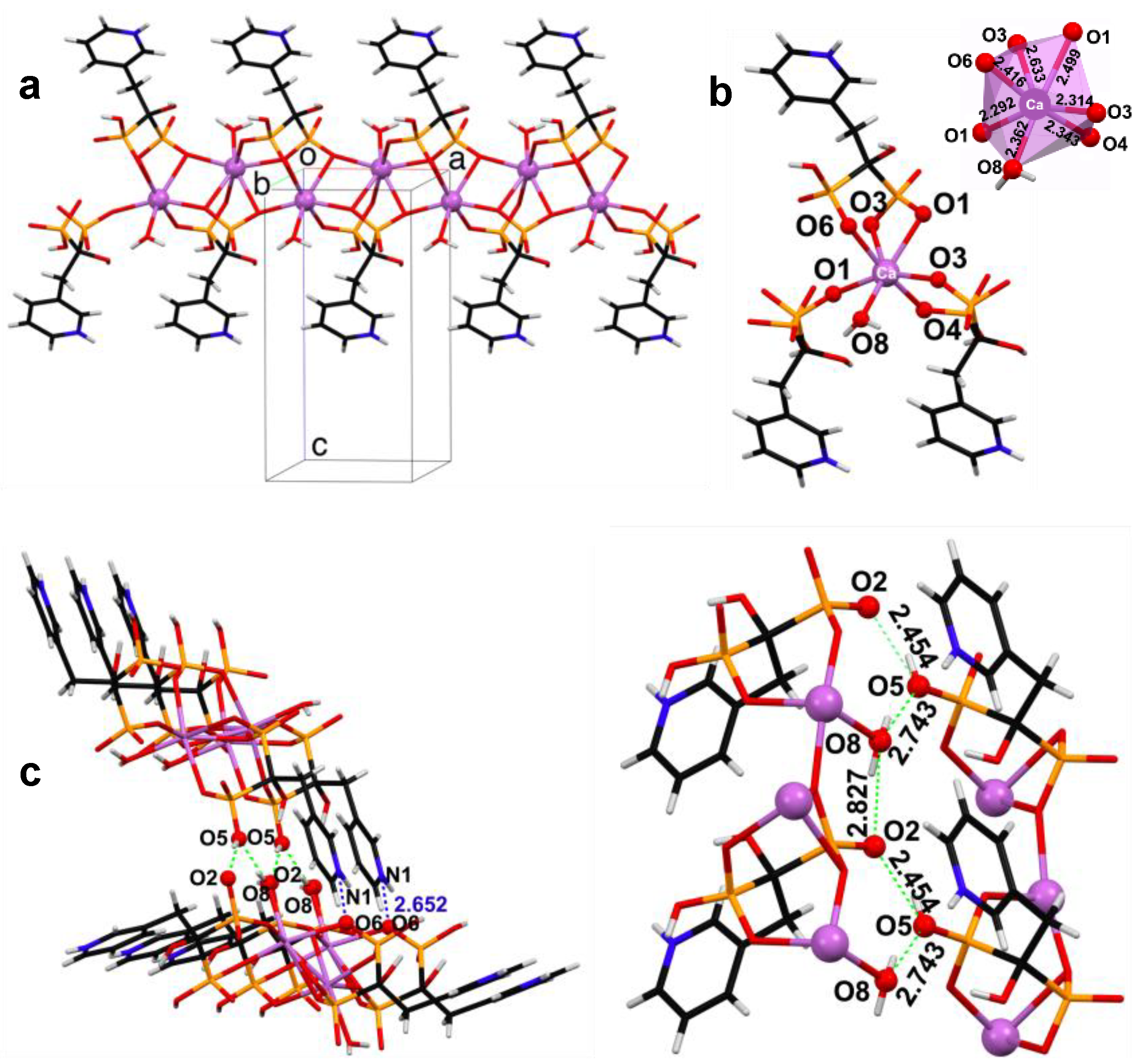
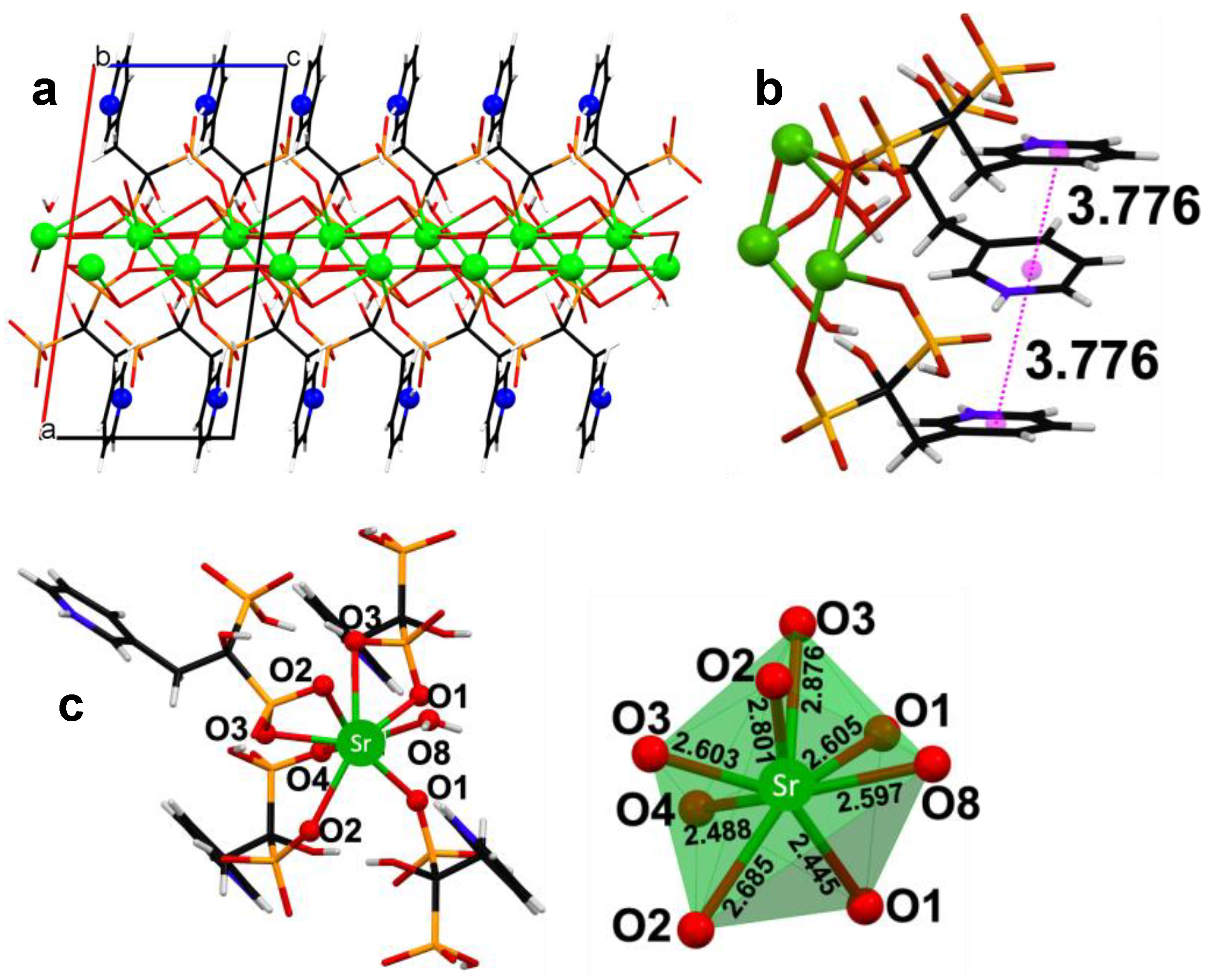
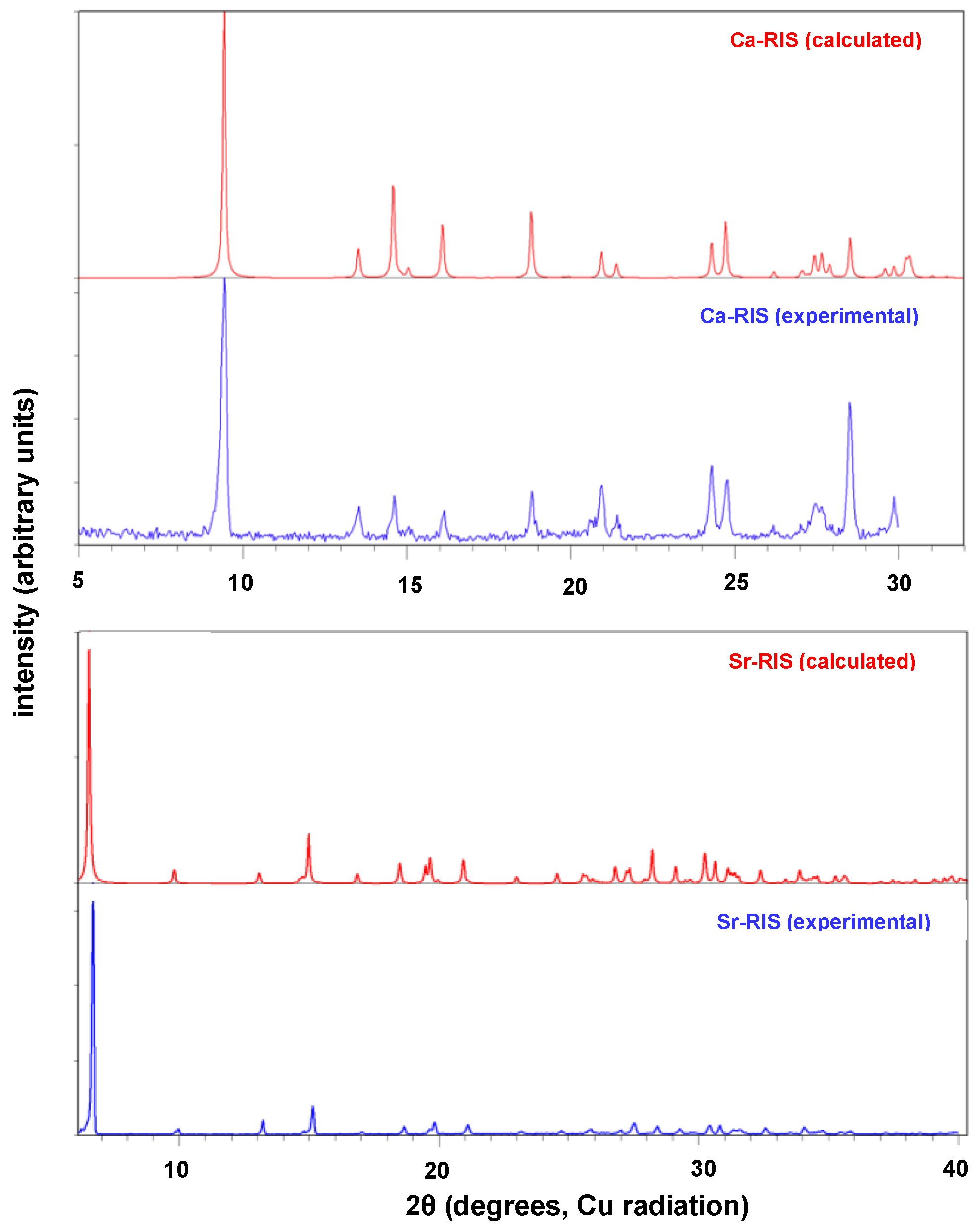
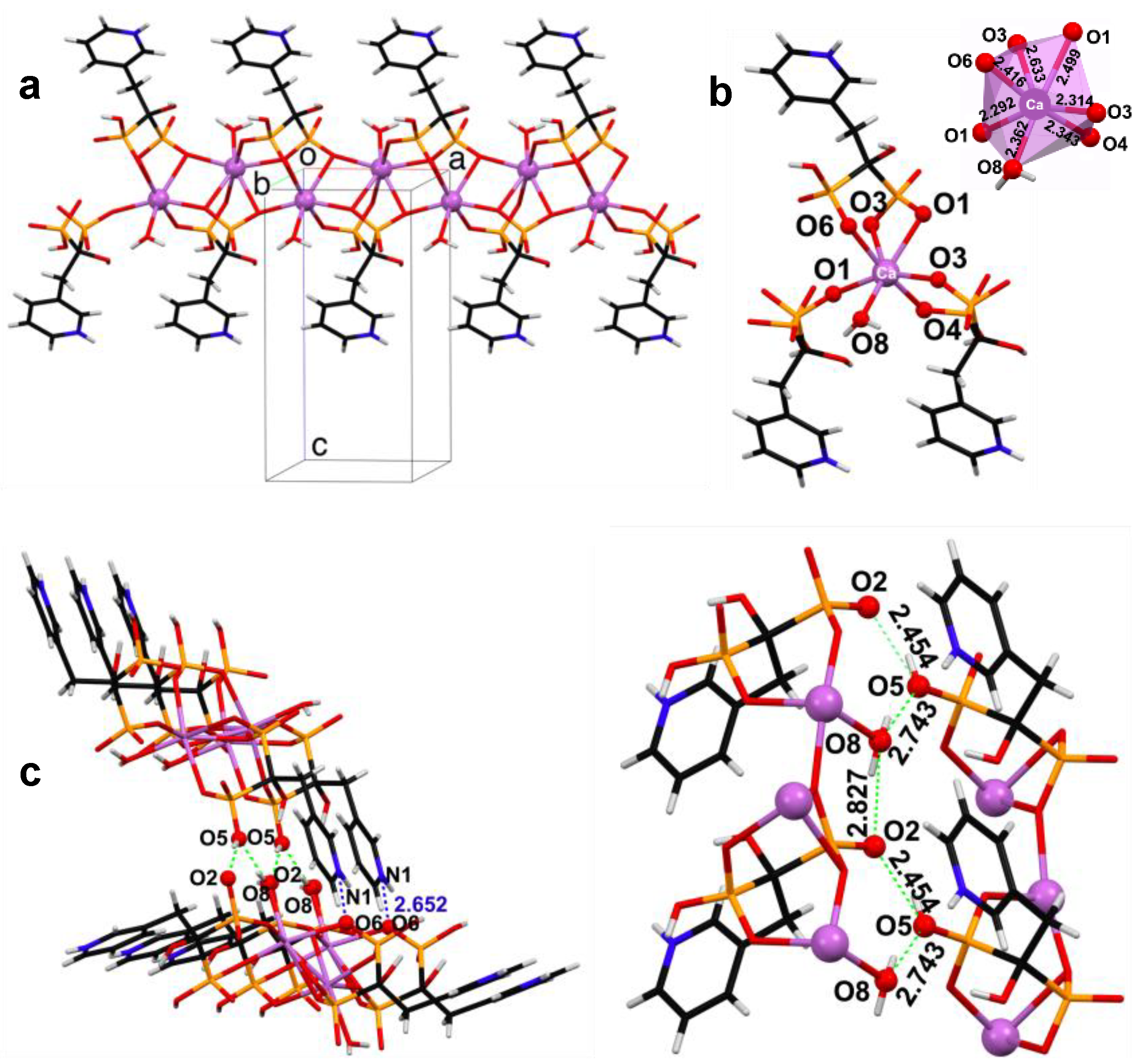
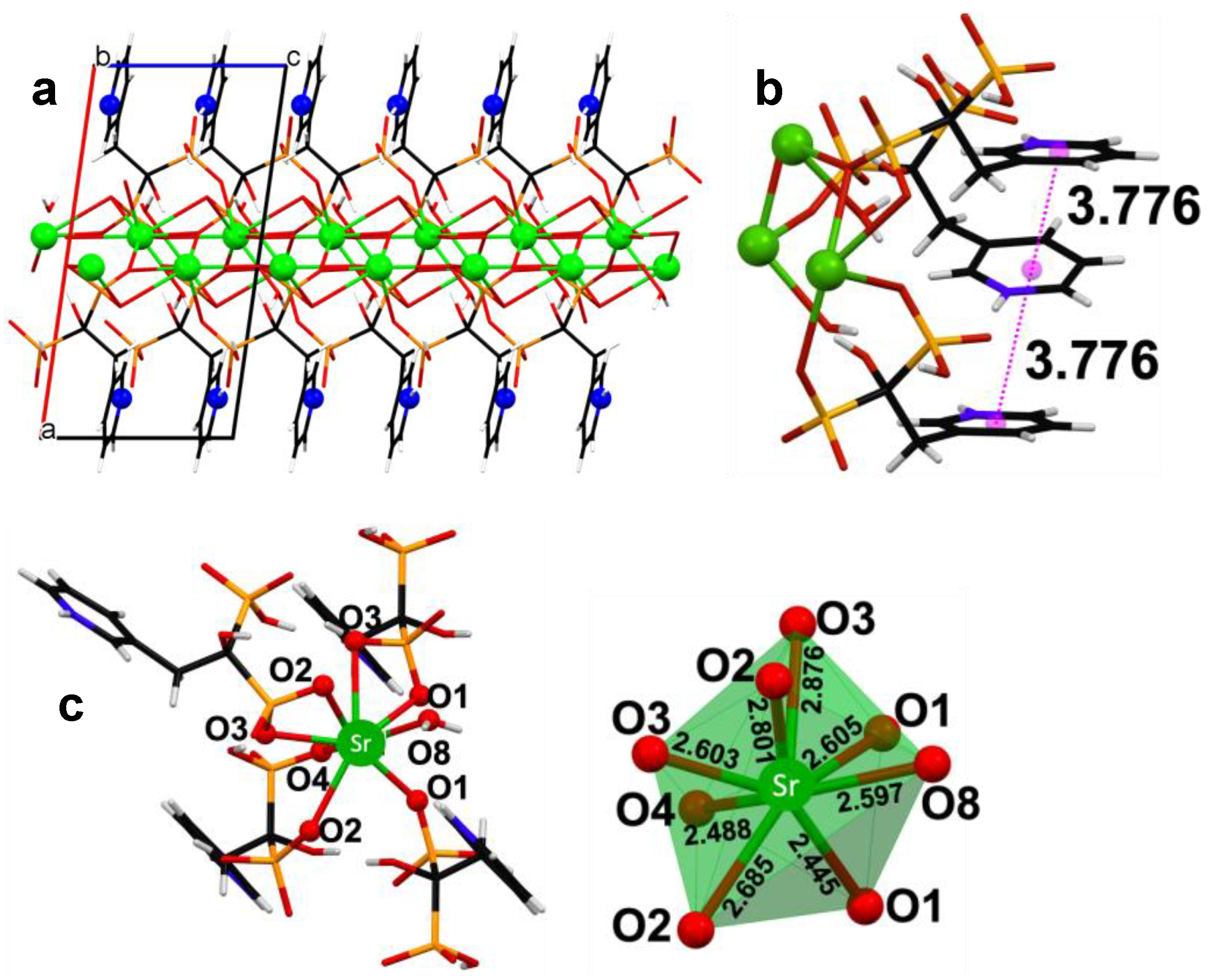
3.2. Powder and Single Crystal X-ray Diffraction Studies of (Ca/Sr)–RIS Compounds
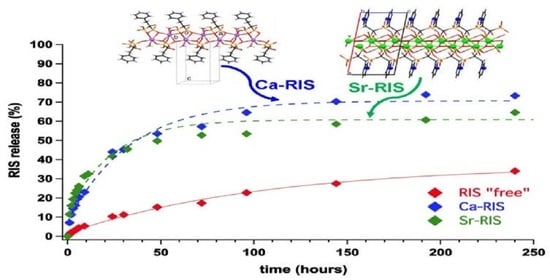
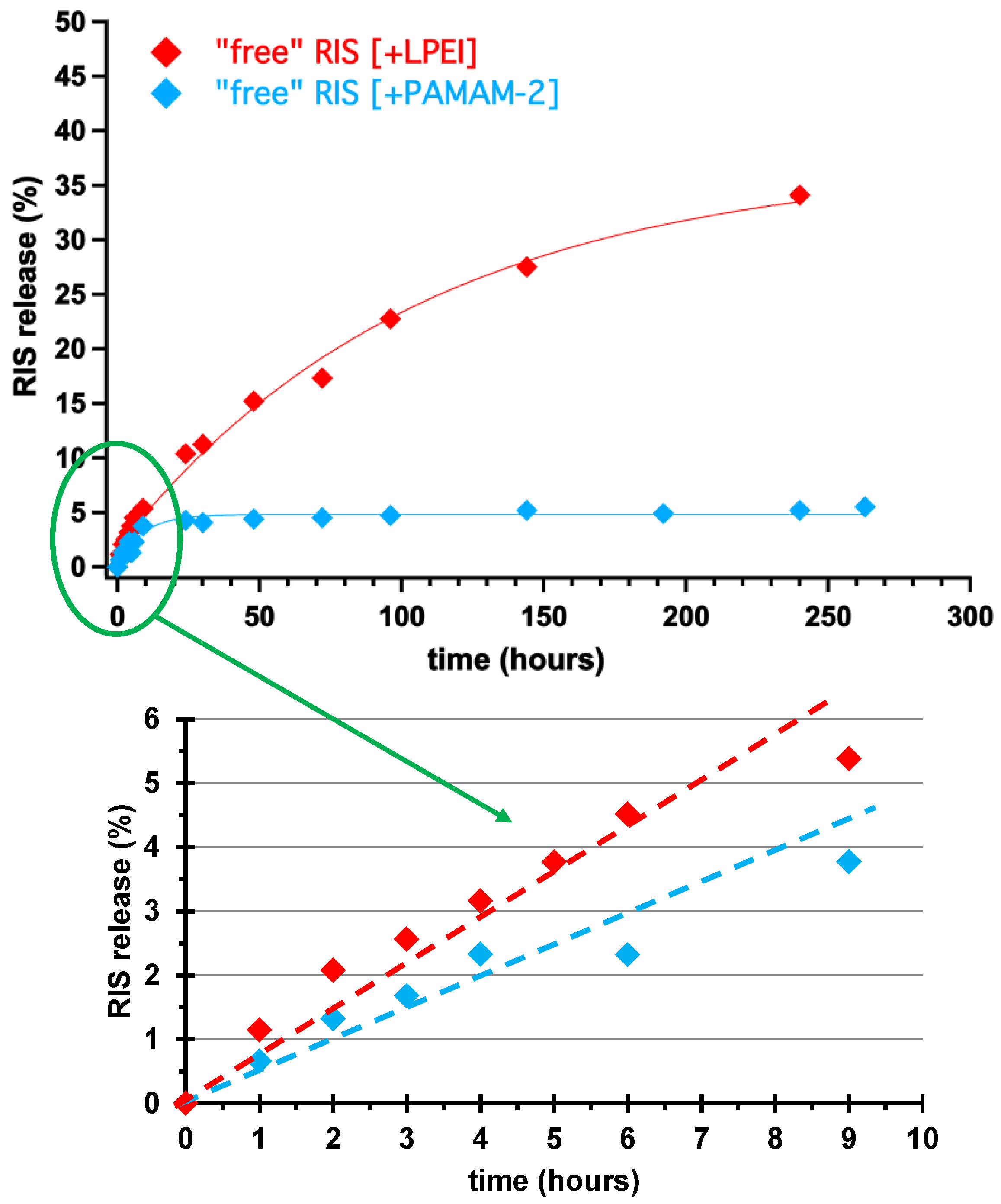
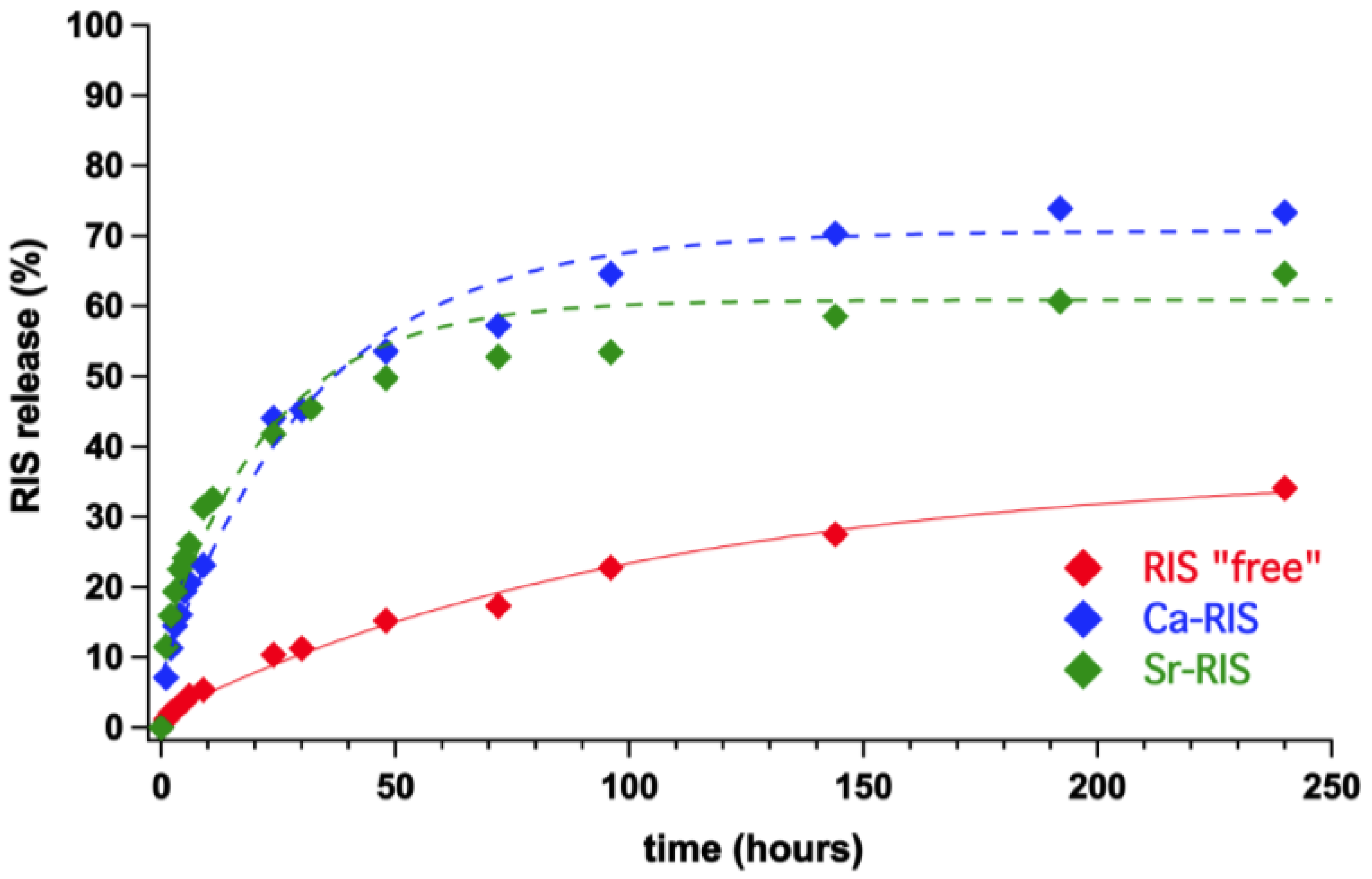
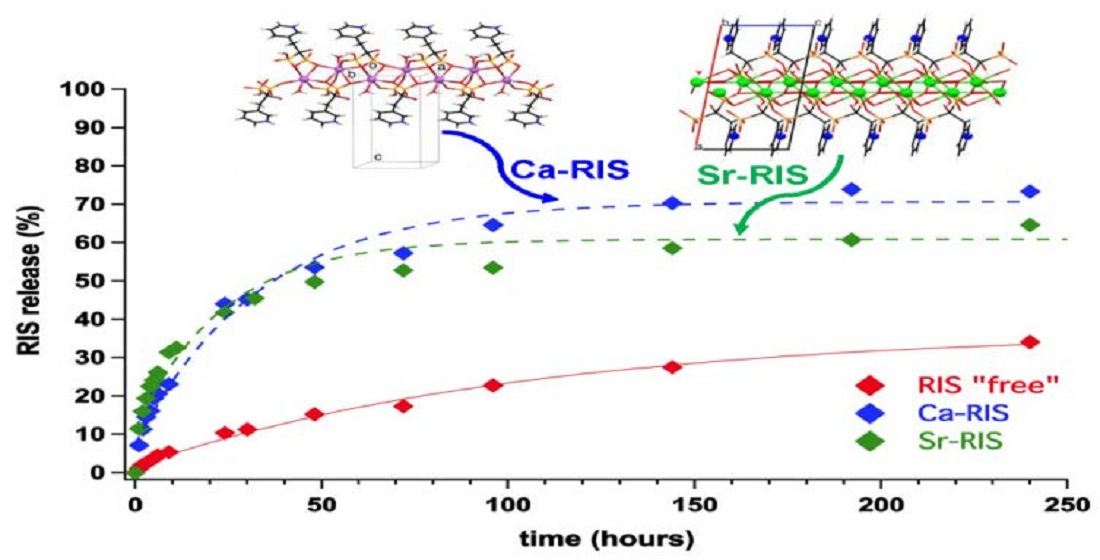
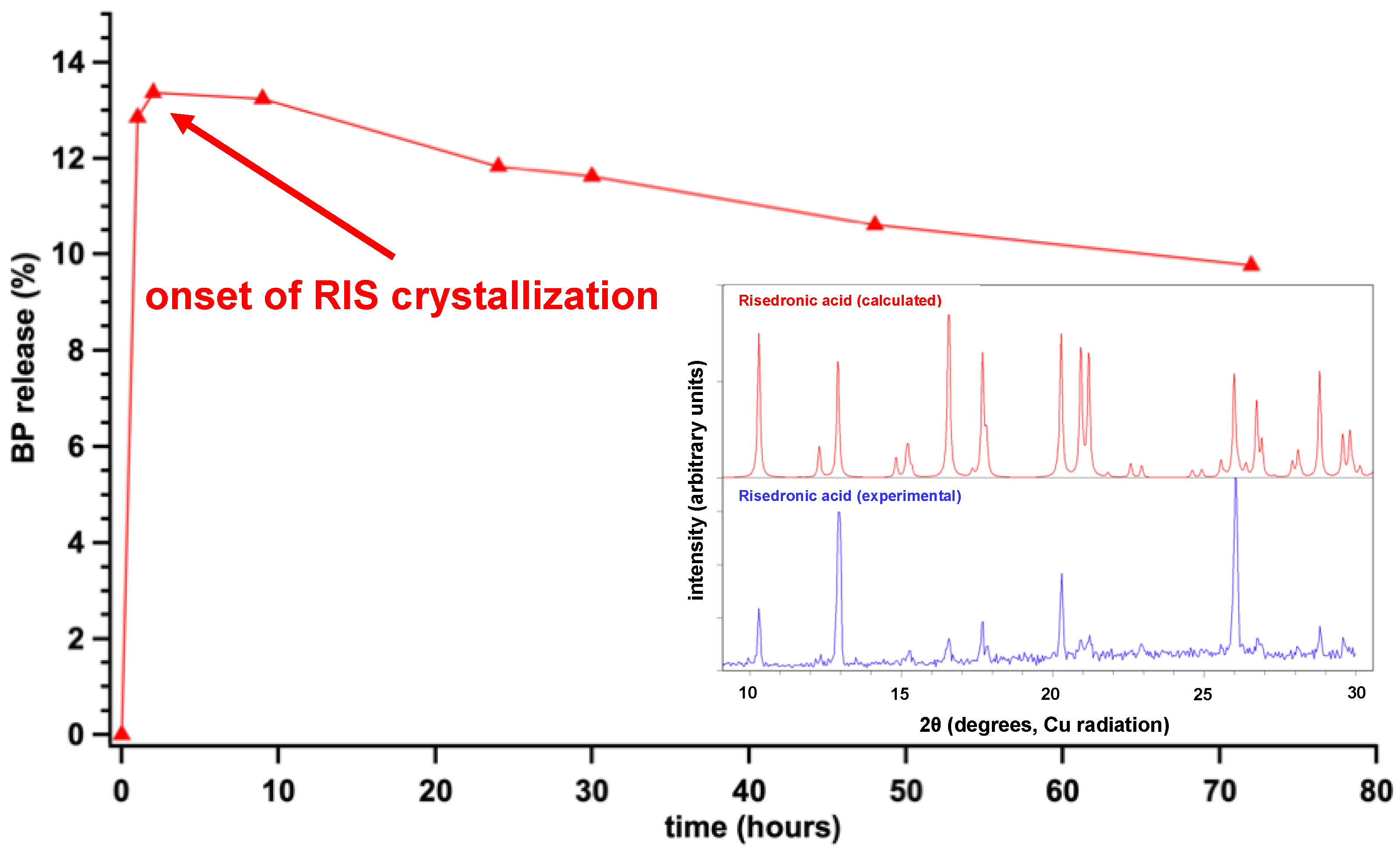
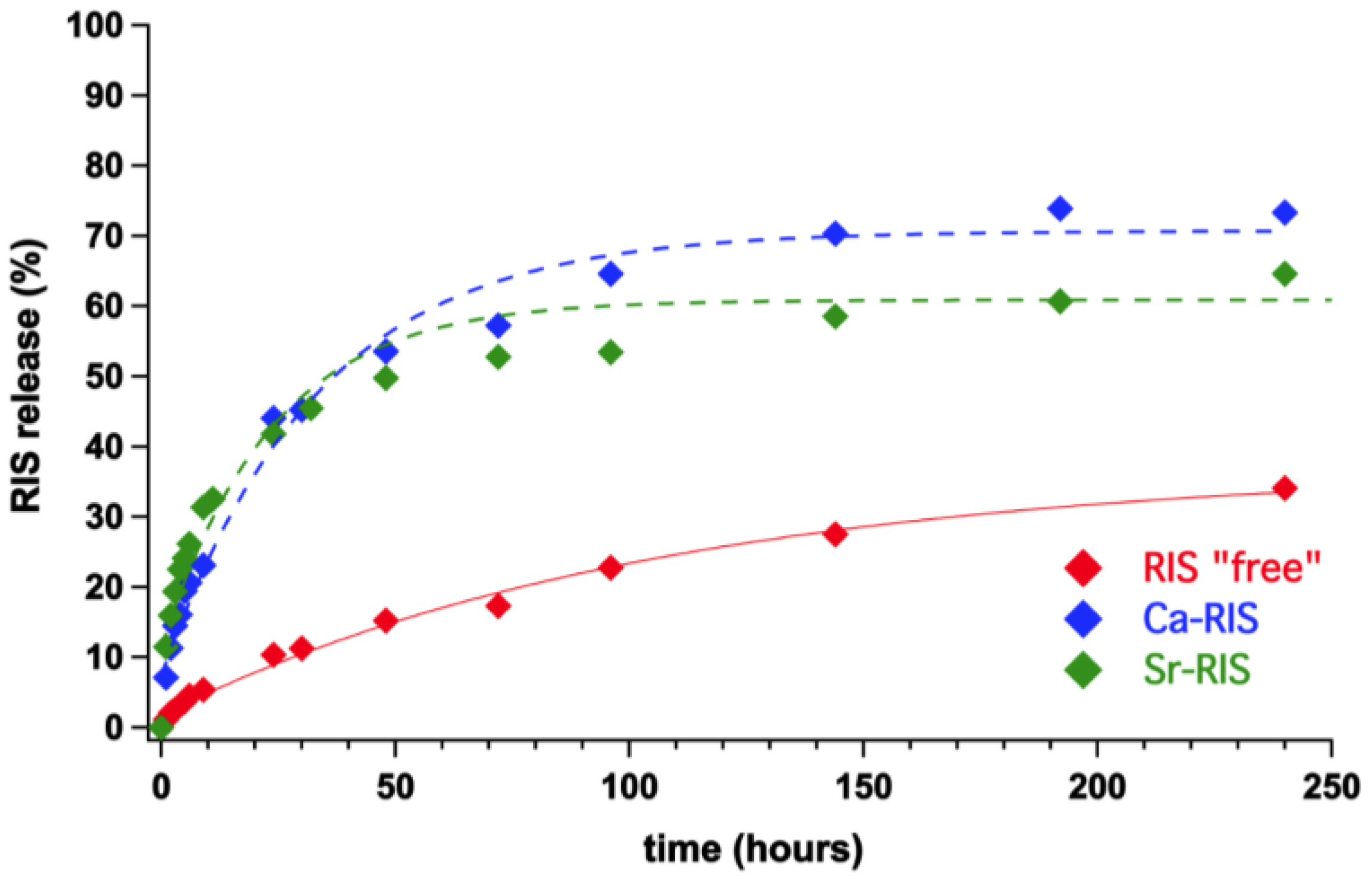
3.3. Controlled Release Study of “Free” RIS, Ca–RIS and Sr–RIS
4. Discussion
5. Conclusions
- (1)
- Two novel coordination polymers containing the alkaline-earth metal ions Ca2+ and Sr2+ and the anti-osteoporotic drug RIS were synthesized and structurally characterized.
- (2)
- Ca–RIS is a 1D coordination polymer, whereas Sr–RIS is a 2D layered coordination polymer.
- (3)
- Both Ca- and Sr–RIS were utilized as controlled release systems (excipient-containing tablets) of the active drug RIS in conditions that mimic the human stomach (pH = 1.2). Because RIS is a sparingly-soluble drug in acidic solutions, it was necessary to use drug solubilizers. The ones tested were the PAMAM-2 amine-terminated cationic dendrimer and the LPEI cationic polymer. Both effected smooth RIS release and prevented its precipitation.
- (4)
- The drug release profiles Ca- and Sr–RIS (in the presence of LPEI) were compared to that of “free” RIS (absence of metals), and it was found that they release RIS 4–5 times faster than the “free” RIS system.
- (5)
- This behavior was rationalized based on the structural idiosyncrasies of each system. The overall drug release profile for each system was the result of several structural factors, such as presence or absence of π–π interactions between the RIS pyridyl rings, H-bonding interactions and strength of the metal–O(phosphonate) bonds.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, X.-L.; Gou, W.-L.; Wang, A.-Y.; Wang, Y.; Guo, Q.-Y.; Lu, Q.; Lu, S.-B.; Peng, J. Basic research and clinical applications of bisphosphonates in bone disease: What have we learned over the last 40 years? J. Transl. Med. 2013, 11, 303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, R.G. Bisphosphonates: From bench to bedside. Ann. N. Y. Acad. Sci. 2006, 1068, 367–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbarosa, J.S.; Paz, F.A.A.; Braga, S.S. Bisphosphonates, Old Friends of Bones and New Trends in Clinics. J. Med. Chem. 2021, 64, 1260–1282. [Google Scholar] [CrossRef] [PubMed]
- Bartl, R.; Frisch, B.; Tresckow, E.; Bartl, C. Bisphosphonates in Medical Practice, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2007; pp. 23–68. [Google Scholar]
- Watts, N.B.; Diab, D.L. Long-Term Use of Bisphosphonates in Osteoporosis. J. Clin. Endocrinol. Metab. 2021, 95, 1555–1565. [Google Scholar] [CrossRef] [Green Version]
- Sandomierski, M.; Zielińska, M.; Voelkel, A. A long-term controlled release of the drug for osteoporosis from the surface of titanium implants coated with calcium zeolite. Mater. Chem. Front. 2021, 5, 5718–5725. [Google Scholar] [CrossRef]
- Park, K.W.; Yun, Y.P.; Kim, S.E.; Song, H.R. The Effect of Alendronate Loaded Biphasic Calcium Phosphate Scaffolds on Bone Regeneration in a Rat Tibial Defect Model. Int. J. Mol. Sci. 2015, 16, 26738–26753. [Google Scholar] [CrossRef] [Green Version]
- Spinthaki, A.; Matheis, J.; Hater, W.; Demadis, K.D. Antiscalant-driven inhibition and stabilization of “magnesium silicate” under geothermal stresses: The role of magnesium-phosphonate coordination chemistry. Energy Fuels 2018, 32, 11749−11760. [Google Scholar] [CrossRef]
- Mozafari, M. Metal-Organic Frameworks for Biomedical Applications; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Barbosa, J.S.; Pinto, M.; Barreiro, S.; Fernandes, C.; Mendes, R.F.; Lavrador, P.; Gaspar, V.M.; Mano, J.F.; Borges, F.; Remião, F.; et al. Coordination Compounds As Multi-Delivery Systems for Osteoporosis. ACS Appl. Mater. Interfaces 2021, 13, 35469−35483. [Google Scholar] [CrossRef]
- Vassaki, M.; Papathanasiou, K.E.; Hadjicharalambous, C.; Chandrinou, D.; Turhanen, P.; Choquesillo-Lazarte, D.; Demadis, K.D. Self-Sacrificial MOFs for Ultra-Long Controlled Release of Bisphosphonate Anti-Osteoporotic Drugs. Chem. Commun. 2020, 56, 5166–5169. [Google Scholar] [CrossRef]
- Nancollas, G.H.; Tang, R.; Phipps, R.J.; Henneman, Z.; Gulde, S.; Wu, W.; Mangood, A.; Russell, R.G.G.; Ebetino, F.H. Novel insights into actions of bisphosphonates on bone: Differences in interactions with hydroxyapatite. Bone 2006, 38, 617−627. [Google Scholar] [CrossRef]
- Bruker APEX3. APEX3 V2019.1; Bruker-AXS: Madison, WI, USA, 2019. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated Space-Group and Crystal-Structure Determination. Acta Crystallogr. Sect. A Found. Crystallogr. 2015, 71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal Structure Refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. [Google Scholar] [CrossRef]
- Kieczykowski, G.R.; Jobson, R.B.; Melillo, D.G.; Reinhold, D.F.; Grenda, V.J.; Shinkai, I. Preparation of (4-Amino-1-Hydroxybutylidene) bisphosphonic Acid Sodium Salt, MK-217 (Alendronate Sodium). An Improved Procedure for the Preparation of 1-Hydroxy-1,1-bisphosphonic Acids. J. Org. Chem. 1995, 60, 8310–8312. [Google Scholar] [CrossRef]
- Gossman, W.L.; Wilson, S.R.; Oldfield, E. Three hydrates of the bisphosphonate risedronate, consisting of one molecular and two ionic structures. Acta Cryst. 2003, C59, m33–m36. [Google Scholar] [CrossRef] [PubMed]
- Neofotistou, E.; Demadis, K.D. Silica Scale Growth Inhibition by Polyaminoamide STARBURST® Dendrimers. Coll. Surf. A Physicochem. Eng. Asp. 2004, 242, 213–216. [Google Scholar] [CrossRef]
- Demadis, K.D. A Structure/Function Study of Polyaminoamide (PAMAM) Dendrimers as Silica Scale Growth Inhibitors. J. Chem. Technol. Biotechnol. 2005, 80, 630–640. [Google Scholar] [CrossRef]
- Demadis, K.D.; Neofotistou, E. Synergistic Effects of Combinations of Cationic Polyaminoamide Dendrimers/Anionic Polyelectrolytes on Amorphous Silica Formation: A Bioinspired Approach. Chem. Mater. 2007, 19, 581–587. [Google Scholar] [CrossRef]
- Demadis, K.D.; Neofotistou, E. Inhibition and Growth Control of Colloidal Silica: Designed Chemical Approaches. Mater. Perform. 2004, 43, 38–42. [Google Scholar]
- Neofotistou, E.; Demadis, K.D. Cationic Polymeric Chemical Inhibitors and Multifunctional Blends for the Control of Silica Scale in Process Waters. Int. J. Corros. Scale Inhib. 2014, 3, 28–34. [Google Scholar] [CrossRef]
- Neofotistou, E.M.A.S.E.; Demadis, K.D. Environmentally benign chemical additives in the treatment and chemical cleaning of process water systems: Implications for green chemical technology. Desalination 2007, 210, 257–265. [Google Scholar]
- Barbey, C.; Lecouvey, M. Crystal structure of (l-hydroxy-l-phosphono-2-pyridin-3-yl-ethyl)-phosphonic acid (risedronate), C7H11NO7P2·H2O, an antiresorptive bones drug. Ζ. Kristallogr. 2002, 217, 137–138. [Google Scholar]
- Stahl, K.; Oddershede, J.; Preikschat, H.; Fischer, E.; Bennekou, J.S. Ammonium 1-hydroxy-2-(2-pyridinio)ethane-1,1-diyldiphosphonate dihydrate and potassium 1-hydroxy-2-(2-pyridinio)ethane-1,1-diyl-diphosphonate dihydrate. Acta Cryst. 2006, C62, m112–m115. [Google Scholar]
- Ma, K.-R.; Zhang, Y.; Kan, Y.-H.; Yang, X.-J.; Cong, M.-H. Three M(II)-diphosphonate coordination polymers with N-heterocyclic group (M = Ni, Fe, Mg): Synthesis, characterization and magnetic properties. Synth. Met. 2013, 182, 40–48. [Google Scholar] [CrossRef]
- Demoro, B.; Caruso, F.; Rossi, M.; Benítez, D.; Gonzalez, M.; Cerecetto, H.; Parajón-Costa, B.; Castiglioni, J.; Galizzi, M.; Docampo, R.; et al. Risedronate metal complexes potentially active against Chagas disease. J. Inorg. Biochem. 2010, 104, 1252–1258. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.-R.; Cao, L.; Cong, M.-H.; Kan, Y.-H.; Li, R.-Q. Three pyridyl modified Cu(II)/Cd(II)-diphosphonates: Syntheses, crystal structures and properties. J. Mol. Struct. 2017, 1139, 67–77. [Google Scholar] [CrossRef]
- Papathanasiou, K.E.; Vassaki, M.; Spinthaki, A.; Alatzoglou, F.-E.G.; Tripodianos, E.; Turhanen, P.; Demadis, K.D. Phosphorus chemistry: From small molecules to polymers to pharmaceutical and industrial applications. Pure Appl. Chem. 2019, 91, 421–441. [Google Scholar] [CrossRef]
- Papathanasiou, K.E.; Turhanen, P.; Brückner, S.I.; Brunner, E.; Demadis, K.D. Smart, programmable and responsive injectable hydrogels for controlled release of cargo osteoporosis drugs. Sci. Rep. 2017, 7, 4743. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tablet | Na-RIS·2.5H2O | Ca–RIS | Sr–RIS |
|---|---|---|---|
| MW (g/mol) | 350.13 | 339.19 | 386.73 |
| Drug mass (g) | 0.298 | 0.288 | 0.329 |
| Lactose (g) | 0.234 | 0.237 | 0.224 |
| Cellulose (g) | 0.234 | 0.237 | 0.224 |
| Silica (g) | 0.234 | 0.237 | 0.224 |
| Ca–RIS | Sr–RIS | |
|---|---|---|
| Empirical formula | C7H11CaNO8P2 | C7H11SrNO8P2 |
| Mr | 339.19 | 386.73 |
| Crystal system | orthorhombic | monoclinic |
| Space group | P212121 | P21/n |
| a (Å) | 6.7990(9) | 13.679(5) |
| b (Å) | 13.091(2) | 12.101(3) |
| c(Å) | 13.446(2) | 6.987(4) |
| α (°) | 90 | 90 |
| β (°) | 90 | 98.170(15) |
| γ (°) | 90 | 90 |
| V (Å3) | 1196.77 | 1144.82 |
| Z | 4 | 4 |
| R factor (%) | 5.71 | 3.19 |
| CCDC code | 2118543 | 2118546 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassaki, M.; Kotoula, C.; Turhanen, P.; Choquesillo-Lazarte, D.; Demadis, K.D. Calcium and Strontium Coordination Polymers as Controlled Delivery Systems of the Anti-Osteoporosis Drug Risedronate and the Augmenting Effect of Solubilizers. Appl. Sci. 2021, 11, 11383. https://doi.org/10.3390/app112311383
Vassaki M, Kotoula C, Turhanen P, Choquesillo-Lazarte D, Demadis KD. Calcium and Strontium Coordination Polymers as Controlled Delivery Systems of the Anti-Osteoporosis Drug Risedronate and the Augmenting Effect of Solubilizers. Applied Sciences. 2021; 11(23):11383. https://doi.org/10.3390/app112311383
Chicago/Turabian StyleVassaki, Maria, Christina Kotoula, Petri Turhanen, Duane Choquesillo-Lazarte, and Konstantinos D. Demadis. 2021. "Calcium and Strontium Coordination Polymers as Controlled Delivery Systems of the Anti-Osteoporosis Drug Risedronate and the Augmenting Effect of Solubilizers" Applied Sciences 11, no. 23: 11383. https://doi.org/10.3390/app112311383
APA StyleVassaki, M., Kotoula, C., Turhanen, P., Choquesillo-Lazarte, D., & Demadis, K. D. (2021). Calcium and Strontium Coordination Polymers as Controlled Delivery Systems of the Anti-Osteoporosis Drug Risedronate and the Augmenting Effect of Solubilizers. Applied Sciences, 11(23), 11383. https://doi.org/10.3390/app112311383