1. Introduction
Monoclonal antibodies (mAbs) are highly versatile biomolecules used manifold in analytical and diagnostic systems for the detection of targets ranging from low molecular weight substances to whole microorganisms [
1,
2,
3]. In point of care (POC) systems such as for contaminated drinking water, antibody-based systems are indispensable tools to monitor contamination with pathogenic microorganisms [
4]. For the establishment of these detection systems, highly specific mAbs recognizing the pathogen of interest are mandatory. In addition to the complexity of pathogens, the generation of specific mAbs is limited by the complex procedure of inducing strain-specific immune responses and the laborious selection of desired hybridoma cell lines [
5]. With this objective, we performed an initial proof of concept study by combining bioinformatic epitope prediction with novel immunization and selection tools to identify antibody candidates that can discriminate
E. coli O157:H7 strain (EHEC) from
Legionella pneumophila and
Bacillus ssp. The strain
E. coli O157:H7 (EHEC) was used as a model target to show the feasibility of our workflow.
The process of generating mAbs against certain bacterial strains is often initiated by using inactivated microorganism fractions for immunization. These fractions display numerous different cell surface structures not solely specific for the target strain. Therefore, the immunization with these fractions leads to antibodies with broad specificity to both, the desired bacterial strain but other strains, too. These undesired cross-reactivities represent a major challenge in the process of antibody generation [
6]. Furthermore, the immunization of chosen epitopes can carry high risks when no or only minor immune reactions take place. An appropriate immune reaction is often dependent on the used carrier proteins and the interval of immunizations.
In addition to this, the quality of the underlying screening process after fusion is key for the selection of appropriate binders and the functionality in the later application. Especially, when microorganisms are used as antigenic targets, it is important to select suitable and unique epitope structures on the cell surface so that the antibody can detect these structures in the native conformation and assay environment [
6]. When hybridoma technology is used, the screening process is mainly performed by limited dilution in combination with numerous enzyme-linked immunosorbent assay (ELISA) screenings, which makes it a time-consuming and expensive procedure. In addition, the presentation of the antigen in cell-based ELISAs is often not related to the final environment in the final detection system.
To circumvent these disadvantages and provide an optimized process, we chose the enterohemorrhagic
E. coli O157:H7 strain (EHEC) as a model organism to evaluate our workflow. The
E. coli O157:H7 serotype is a food-borne pathogen first identified in 1982 during an outbreak in Michigan [
7]. It is now the dominant hemorrhagic serotype, infecting around 100,000 patients every year in the U.S. [
8], inducing gastrointestinal indications up to life-threatening complications as Hemolytic Uremic Syndrome (HUS) or Hemorrhagic Colitis induced by the production of Shiga toxin (Stx). The state-of-the-art detection of EHEC infections is performed mainly by PCR and conventional plate counts [
9]. These methods are reliable but they need both specialized equipment and time. Ready-to-use POC devices represents a fast and on-site alternative to monitor outbreaks in the field where no laboratory access is suitable. Specific antibodies are the centerpiece for POC applications. However, false-positive results may occur due to cross-reactivities of the antibody to other bacterial strains in the sample. Therefore, a focused antibody generation against a targeted structure located only on the desired bacterial strain is of great importance.
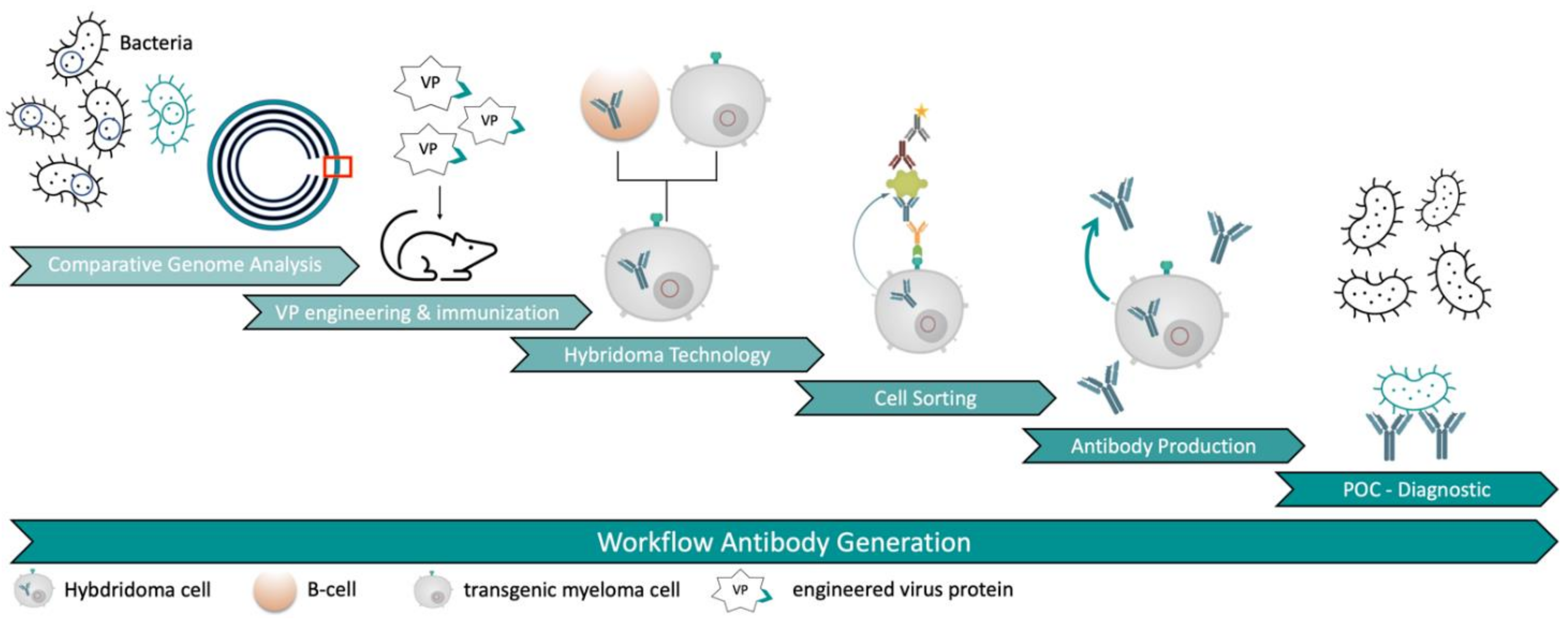
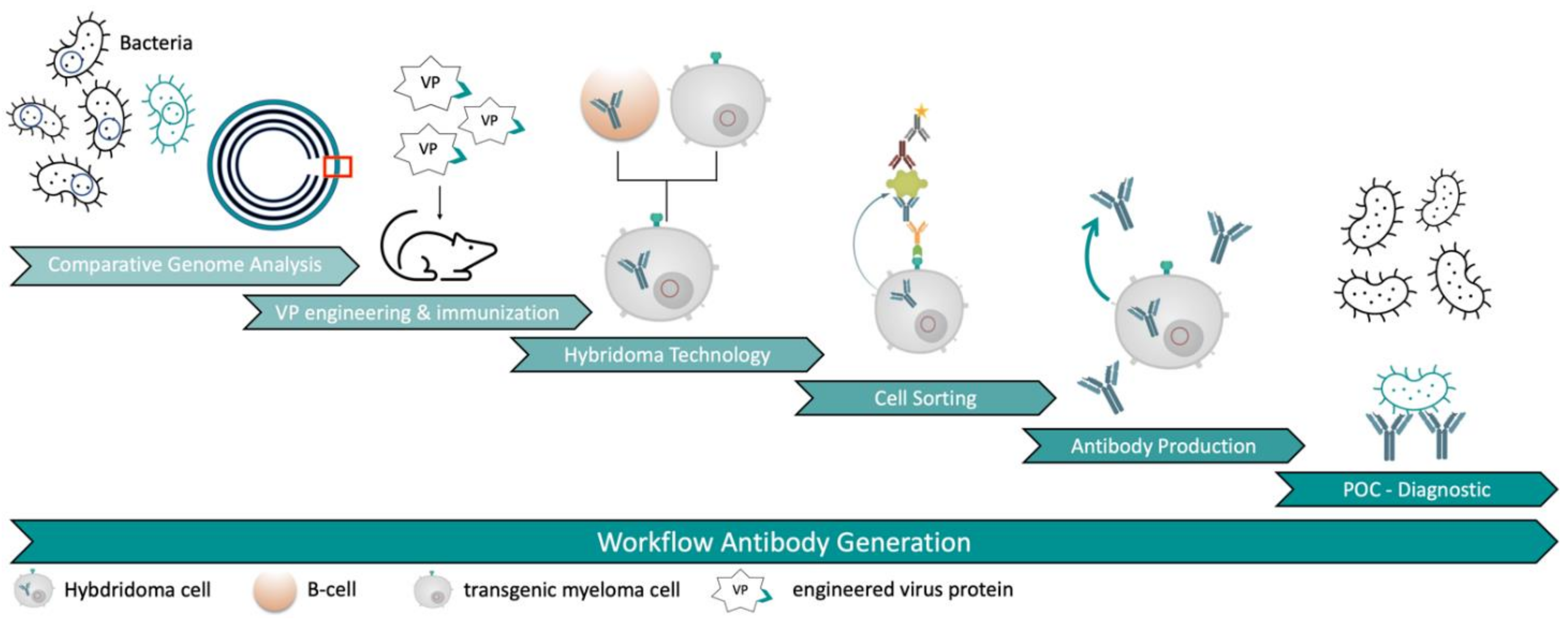
Our new approach led to the generation of several mAbs against
E. coli O157:H7 serotype without the need of immunizing whole bacteria cells. It was possible to generate specific mAbs in a very short time frame due to the combination of a very effective immunization with viral carrier proteins and a novel hybridoma selection system (
Figure 1). This approach could serve as an improvement for antibody discovery in the future.
2. Materials and Methods
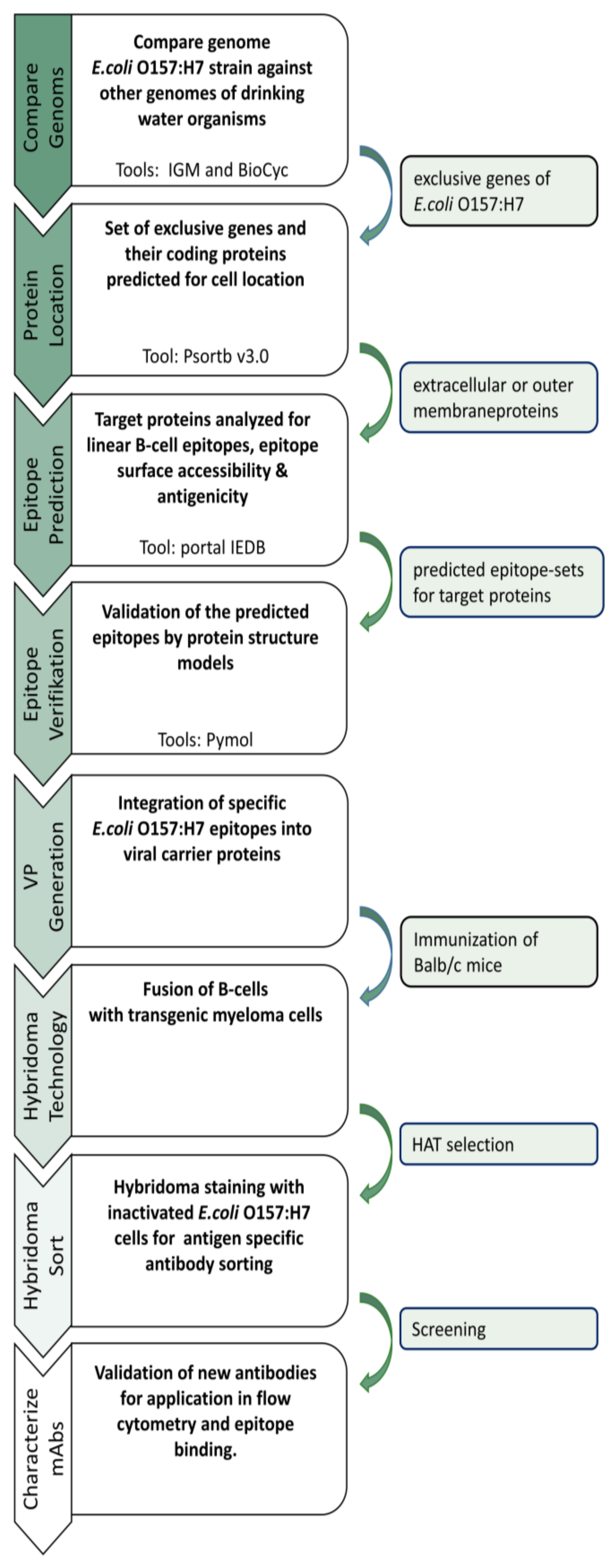
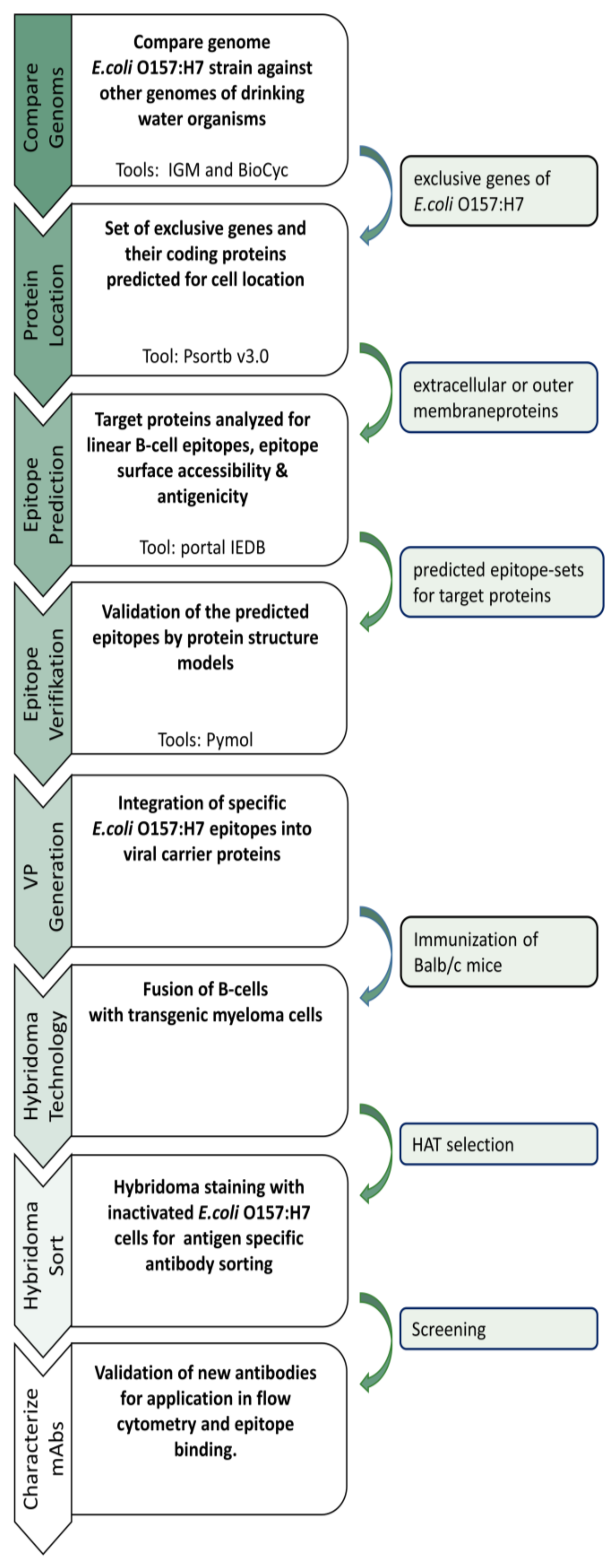
2.1. Comparative Genome Analysis of E. coli and Non-E. coli Strains
To identify strain-specific epitopes suitable for antibody generation, the genomes of different microorganisms were analyzed (
Table 1). The genome of
E. coli O157:H7 str. EDL933 (NZ_CP015855.1) was set as a reference genome and compared against eight other non-
E. coli strains, along with the pan-genome of
E. coli (
Table 1). We used the tools from the IMG/M system [
10] as well as the comparative genome tools from BioCyc [
11] with the default settings. The analysis excludes all genes that show an E-value greater than 0.5 and a sequence identity over 70%. In addition, genes that encode proteins with a length of 100 amino acids (aa) or less are discarded from the results. The average protein length is indicated with 267 aa [
12]. The analysis results in a set of genes that is unique to the reference bacterial strain.
2.2. Protein Localization
The identified set of genes was further analyzed to specify the localization of the corresponding proteins. In terms of antibody generation, proteins or epitopes have to be located on the cell surface of the microorganisms so that the generated antibody can bind the epitope. For this, the exclusive non-homologous
E. coli O157:H7 proteins have been subjected to PSORTb version 3.0 [
13]. This tool allows the prediction of the cellular localization of proteins using a Bayesian network model to calculate the associated probability for each localization site. The prediction was carried out with the default settings for Gram-negative bacteria. The outer membrane proteins and the extracellular proteins were filtered from the results and further analyzed for antibody generation.
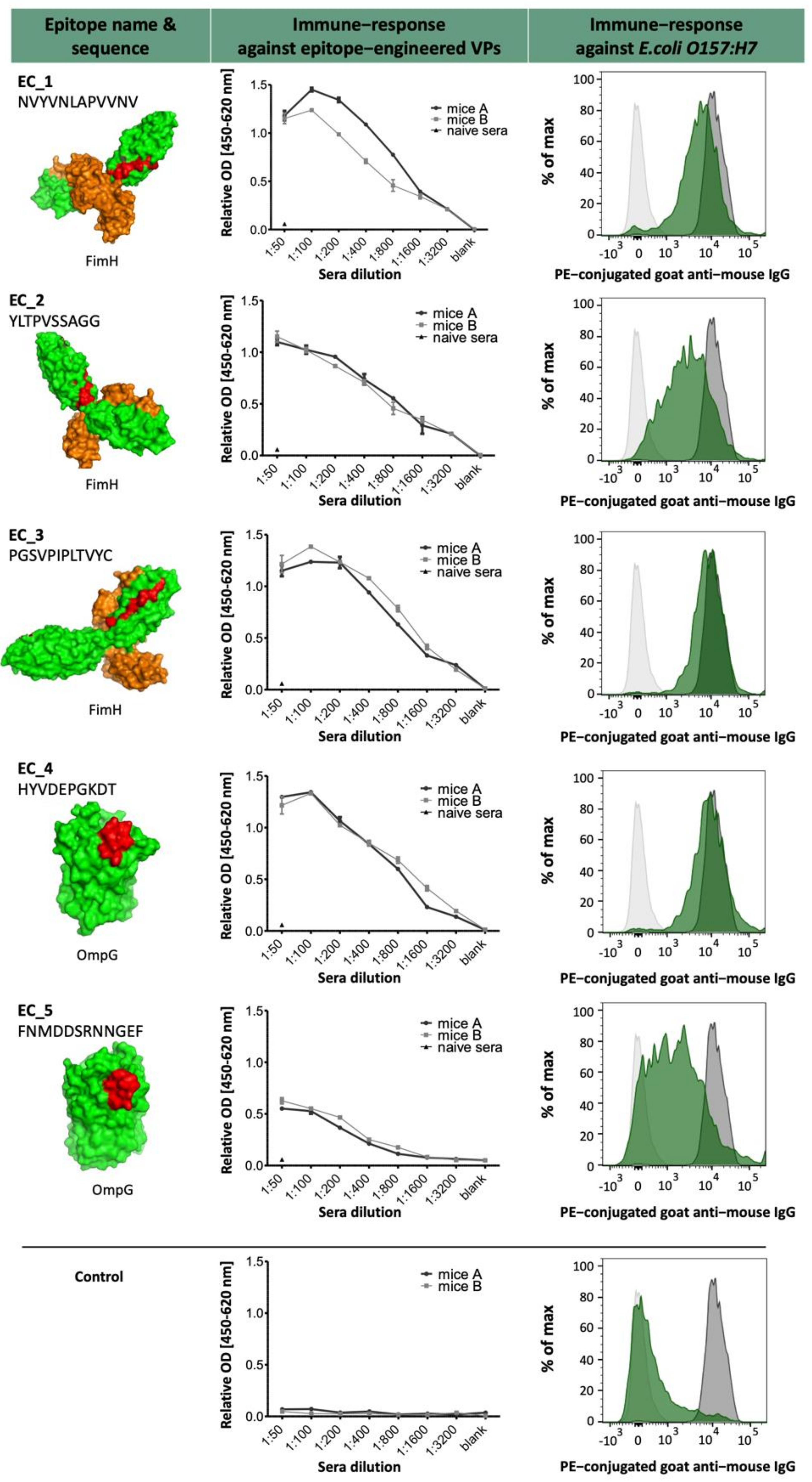
2.3. B-Cell Epitope Prediction
The proteins FimH and OmpG were examined for the presence of linear B-cell epitopes using BepiPred-2.0 from the IEDB portal [
14]. The prediction of specific epitope sequences allows us to engineer our viral carrier proteins used for immunization (VPs). The epitope sequences were further analyzed using the Emini-surface accessibility prediction algorithm and the antigenicity prediction according to Kolaskar and Tongaonkar scale (
Table 2). A successful generation of mAbs is achievable only if the predicted linear epitopes are accessible binding sites for the antibody on the protein surface.
2.4. Preparation of Immunogenic VPs
For immunization, the selected epitope sequences from
Supplementary Table S1 were genetically inserted into a VP as described previously [
15]. Briefly, oligonucleotides of the epitope sequences were synthesized and hybridized. Subsequently, these oligonucleotides were cloned into the vector pET22b with the coding VP sequence. The epitope-engineered VPs were produced recombinantly in
E. coli and purified via NTA affinity chromatography. The purified epitope-engineered VPs were dialyzed against PBS and used for immunization.
2.5. Immunization of Balb/c Mice
For each epitope, two six-month-old mice (Balb/c strain) were immunized with the corresponding epitope-engineered VPs. For this purpose, 50 µg epitope-engineered VP in PBS were injected intraperitoneally on days 0, 7, 14, and 21. Blood samples were taken on day 29 to test the immune response in ELISA. Immunization was conducted following the relevant national and international guidelines. The study was approved by the Ministry of Environment, Health, and Consumer Production of the Federal State of Brandenburg, Germany (reference number V3-2347-A16-4-2012) and carried out in compliance with the ARRIVE guidelines.
2.6. Serum ELISA
To prove successful immunization, 5 µg/mL epitope-engineered VPs in PBS were coated (50 µL per well) overnight at 4 °C in a 96-well plate as described previously [
15]. In the next step, wells were washed three times with tap water. Blocking was performed by using 100 μL PBS/NCS for 30 min at room temperature (RT). After a further washing step, sera of immunized mice were diluted in 50 µL PBS/NCS in a dilution series of 1:50 to 1:3200. Following incubation for 1 h at RT, the wells were washed again, and a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG was used for detection (1:5000, Dianova GmbH, Hamburg, Germany). After 45 min incubation, the substrate solution (50 µL per well, 0.12 mg/mL tetramethylbenzidine (Carl Roth GmbH, Karlsruhe, Germany), in 50 mM NaH
2PO4 with 0.04% CH
4N
2O × H
2O
2) was added. The substrate reaction was stopped after 15 min by using 50 µL 1 M H
2SO
4 solution. The optical density (OD) was measured at 450 nm with a reference at 620 nm. Preimmune sera from the same mice served as negative control. Positively tested mice were injected with a final boost of 50 µg epitope-engineered VPs at day 36. The spleen was removed on day 40. Splenocytes were isolated and used for fusion with transgenic myeloma cells as described previously [
16].
2.7. Fusion and HAT Selection
The fusion of splenocytes with a transgenic myeloma cell line was conducted according to Listek et al. [
16]. The fused cells were cultivated for 14 days in hypoxanthine–aminopterin–thymidine (HAT) supplemented RPMI full growth medium in three T75 culture flasks together with feeder cells under 6% CO
2, at 37 °C and 95% humidity. Subsequently, the cells were collected and prepared for cell staining and sorting.
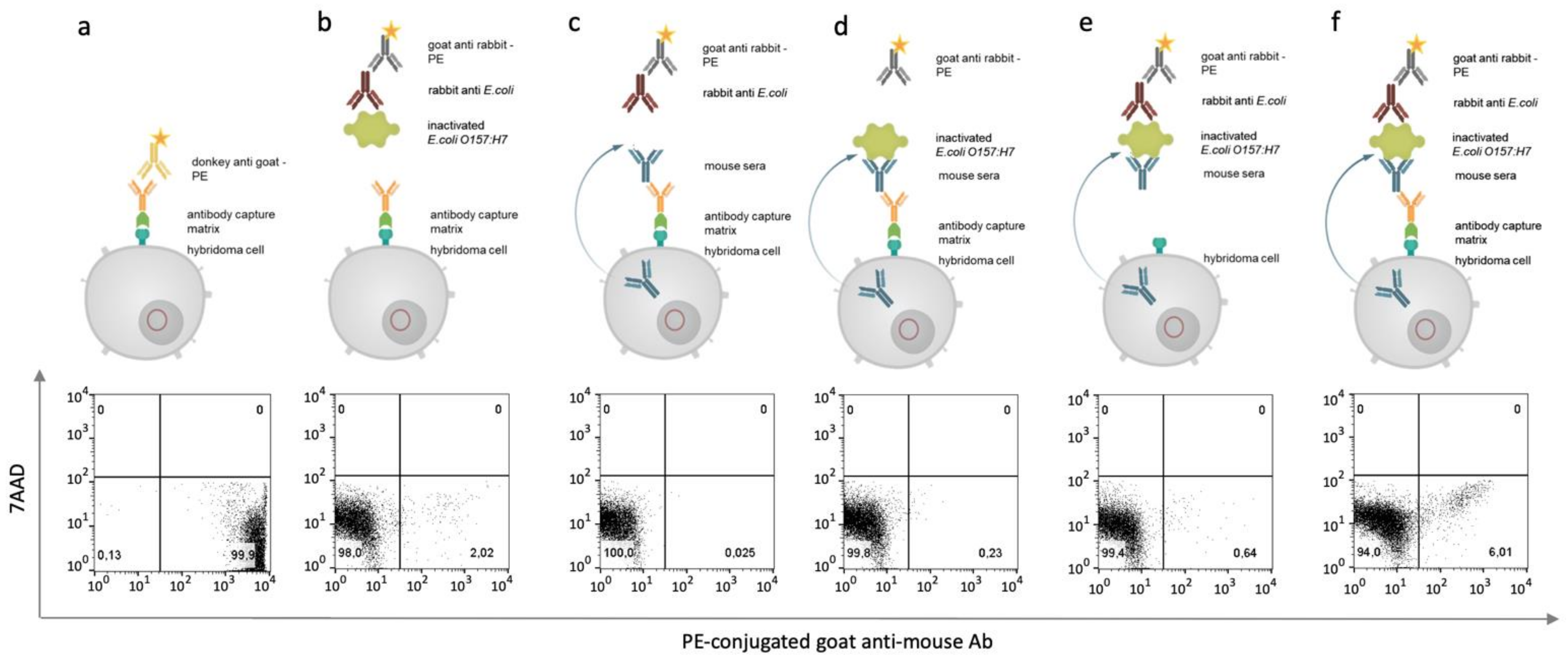
2.8. Hybridoma Cell Staining and Flow Cytometry Sorting
After 14 days of HAT selection, transgenic hybridoma cells were prepared for antigen-specific sorting. Therefore, an antibody capture matrix was set up as described previously [
16]. Essential for the establishment of a successful antibody capture matrix is the biotinylation of the artificial surface receptor, which was checked flow cytometrically by using 3 µL of phycoerythrin-conjugated streptavidin (SAV, Merck, Darmstadt, Germany). Next, the antibody capture matrix was formed by adding SAV-conjugated goat anti mouse IgG antibody (1 mg/mL) to biotinylated hybridoma cells and incubating it for 20 min. The binding was proved flow cytometrically by using a FACS Melody (BD Biosciences, Franklin Lakes, USA).
Following this, the cells were washed with 10 mL MACS buffer. The pellet was resuspended in 300 μL MACS buffer, and 2 million inactivated E. coli O157:H7 cells were added. The sample was incubated for 20 min at 4 °C and then washed three times with 10 mL of MACS buffer. Again, the pellet was resuspended in 300 µL MACS buffer and incubated with a polyclonal rabbit anti-E. coli antibody (5 µg/mL, Lot: GR3215121-2, Abcam). For 20 min at 4 °C, the cells were incubated and then washed with 10 mL of MACS buffer. In the last staining step, the resuspended cells were stained with a phycoerythrin (PE)-conjugated goat anti-rabbit Fab Fragment (1:500, Lot:121745, Jackson ImmunoResearch, West Grove, PA, USA). Once again, the cells were washed twice with 10 mL MACS buffer. The pellet was resuspended in 500 µL MACS buffer. Finally, 3 µL 7-AAD (1 mg/mL, ThermoFisher, Waltham, MA, USA) was added, and the cell solution was incubated for 15 min at 4 °C. The samples were measured and sorted by flow cytometry (BD FACSMelody). The analysis was carried out by using FlowJo™ Software (FlowJo™ Software (Windows) Version 10.0.7. Ashland).
2.9. Screening of Antigen-Specific Antibody-Producing Hybridomas
The sorted hybridoma cells were seeded in 96-well plates (Greiner bio-one) and cultivated under 6% CO2 at 37 °C and 95% humidity in RPMI full-growth medium supplemented with 10% fetal calf serum (FCS), 2 mM glutamine, 50 μM ß-mercaptoethanol, and 0.1 mg/mL gentamycine. The production of murine antibodies in general was tested after 7 days in a sandwich ELISA by using a goat anti-mouse IgG as a catcher (Lot: 127281, Jackson ImmunoResearch) and a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Lot: 138862, Dianova GmbH, Hamburg, Germany) as a detector. The specificity of the produced antibodies was analyzed by flow cytometry by incubating 2 million inactivated E. coli O157:H7 cells with 100 µL hybridoma culture supernatant. The sample was incubated at 4 °C for 30 min. For the washing step, the cells were mixed with 500 µL MACS buffer and centrifuged at 8000× g for 5 min. Then, the pellet was resuspended in 100 µL MACS buffer. Next, 100 µL PE-conjugated rabbit anti-mouse antibody (1:500, Lot:GR3236250-1, Abcam, Cambridge, UK) was added to the cells, and the mixture was incubated for 30 min at 4 °C. Again, the cells were washed with 500 µL MACS buffer and resuspended in 200 µL MACS buffer for further analysis. A rabbit polyclonal anti-E. coli antibody (5 µg/mL, Lot: GR3215121-2, Abcam) served as positive control. Flow cytometric analyses were performed using the FACS Melody (Becton Dickinson). Measurements were analyzed by using FlowJo™ Software (FlowJo™ Software (Windows) Version 10.0.7. Ashland).
2.10. Antibody Purification
Positively tested monoclonal hybridomas were cultivated in T75 culture flasks (Greiner Bio-One) to collect cell culture supernatant for antibody purification. Antibodies were purified by using protein A-mediated affinity chromatography as described previously by Lütkecosmann et al. [
15]. Briefly, the culture supernatant was centrifuged (13,000×
g, 15 min, 4 °C), filtered (0.45 mm), and transferred to the protein A column. Elution was performed by using 0.1 M citrate with a pH of 3.5. Eluted antibodies were immediately neutralized with 500 µL 1 M Tris HCl, pH = 9.0.
4. Discussion
The generation of antibodies for the detection of MOs is associated with several issues. Identifying suitable hybridomas whose antibodies bind solely to strain-specific targets is extremely challenging. In particular, for MOs, it is difficult to identify species-specific epitopes due to a high overlap between highly conserved regions and the lack of appropriate cross-reactivity screens [
19]. Screenings for suitable binders generally use ELISA/ELISPOT techniques and Western or dot blotting methods. However, these methods also possess certain disadvantages related to the purity of used components, unspecific binding to plastic, or the high levels of cross-reactivity between the strains. A flow cytometry approach is a useful and sensitive alternative to screen for potential binders due to its ability to gate on whole bacteria and identify mAbs that bind on cell surface-related targets [
20]. In addition, flow cytometry staining of bacteria is a simple and reliable process.
In this study, we focused on the combination of three aspects to establish a comprehensive workflow for MO-specific mAbs—first, a bioinformatic one for the identification of unique and strain-specific epitopes, a second one for an epitope-specific immunization with engineered VPs, and a third one with a flow cytometry-based selection system for the identification of specific antibody-producing hybridomas.
For the bioinformatics analysis, we compared the genomes of eight indicator MOs that are commonly associated with contaminated drinking water [
21], as well as the pan-genome of
E. coli with the reference strain
E. coli O157:H7. The panel of MOs used for this proof-of-concept study provides a valid database to demonstrate our workflow. However, for future applications, we would like to point out that the panel can still be adapted and extended.
To guarantee the accessibility of these epitopes, we combined several prediction and localization tools such as IEDB, PSORTb, and BioCyc. The combination of these different tools has already been proven in several studies to identify new vaccines against MOs [
22,
23].
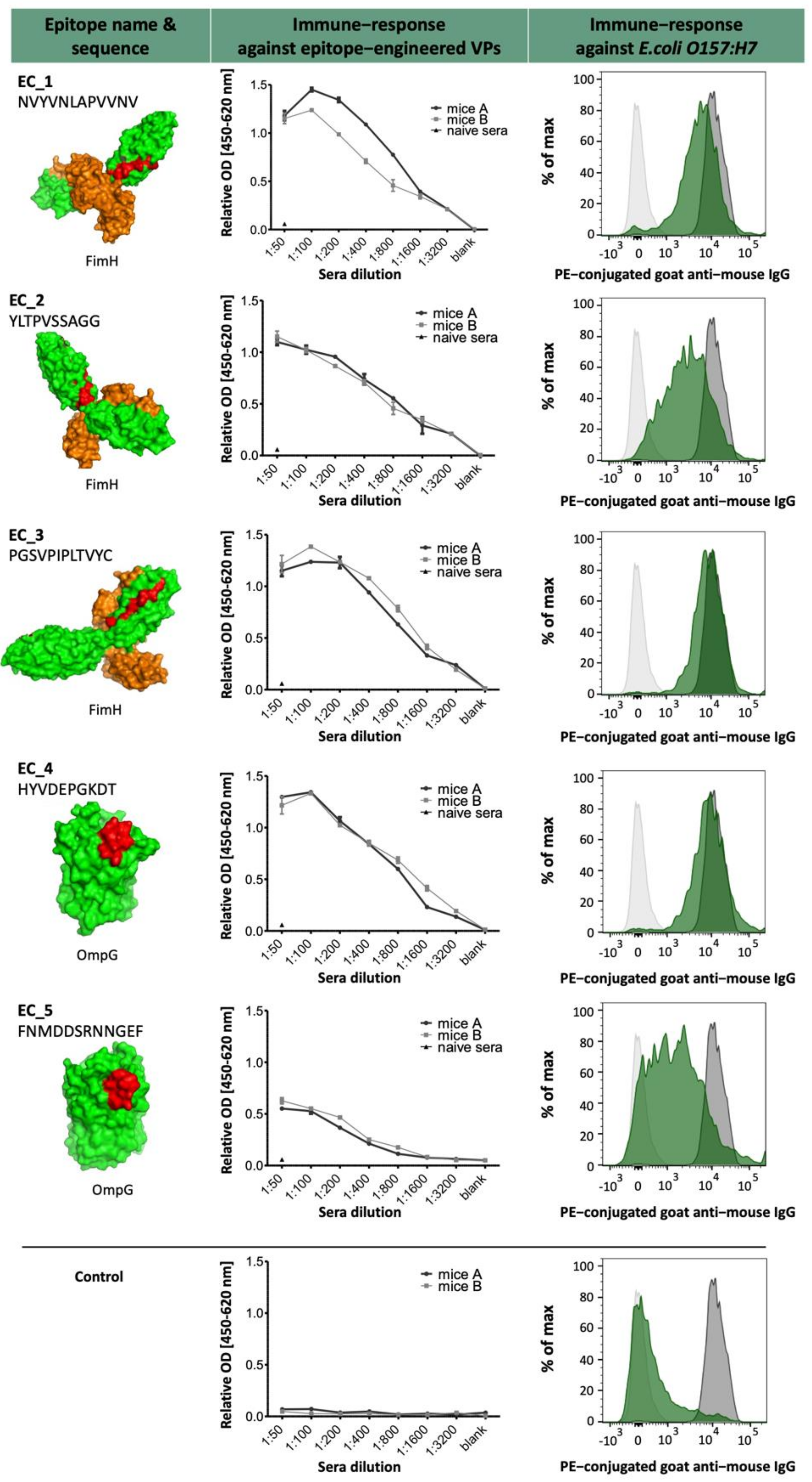
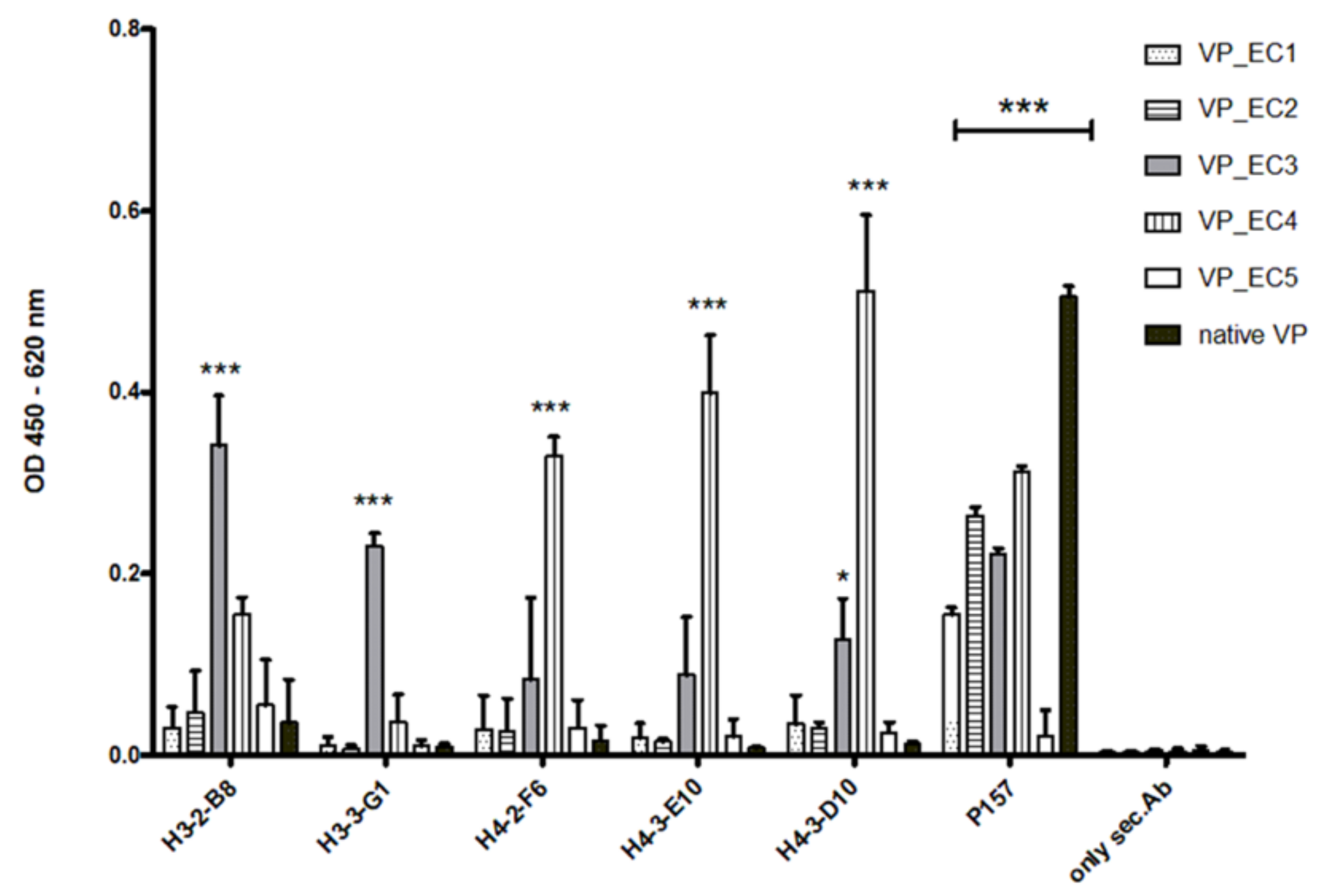
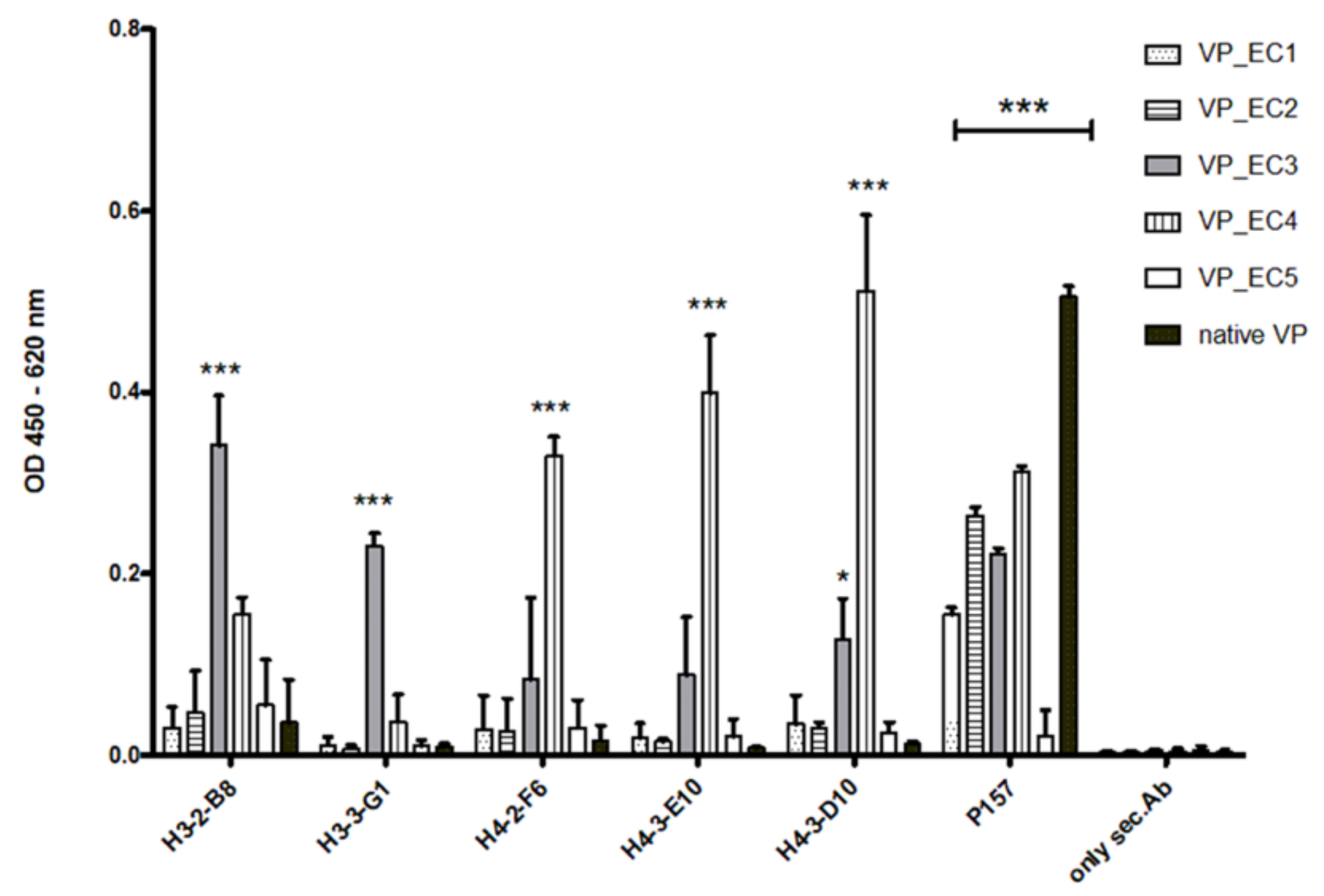
The selected epitopes were cloned into our VP system, which allows a surface-related presentation of the epitope sequence on a virus-derived carrier protein. Another advantage of using such a system for immunization is a very rapid and specific immune response to the inserted epitopes, as shown in
Figure 3 and
Figure 7. Four out of five selected epitopes were able to induce a very specific titer within 4 weeks without the need of immunizing whole bacterial cell extracts and adjuvant. The use of inactivated bacteria or bacterial cell extracts for immunization is accompanied by certain pitfalls, as it was described earlier in van der Woude et al. 2011 [
24]. Many bacterial species show phenotypic alterations dependent of their growth phase, culture media, or other environmental factors, which is influencing the presentation of specific epitope sequences. Using the bioinformatic workflow described in this study in combination with the VP system for immunization, a targeted and reliable induction of strain-specific mAbs can be achieved. Although we choose characterized and well-defined protein targets from
E. coli in this pilot study, we postulate that by using structural prediction tools such as iTasser [
25], it will be possible to select surface-related epitopes from less characterized target proteins as well. We previously started with the first experiments in this direction to see if our workflow is suitable for rare characterized targets as well.
In the next part of the study, we developed a bacteria-specific staining and sorting protocol for antibody-producing hybridomas based on the selection principle that was published recently [
16]. In this method, we used our novel transgenic myeloma cell lines, which express an artificial cell surface marker. This marker allows an antigen- or isotype-specific screening of hybridomas by linking the produced antibody to the corresponding producing mother cell. This selection principle can be performed after two weeks of HAT selection and displays an innovative new technology to identify and select specific mAb-producing hybridomas. In comparison to conventional screening methods via ELISA and the limited dilution of cells, combined with existing problems of coating ELISA plates with whole microorganisms or unspecific binding to plastic, this new approach can turn the selection process into a highly efficient task. The setup enabled a selection of nearly 1 × 10
5 positive hybridomas specific for
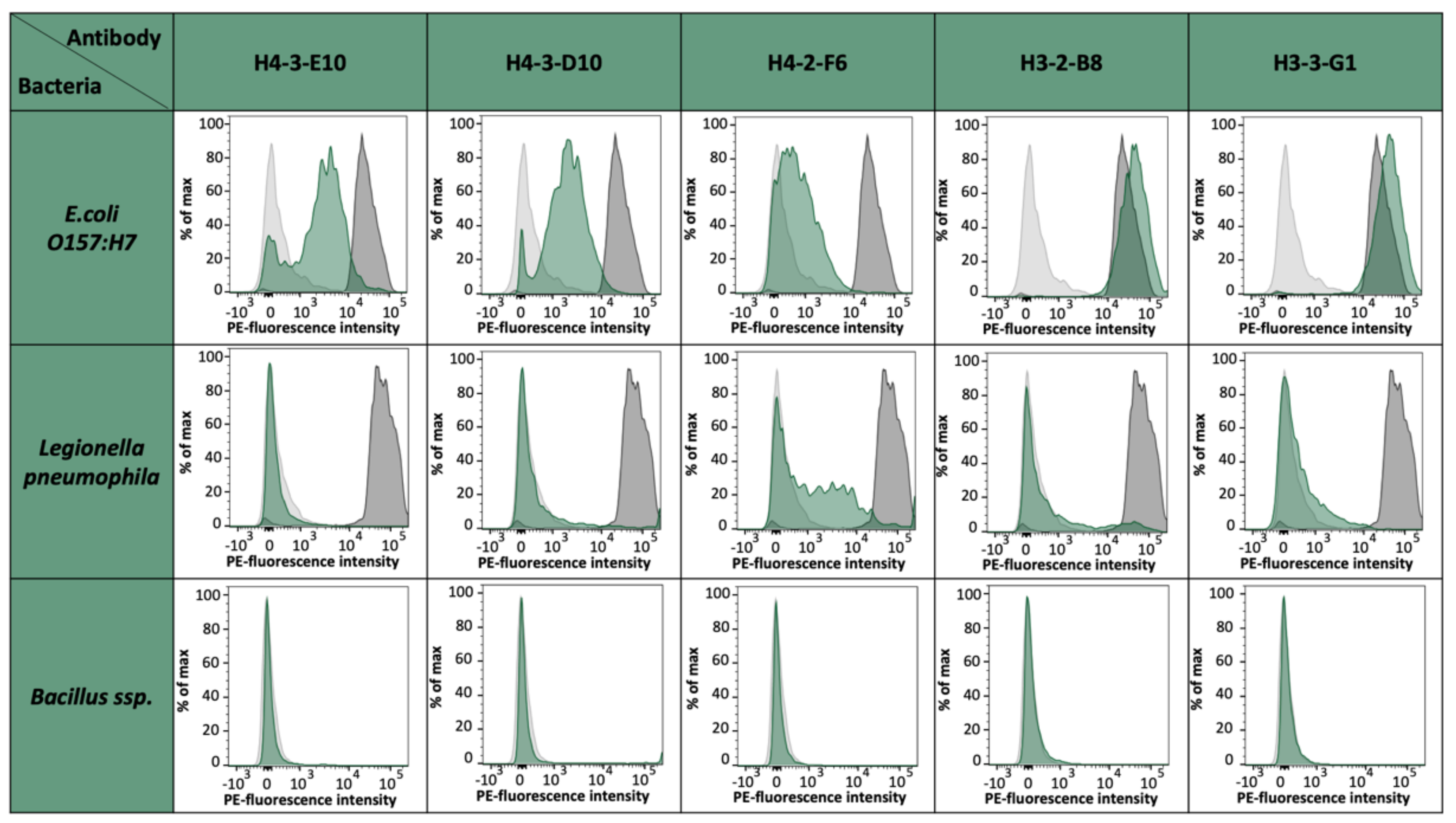
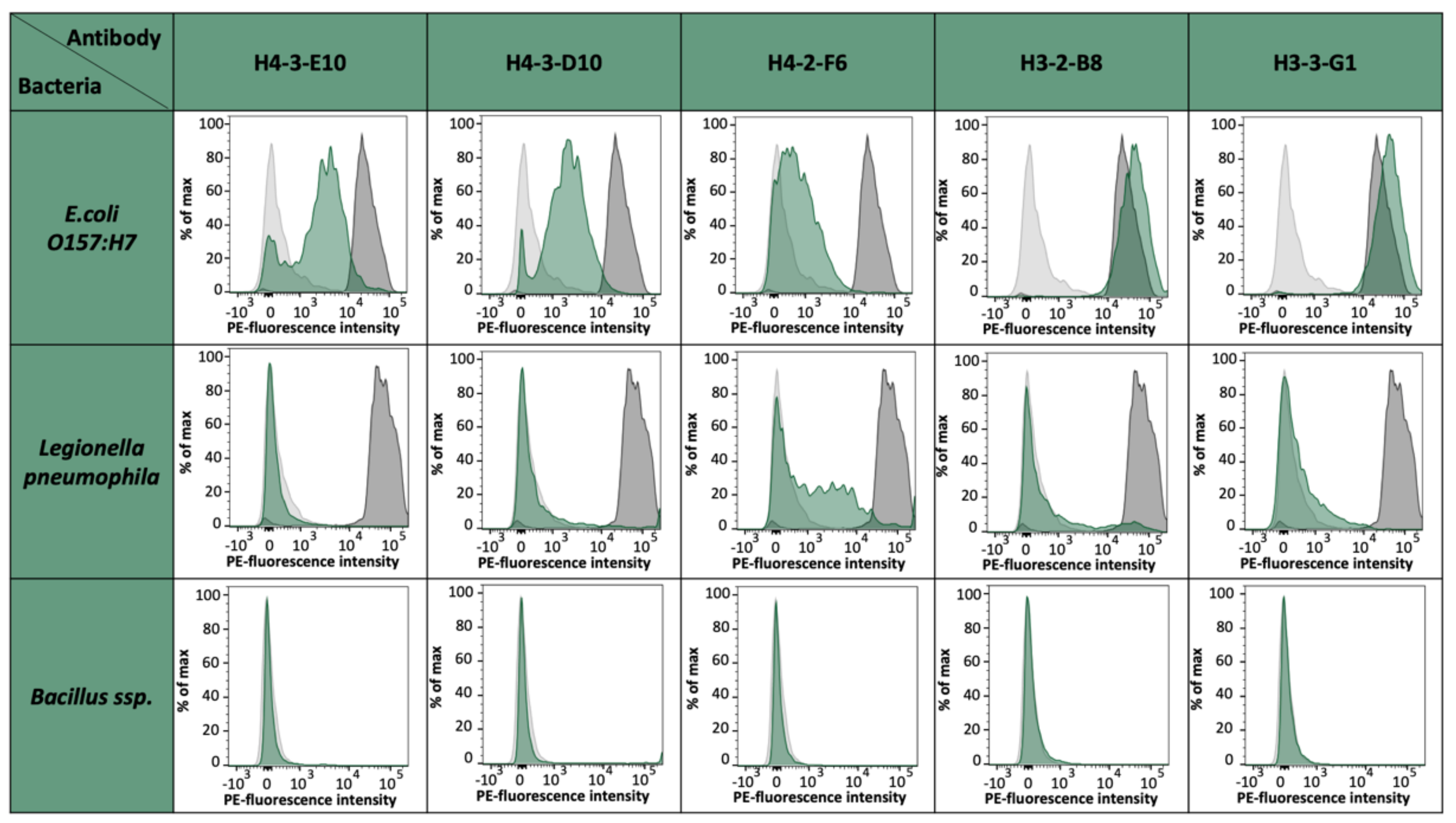
E. coli strain O157:H7 epitopes. For the fusion of EC4, we could select up to 75,000 specific hybridomas. From 55 monoclonals and 156 polyclonals plated on 96-well plates, 44 monoclonals and 154 polyclonals could be established as specific, which represents an optimal case. For the fusions EC2 and EC3, an output of 18% positive hybridoma cells could be achieved. With these results, we saw differences between the single epitopes and also the general instabilities after fusion and selection, but with this fast sorting, a high number of potential mAb candidates can be selected shortly and effectively. The obtained antibody candidates were further characterized for a possible cross-reactivity against other bacterial strains present in drinking water such as
Legionella pneumophila,
Bacillus ssp., and
E. coli K12 [data not shown]. Except for H4-2-F6, which shows a slight cross-reactivity to
L. pneumophila cells, all other antibody candidates were negative for tested bacterial species and solely positive for
E. coli O157:H7. It has to be noted that
E. coli K12 has coding genes for OmpG. However, previous studies have already shown that the expression is almost non-existent under laboratory conditions [
26].
With the pilot approach shown in this study, the generation and selection of MO-specific antibodies seem to be a reliable and fast alternative without the necessity of using whole bacterial cell extracts or cell-based ELISA screenings. The generated antibodies could serve as potential candidates for POC systems in the field of food or drinking water analyses and are currently validated for upcoming developments.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}