
QuEChERS-Based Methodology for the Screening of Alkylphenols and Bisphenol A in Dairy Products Using LC-LTQ/Orbitrap MS





Abstract
1. Introduction
2. Materials and Methods
2.1. Standards and Materials
2.2. Dairy Samples
2.3. Sample Preparation
2.4. LC-LTQ/Orbitrap MS Conditions
2.5. Statistics
3. Results and Discussion
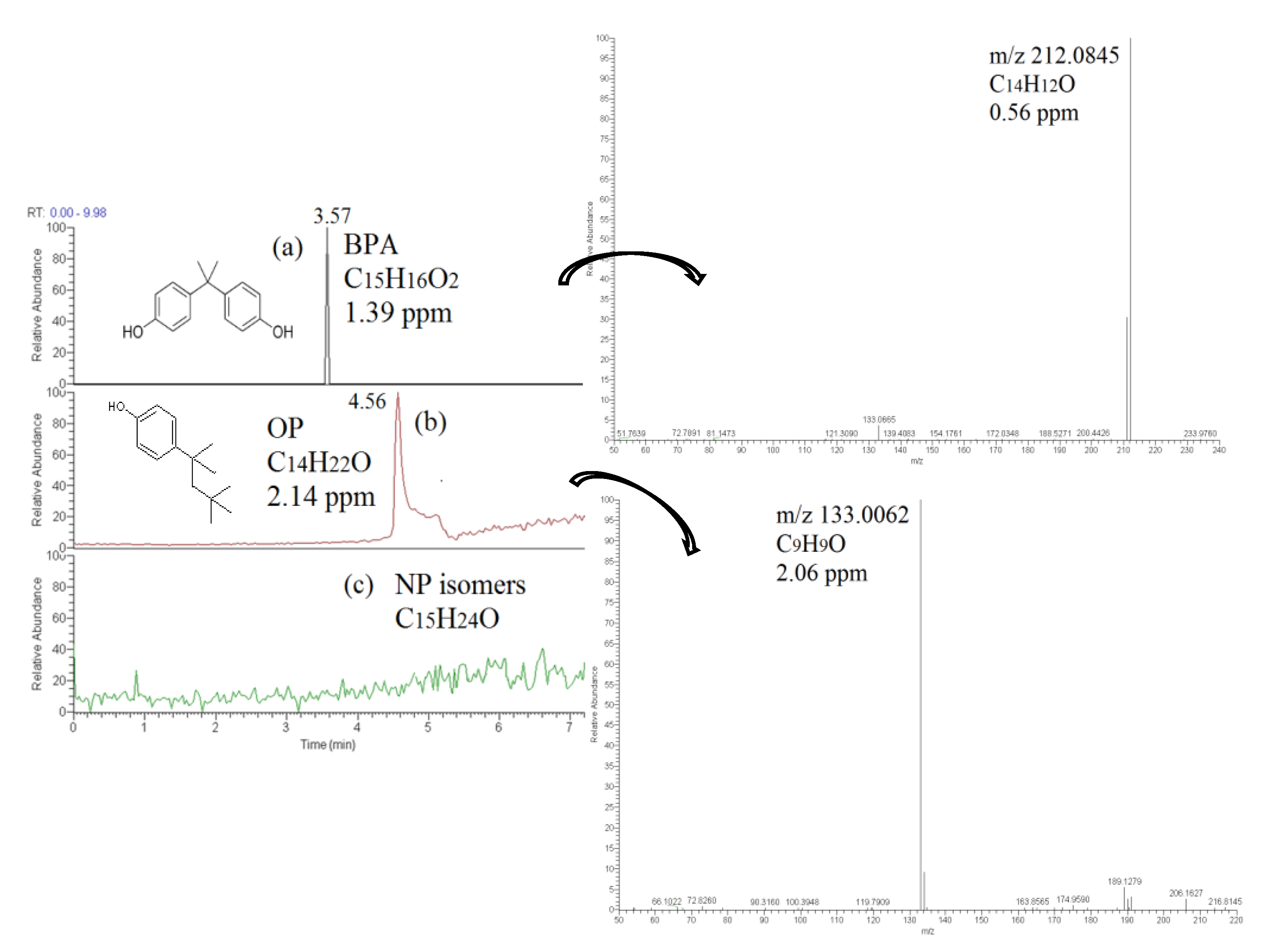
3.1. LC Separation-LTQ/Orbitrap MS Determination
3.2. Optimization of QuEChERS Extraction Conditions
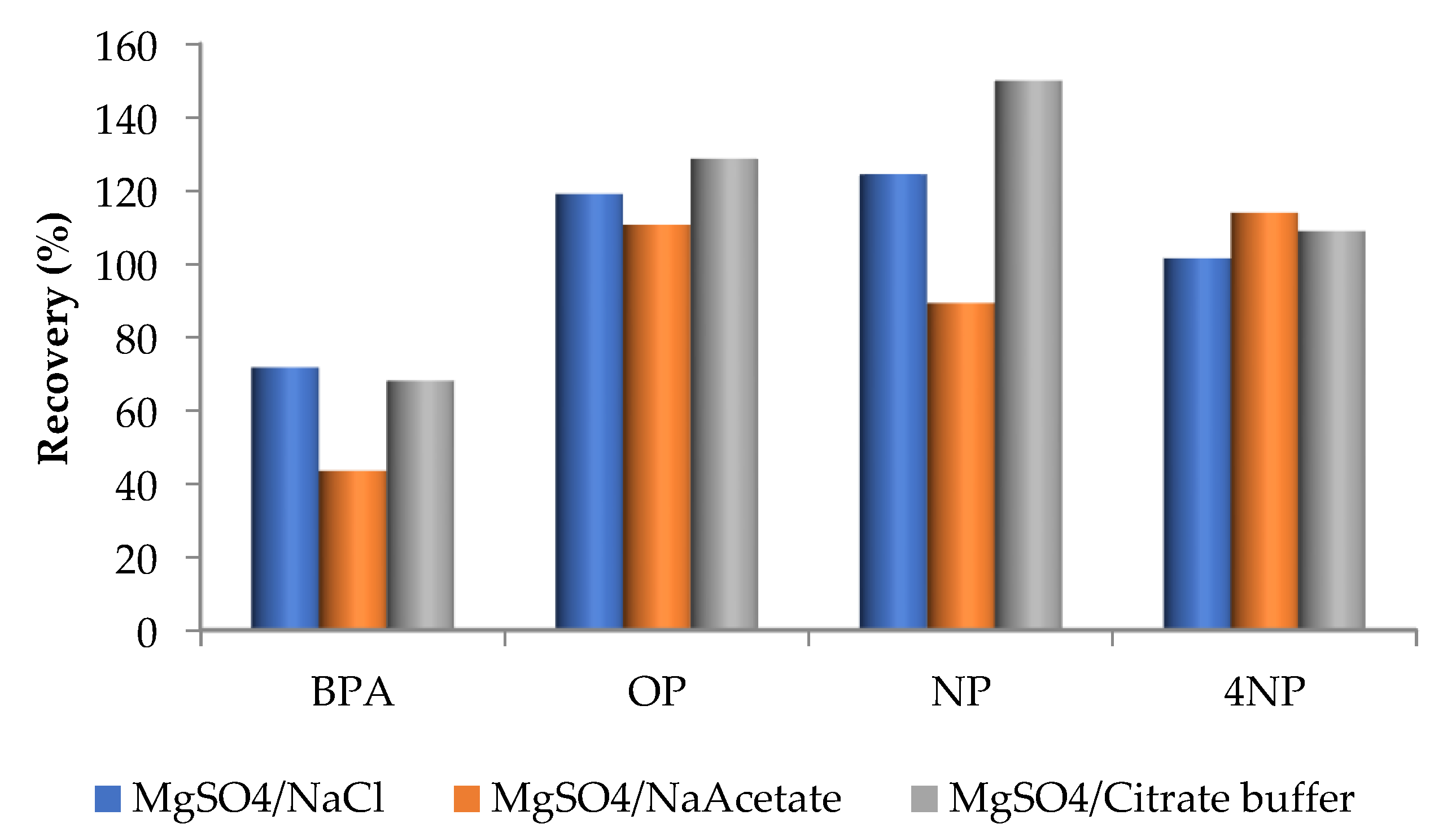
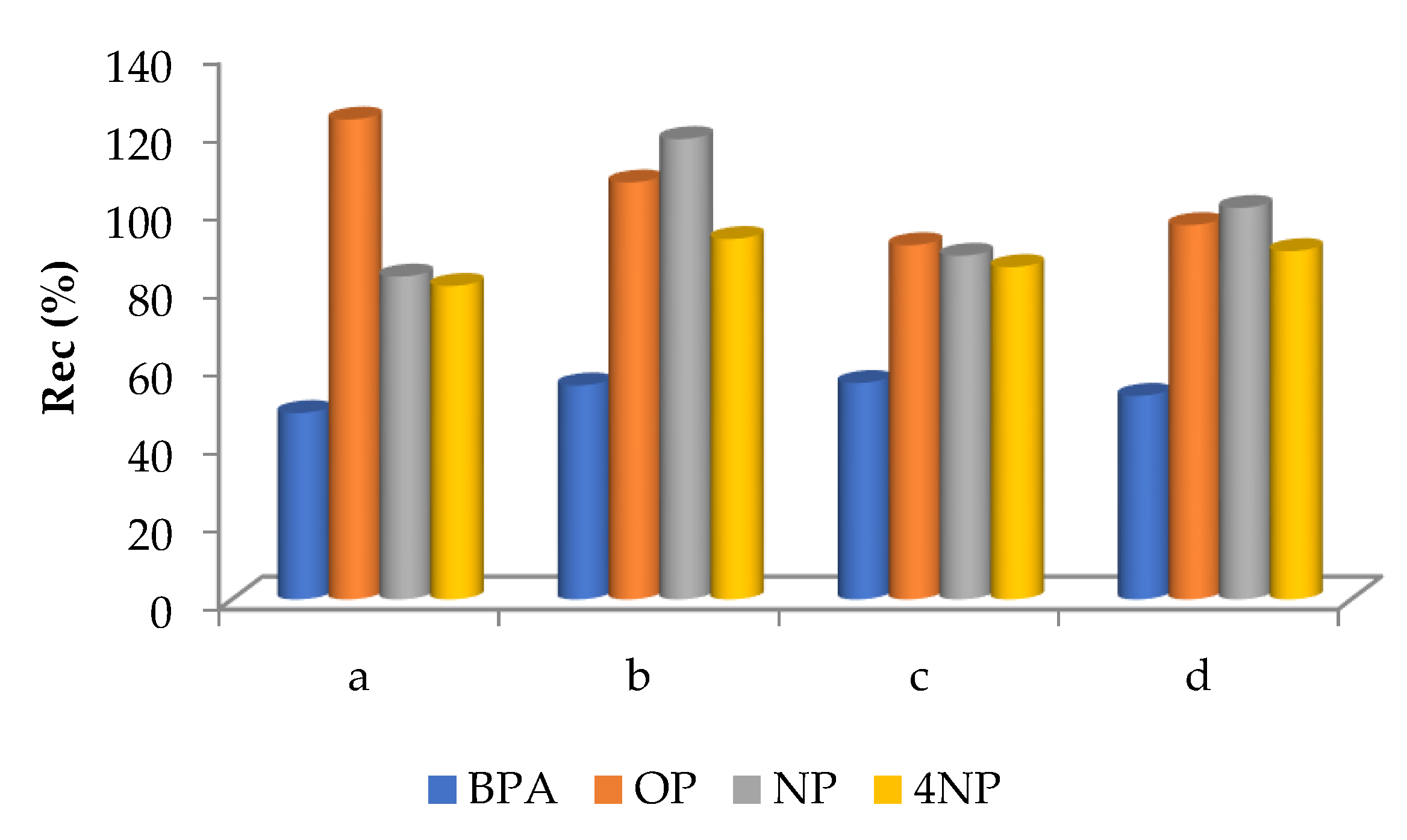
3.2.1. Preliminary Experiments
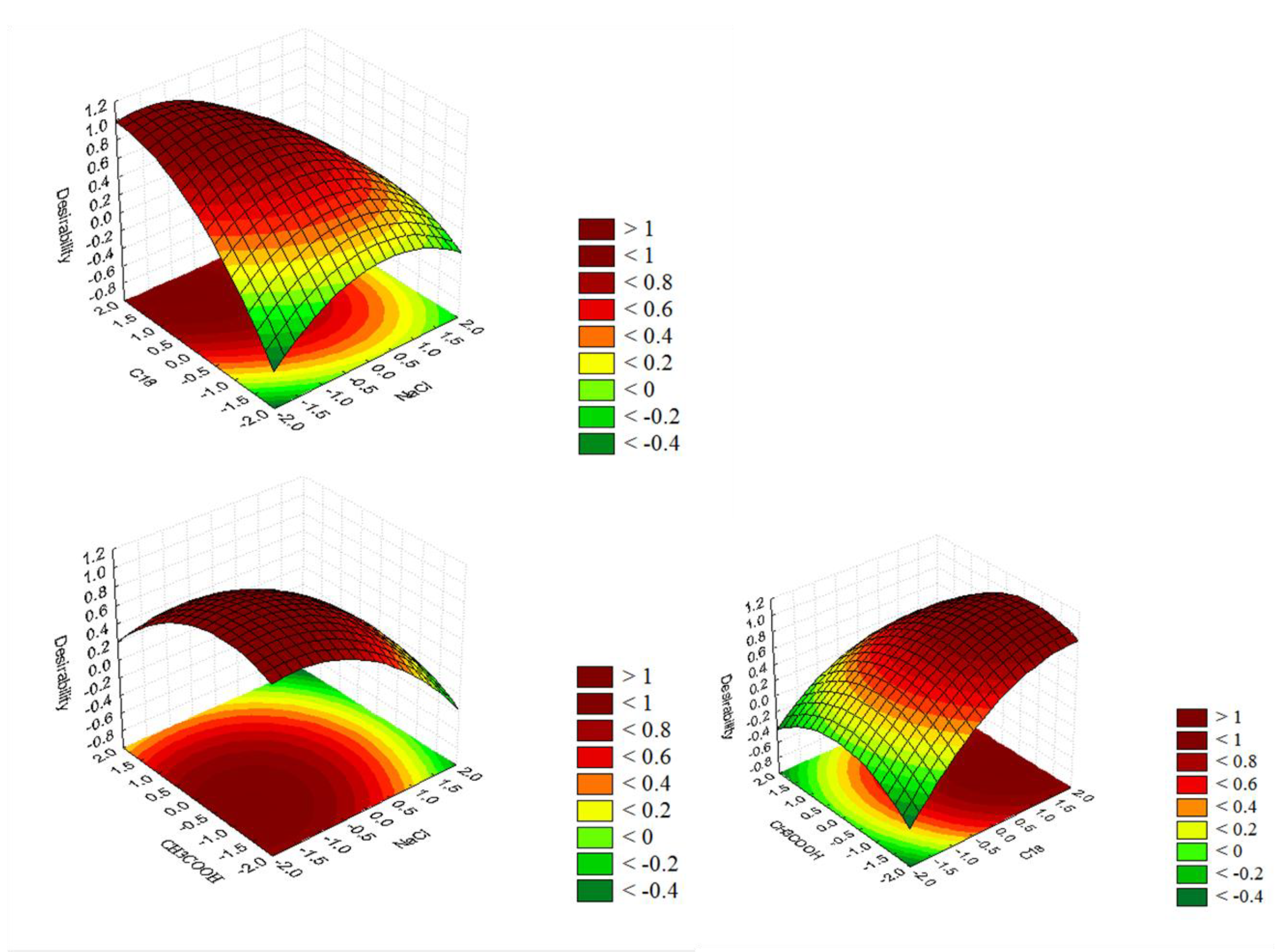
3.2.2. Multivariable Method
3.3. Analytical Performance of the Method
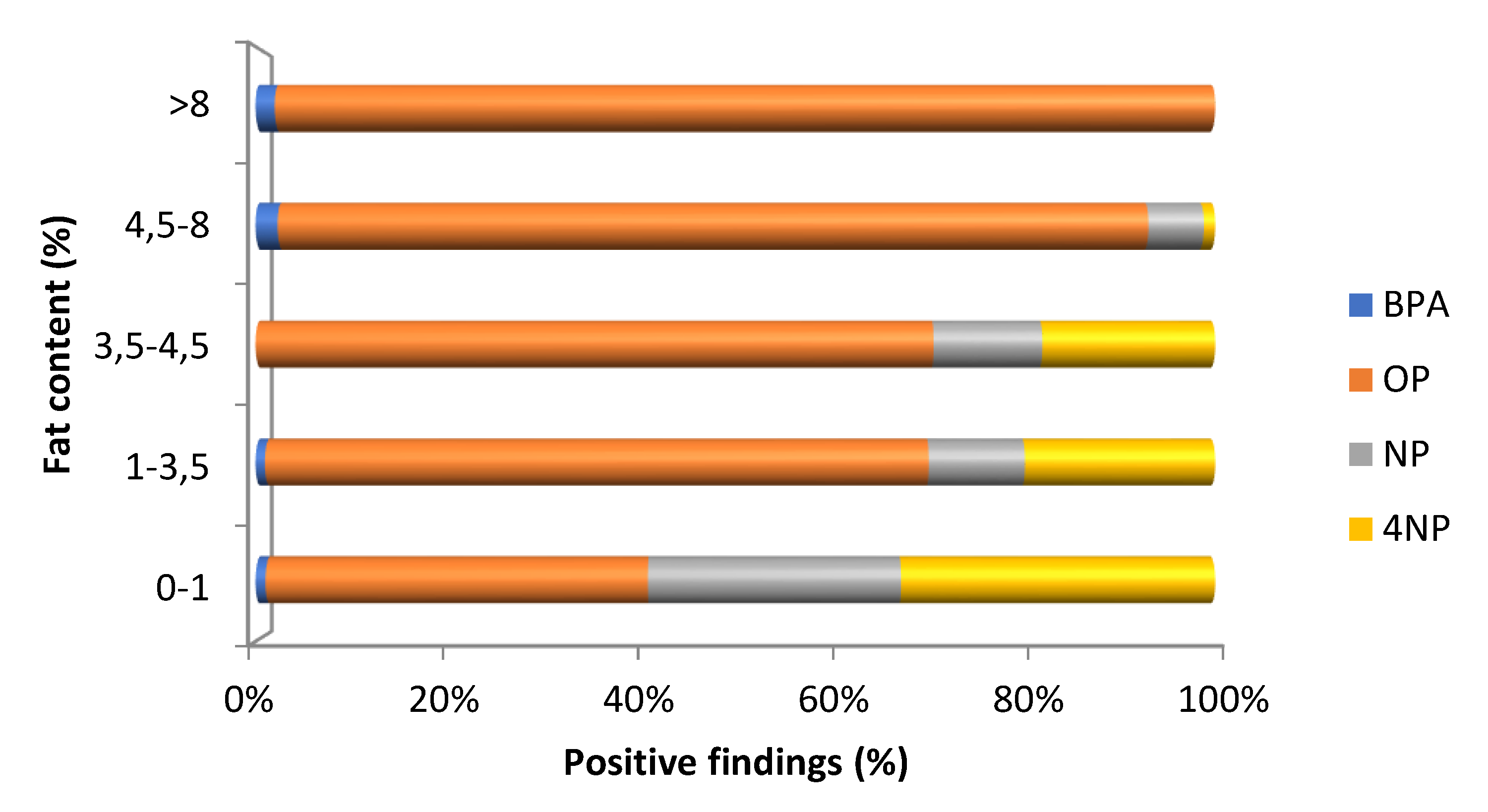
3.4. Application to Real Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colborn, T.; Dumanoski, D.; Myers, J.P. Our Stolen Future: Are We Threatening our Fertility, Intelligence and Survival? A Scientific Detective Story; Dutton: New York, NY, USA, 1997. [Google Scholar]
- Guillete, L.; Crain, D.A. Environmental Endocrine Disrupters: An Evolutionary Perspective; Taylor & Francis, Inc.: Abingdon, UK, 2000. [Google Scholar]
- Shelby, M.D. NTP-CERHR monograph on the potential human reproductive and developmental effects of bisphenol A. NTP CERHR MON. 2008, 22, vii–ix, 1–64. [Google Scholar]
- Zhou, R.Z.; Jiang, J.; Mao, T.; Zhao, Y.S.; Lu, Y. Multiresidue analysis of environmental pollutants in edible vegetable oils by gas chromatography–tandem mass spectrometry. Food Chem. 2016, 207, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Vazquez-Duhalt, R.; Marquez-Rocha, F.; Ponce, E.; Licea, A.F.; Viana, M.T. Nonyphenol, an integrated vision of a pollutant. Appl. Ecol. Environ. Res. 2005, 4, 1–25. [Google Scholar] [CrossRef]
- Commission Directive 2013/39/EU amending Directives 2000/60/EC and 2008/105/EC as regards priority substances in the field of water policy. Off. J. Eur. Union 2013, L226, 1–17.
- Commission Directive 2003/53/EC. Amending for the 26th time the Council directive 76/769/EEC relating to restrictions on the marketing and use of certain dangerous substances and preparations (nonylphenol, nonylphenol ethoxylate and cement). Off. J. Eur. Union 2003, L178, 24–27.
- Chevolleau, S.; Debrauwer, L.; Stroheker, T.; Viglino, L.; Mourahib, I.; Meireles, M.H.; Grimaldi, M.; Balaguer, P.; Di Gioia, L. A consolidated method for screening the endocrine activity of drinking water. Food Chem. 2016, 213, 274–283. [Google Scholar] [CrossRef]
- Ferrer, E.; Santoni, E.; Vittori, S.; Font, G.; Mañes, J.; Sagratini, G. Simultaneous determination of bisphenol A, octylphenol, and nonylphenol by pressurised liquid extraction and liquid chromatography–tandem mass spectrometry in powdered milk and infant formulas. Food Chem. 2011, 126, 360–367. [Google Scholar] [CrossRef]
- Fairbairn, D.J.; Arnold, W.A.; Barber, B.L.; Kaufenberg, E.F.; Koskinen, W.C.; Novak, P.J.; Rice, P.J.; Swackhamer, D.L. Contaminants of Emerging Concern: Mass Balance and Comparison of Wastewater Effluent and Upstream Sources in a Mixed-Use Watershed. Environ. Sci. Technol. 2016, 50, 36–45. [Google Scholar] [CrossRef]
- Salgueiro-González, N.; Turnes-Carou, I.; Besada, V.; Muniategui-Lorenzo, S.; López-Mahía, P.; Prada-Rodríguez, D. Occurrence, distribution and bioaccumulation of endocrine disrupting compounds in water, sediment and biota samples from a European river basin. Sci. Total Environ. 2015, 529, 121–130. [Google Scholar] [CrossRef]
- Albero, B.; Sánchez-Brunete, C.; Miguel, E.; Tadeo, J.L. Application of matrix solid-phase dispersion followed by GC–MS/MS to the analysis of emerging contaminants in vegetables. Food Chem. 2017, 217, 660–667. [Google Scholar] [CrossRef]
- Chen, M.; Fan, Z.; Zhao, F.; Gao, F.; Mu, D.; Zhou, Y.; Shen, H.; Hu, J. Occurrence and Maternal Transfer of Chlorinated Bisphenol A and Nonylphenol in Pregnant Women and Their Matching Embryos. Environ. Sci. Technol. 2016, 50, 970–977. [Google Scholar] [CrossRef]
- Cunha, S.C.; Fernandes, J.O. Assessment of bisphenol A and bisphenol B in canned vegetables and fruits by gas chromatography-mass spectrometry after QuEChERS and dispersive liquid-liquid microextraction. Food Control 2013, 33, 549–555. [Google Scholar] [CrossRef]
- Khedr, A. Optimized extraction method for LC–MS determination of bisphenol A, melamine and di(2-ethylhexyl) phthalate in selected soft drinks, syringes, and milk powder. J. Chromatogr. B 2013, 930, 98–103. [Google Scholar] [CrossRef]
- EFSA Panel on Food Contact Materials, Enzymes, Flavourings and Processing Aids (CEF), Scientific Opinion on the risks to public health related to the presence of bisphenol A (BPA) in foodstuffs. EFSA J. 2015, 13, 3978. [CrossRef]
- Chen, J.; Mullin, C.A. Determination of nonylphenol ethoxylate and octylphenol ethoxylate surfactants in beehive samples by high performance liquid chromatography coupled to mass spectrometry. Food Chem. 2014, 158, 473–479. [Google Scholar] [CrossRef]
- Fasano, E.; Cirillo, T.; Esposito, F.; Lacorte, S. Migration of monomers and plasticizers from packed foods and heated microwave foods using QuEChERS sample preparation and gas chromatography/mass spectrometry. Food Sci. Technol. 2015, 64, 1015–1021. [Google Scholar] [CrossRef]
- Munaretto, J.S.; Ferronato, G.; Ribeiro, L.C.; Martins, M.L.; Adaime, M.B.; Zanella, R. Development of a multiresidue method for the determination of endocrine disrupters in fish fillet using gas chromatography–triple quadrupole tandem mass spectrometry. Talanta 2013, 116, 827–834. [Google Scholar] [CrossRef]
- Pastor-Belda, M.; Viñas, P.; Campillo, N.; Hernández-Córdoba, M. Magnetic solid phase extraction with CoFe2O4/oleic acid nanoparticles coupled to gas chromatography-mass spectrometry for the determination of alkylphenols in baby foods. Food Chem. 2017, 221, 76–81. [Google Scholar] [CrossRef]
- Wu, H.; Li, G.; Liu, S.; Hu, N.; Geng, D.; Chen, G.; Sun, Z.; Zhao, X.; Xia, L.; You, J. Monitoring the contents of six steroidal and phenolic endocrine disrupting chemicals in chicken, fish and aquaculture pond water samples using pre-column derivatization and dispersive liquid–liquid microextraction with the aid of experimental design methodology. Food Chem. 2016, 192, 98–106. [Google Scholar]
- Shao, B.; Han, H.; Tu, X.; Huang, L. Analysis of alkylphenol and bisphenol A in eggs and milk by matrix solid phase dispersion extraction and liquid chromatography with tandem mass spectrometry. J. Chromatogr. B 2007, 850, 412–416. [Google Scholar] [CrossRef]
- Gallart-Ayala, H.; Moyano, E.; Galceran, M.T. Analysis of bisphenols in soft drinks by on-line solid phase extraction fast liquid chromatography–tandem mass spectrometry. Anal. Chim. Acta 2011, 683, 227–233. [Google Scholar] [CrossRef]
- Perez, S.; Eichhorn, P.; Barcelo, D. Applications of Time-of-Flight and Orbitrap Mass Spectromentry in Environmental, Food, Doping and Forensic Analysis. In Wilson and Wilson’s Comprehensive Analytical Chemistry; Damia Barcelo, Ed.; Elsevier: Amsterdam, The Netherlands, 2016; Volume 71. [Google Scholar]
- Commission Directive 2002/63/EC establishing Community methods of sampling for the official control of pesticide residues in and on products of plant and animal origin and repealing Directive 79/700/EEC. Off. J. Eur. Communities 2002, L187, 30.
- Yan, H.; Liu, X.; Cui, F.; Yun, H.; Li, J.; Ding, S.; Yang, D.; Zhang, Z. Determination of amantadine and rimantadine in chicken muscle by QuEChERS pretreatment method and UHPLC coupled with LTQ Orbitrap mass spectrometry. J. Chromatogr. B 2013, 938, 8–13. [Google Scholar] [CrossRef]
- Zhao, H.; Xiang, L.; Li, J.; Yang, Z.; Fang, J.; Zhao, C.; Xu, S.; Cai, Z. Investigation on fragmentation pathways of bisphenols by using electrospray ionization Orbitrap mass spectrometry. Rapid Commun. Mass Spectrom. 2016, 30, 1901–1913. [Google Scholar] [CrossRef]
- Paíga, P.; Morais, S.; Oliva-Teles, T.; Correia, M.; Delerue-Matos, C.; Duarte, S.C.; Pena, A.; Lino, C.M. Extraction of ochratoxin A in bread samples by the QuEChERS methodology. Food Chem. 2012, 135, 2522–2528. [Google Scholar] [CrossRef][Green Version]
- Anastassiades, M.; Lehotay, S.J.; Stajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and dispersive solid-phase extraction for the determination of pesticide residues in produce. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef]
- Lehotay, S.J. Determination of pesticide residues in foods by acetonitrile extraction and partitioning with magnesium sulfate: Collaborative study. J. AOAC Int. 2007, 90, 485–520. [Google Scholar] [CrossRef]
- CEN. Foods of Plant Origin—Determination of Pesticide Residues Using GC-MS and/or LC-MS/MS Following Acetonitrile Extraction/Partitioning and Clean-Up by Dispersive SPE. QuEChERS-Method, EN 15662; British Standards: London, UK, 2008. [Google Scholar]
- European Commission. Guidance Document on Analytical Quality Control and Method Validation Procedures for Pesticides Residues Analysis in Food and Feed. SANTE/11813/2017. European Commission Directorate-General for Health and Food Safety; European Commission: Brussels, Belgium, 2017; pp. 1–46. [Google Scholar]
- European Commission. Analytical Quality Control and Method Validation for Pesticide Residues Analysis in Food and Feed (SANTE/12682/2019); European Commission: Brussels, Belgium, 2019; pp. 1–48. [Google Scholar]
- Guenther, K.; Heinke, V.; Thiele, B.; Kleist, E.; Prast, H.; Raecker, T. Endocrine Disrupting Nonylphenols are ubiquitous in food. Environ. Sci. Technol. 2002, 36, 1676–1680. [Google Scholar] [CrossRef]
- Raecker, T.; Thiele, B.; Boehme, R.M.; Guenther, K. Endocrine disrupting nonyl- and octylphenol in infant food in Germany: Considerable daily intake of nonylphenol for babies. Chemosphere 2011, 82, 1533–1540. [Google Scholar] [CrossRef]
- Casajuana, N.; Lacorte, S. New Methodology for the Determination of Phthalate Esters, Bisphenol A, Bisphenol A Diglycidyl Ether, and Nonylphenol in Commercial Whole Milk Samples. J. Agric. Food Chem. 2004, 52, 3702–3707. [Google Scholar] [CrossRef] [PubMed]
- Mezcua, M.; Martınez-Uroz, M.A.; Gomez-Ramos, M.M.; Gomez, M.J.; Navas, J.M.; Fernandez-Alba, A.R. Analysis of synthetic endocrine-disrupting chemicals in food: A review. Talanta 2012, 100, 90–106. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Zhang, J.; Duan, H.; Wub, Y.; Shao, B. Bisphenol A and nonylphenol in foodstuffs: Chinese dietary exposure from the 2007 total diet study and infant health risk from formulas. Food Chem. 2015, 167, 320–325. [Google Scholar] [CrossRef] [PubMed]
- Mercogliano, R.; Santonicola, S. Investigation on bisphenol A levels in human milk and dairy supply chain: A review. Food Chem. Toxicol. 2018, 114, 98–107. [Google Scholar] [CrossRef]
- EFSA. Opinion of the Scientific Panel on Food Additives, Flavourings, Processing Aids and Materials in Contact with Food on a request from the Commission related to 2,2-bis(4-hydroxyphenyl)propane (bisphenol A). EFSA J. 2006, 428, 1–75. [Google Scholar]
- Santonicola, S.; Carmela Ferrante, M.; Genni di Leo, N.; Murru, A.; Anastasio, R. Mercogliano Study on endocrine disruptors levels in raw milk from cow’s farms: Risk assessment. Ital. J. Food Saf. 2018, 7, 7668. [Google Scholar]
- Santonicola, S.; Carmela Ferrante, M.; Murru, N.; Galloand, R.P. Mercogliano Hot topic: Bisphenol A in cow milk and dietary exposure at the farm level. J. Dairy Sci. 2019, 102, 1007–1013. [Google Scholar] [CrossRef]
- National Dietary Guidelines for Adults. Institute of Preventive Medicine Environmental & Occupational Health; Prolepsis: Athens, Greek, 2014; ISBN 9789605035594. (In Greek) [Google Scholar]
- EFSA Panel on Food Contact Materials Enzymes Flavourings and Processing Aids (CEF), Scientific Opinion on Bisphenol A: Evaluation of a study investigating its neurodevelopmental toxicity, review of recent scientific literature on its toxicity and advice on the Danish risk assessment of Bisphenol, A. EFSA J. 2010, 8, 1829.
- Regulation (EU) 2018/213 of 12 February 2018 on the Use of Bisphenol A in Varnishes and Coatings Intended to Come into Contact with Food and Amending Regulation (EU) No 10/2011 as Regards the Use of that Substance in Plastic Food Contact Materials; EU Comission: Brussels, Belgium, 2018.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EDCs | Linearity Range (ng g−1) | R2 | Rec ± RSD (%) 5 ng g−1 | Rec ± RSD (%) 50 ng g−1 | LOQ ng g−1 | LOD ng g−1 |
|---|---|---|---|---|---|---|
| BPA | 0.1–200 | 0.9987 | 101 ± 4.2 | 97 ± 0.9 | 0.1 | 0.05 |
| OP | 0.1–10/10–200 | 0.9998 | 101 ± 11.7 | 91 ± 6.3 | 0.1 | 0.05 |
| NP | 5–200 | 0.9966 | 99 ± 6.7 | 96 ± 2 | 5 | 1.5 |
| 4NP | 20–200 | 0.9999 | 108 ± 6.4 | 100 ± 3 | 20 | 5 |
| EDCs | Average Concentration a (μg Κg−1) (±SD) | Concentration Range (μg Kg−1) | Positive Detections b (n) c | n.d.% |
|---|---|---|---|---|
| BPA | 9.4 (±4.2) | 9.1–10.4 | 7 (0) | 74.1 |
| OP | 174.1 (±128.0) | 0.5–301.4 | 26 (1) | 3.7 |
| NP | 137.7 (±93.0) | 36.7–398.8 | 8 (0) | 70.4 |
| 4NP | 296.7 (±130.3) | 66.1–428.7 | 5 (3) | 70.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boti, V.; Kobothekra, V.; Albanis, T.; Konstantinou, I. QuEChERS-Based Methodology for the Screening of Alkylphenols and Bisphenol A in Dairy Products Using LC-LTQ/Orbitrap MS. Appl. Sci. 2021, 11, 9358. https://doi.org/10.3390/app11209358
Boti V, Kobothekra V, Albanis T, Konstantinou I. QuEChERS-Based Methodology for the Screening of Alkylphenols and Bisphenol A in Dairy Products Using LC-LTQ/Orbitrap MS. Applied Sciences. 2021; 11(20):9358. https://doi.org/10.3390/app11209358
Chicago/Turabian StyleBoti, Vasiliki, Vasiliki Kobothekra, Triantafyllos Albanis, and Ioannis Konstantinou. 2021. "QuEChERS-Based Methodology for the Screening of Alkylphenols and Bisphenol A in Dairy Products Using LC-LTQ/Orbitrap MS" Applied Sciences 11, no. 20: 9358. https://doi.org/10.3390/app11209358
APA StyleBoti, V., Kobothekra, V., Albanis, T., & Konstantinou, I. (2021). QuEChERS-Based Methodology for the Screening of Alkylphenols and Bisphenol A in Dairy Products Using LC-LTQ/Orbitrap MS. Applied Sciences, 11(20), 9358. https://doi.org/10.3390/app11209358