
Development of a Microwave-Assisted Extraction Protocol for the Simultaneous Determination of Mycotoxins and Pesticide Residues in Apples by LC-MS/MS

 ,
,  ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Preparation of Stock and Working Standard Solutions
2.3. Instrumentation
2.4. Microwave-Assisted Extraction Optimization
2.5. Sample Preparation
2.6. QuEChERS Extraction
2.7. Method Validation
2.8. Statistics
3. Results
3.1. Microwave-Assisted Extraction Optimization Results
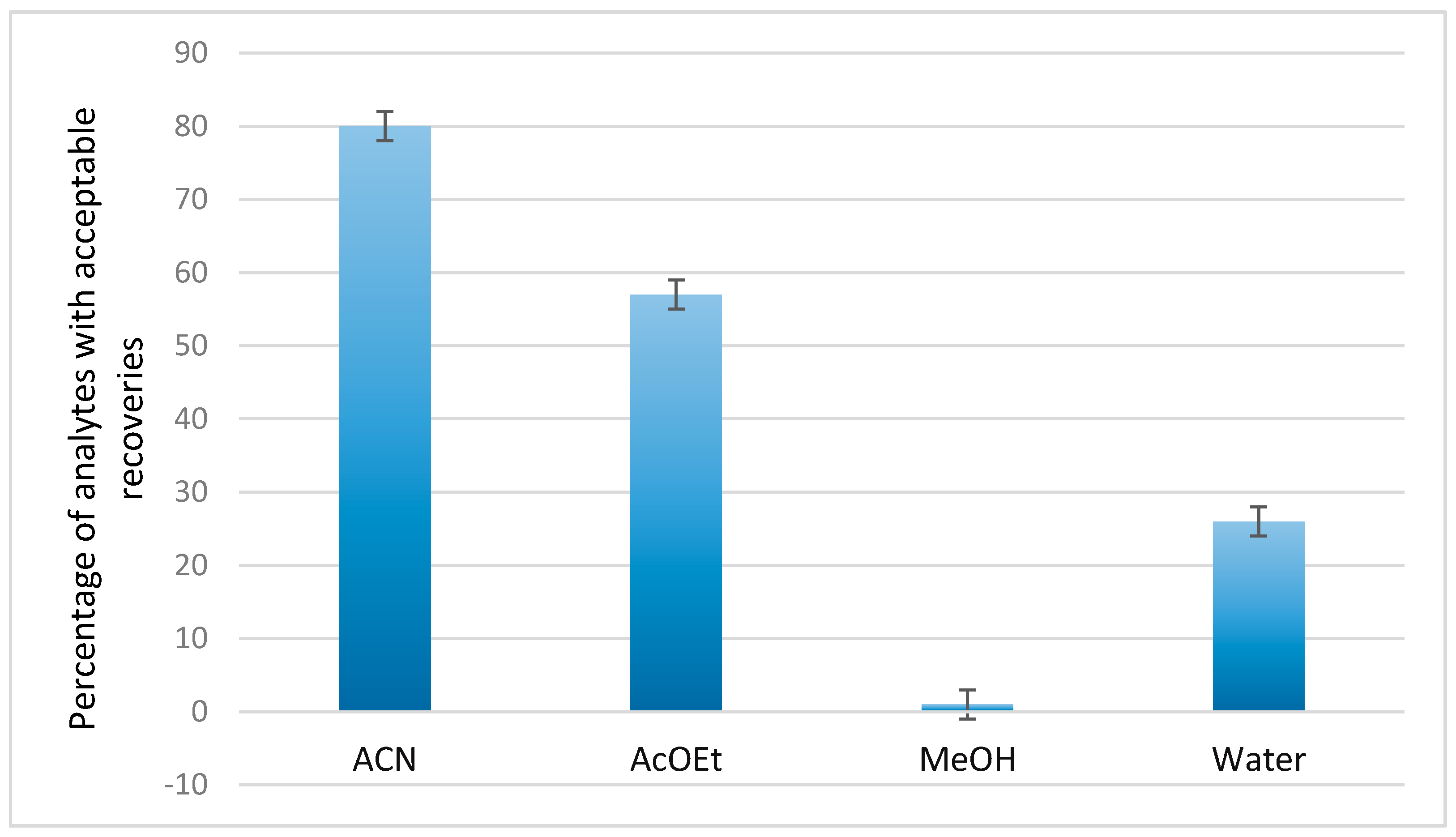
3.1.1. Extraction Solvent
3.1.2. Extraction Temperature
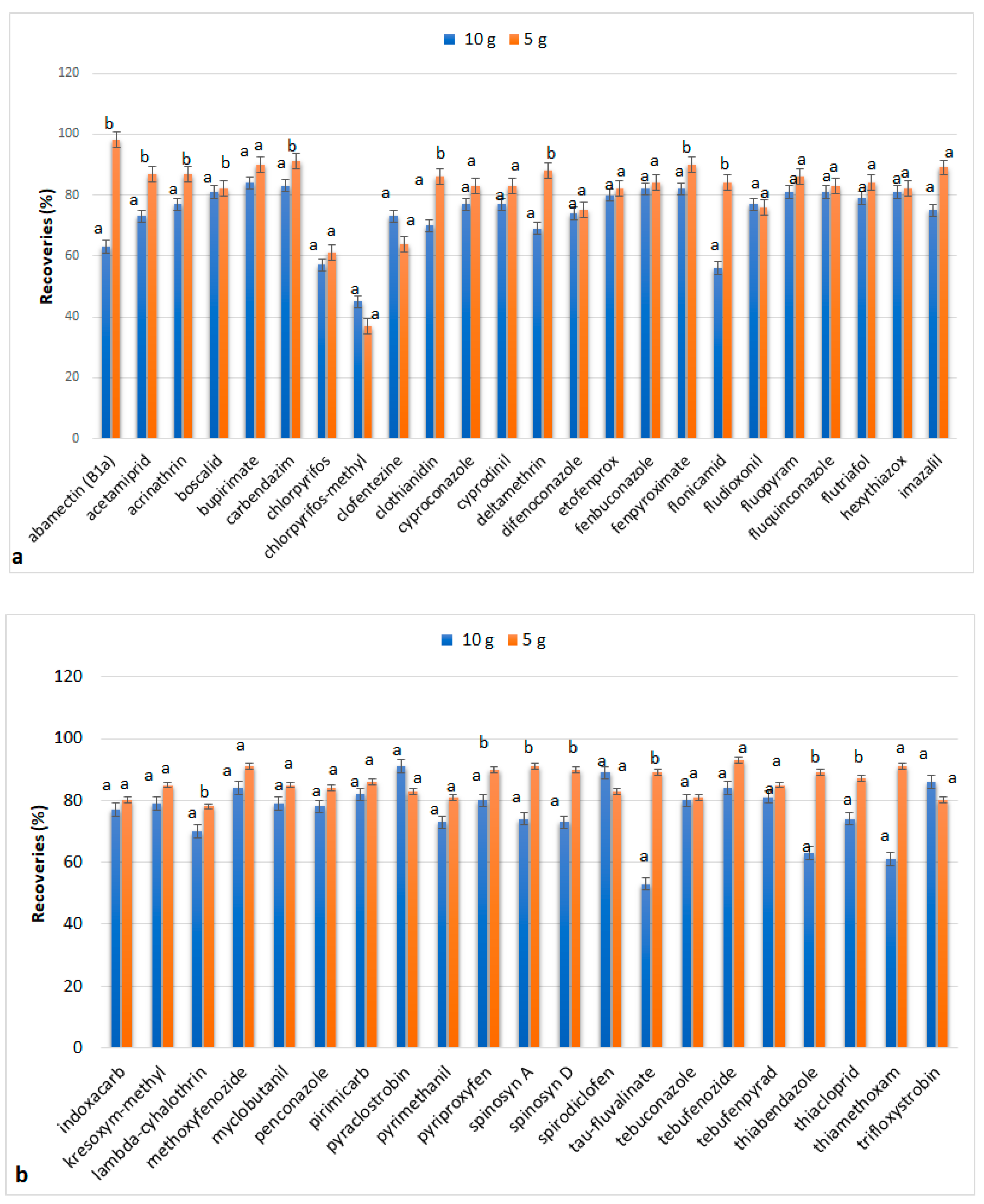
3.1.3. Sample Mass
3.1.4. Extraction Time
3.1.5. Acid Addition
3.2. Method Validation Results
3.3. Comparison with QuEChERS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Conflicts of Interest
References
- Patra, J.K.; Das, G.; Shin, H.S. Microbial Biotechnology; Springer: Singapore, 2018; Volume 2, ISBN 9789811071409. [Google Scholar]
- Kovač, M.; Bulaić, M.; Jakovljević, J.; Nevistić, A.; Rot, T.; Kovač, T.; Šarkanj, I.D.; Šarkanj, B. Mycotoxins, pesticide residues, and heavy metals analysis of croatian cereals. Microorganisms 2021, 9, 216. [Google Scholar] [CrossRef]
- European Commission Regulation (EC) No 396/2005, Maximum residue levels of pesticides in/on food and feed of plant and animal. Off. J. Eur. Union 2005, L70, 1–16.
- Konstantinou, S.; Karaoglanidis, G.S.; Bardas, G.A.; Minas, I.S.; Doukas, E.; Markoglou, A.N. Postharvest fruit rots of apple in Greece: Pathogen incidence and relationships between fruit quality parameters, cultivar susceptibility, and patulin production. Plant Dis. 2011, 95, 666–672. [Google Scholar] [CrossRef] [Green Version]
- Arcella, D.; Eskola, M.; Gómez Ruiz, J.A. Dietary exposure assessment to Alternaria toxins in the European population. EFSA J. 2016, 14, e04654. [Google Scholar] [CrossRef]
- Kalogiouri, N.P.; Thomaidis, N.S. Screening and High-Throughput Multi-Contaminants Methods. In Food Authentication: Management, Analysis and Regulation; John Wiley & Sons: Hoboken, NJ, USA, 2017. [Google Scholar]
- Reichert, B.; de Kok, A.; Pizzutti, I.R.; Scholten, J.; Cardoso, C.D.; Spanjer, M. Simultaneous determination of 117 pesticides and 30 mycotoxins in raw coffee, without clean-up, by LC-ESI-MS/MS analysis. Anal. Chim. Acta 2018, 1004, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yang, F.; Zhang, Y.; Zhou, K. Simultaneous Determination of Seven Carbamate Pesticide Residues in Vegetable by Capillary Electrophoresis with Solid Phase Microextraction. Int. J. Electrochem. Sci. 2021, 16, 1–14. [Google Scholar] [CrossRef]
- Mota, M.F.S.; Waktola, H.D.; Nolvachai, Y.; Marriott, P.J. Gas chromatography—Mass spectrometry for characterisation, assessment of quality and authentication of seed and vegetable oils. TrAC—Trends Anal. Chem. 2021, 138, 116238. [Google Scholar] [CrossRef]
- Vargas Medina, D.A.; Bassolli Borsatto, J.V.; Maciel, E.V.S.; Lanças, F.M. Current role of modern chromatography and mass spectrometry in the analysis of mycotoxins in food. TrAC—Trends Anal. Chem. 2021, 135. [Google Scholar] [CrossRef]
- Yang, Y.; Li, G.; Wu, D.; Liu, J.; Li, X.; Luo, P.; Hu, N.; Wang, H.; Wu, Y. Recent advances on toxicity and determination methods of mycotoxins in foodstuffs. Trends Food Sci. Technol. 2020, 96, 233–252. [Google Scholar] [CrossRef]
- Leite, M.; Freitas, A.; Silva, A.S.; Barbosa, J.; Ramos, F. Maize (Zea mays L.) and mycotoxins: A review on optimization and validation of analytical methods by liquid chromatography coupled to mass spectrometry. Trends Food Sci. Technol. 2020, 99, 542–565. [Google Scholar] [CrossRef]
- Katerere, D.R.; Stockenström, S.; Balducci, G.; Shephard, G.S. Determination of patulin in apple juice: Comparative evaluation of four analytical methods. J. AOAC Int. 2007, 90, 162–166. [Google Scholar] [CrossRef] [Green Version]
- Bletsou, A.A.; Jeon, J.; Hollender, J.; Archontaki, E.; Thomaidis, N.S. Targeted and non-targeted liquid chromatography-mass spectrometric workflows for identification of transformation products of emerging pollutants in the aquatic environment. TrAC—Trends Anal. Chem. 2015, 66, 32–44. [Google Scholar] [CrossRef] [Green Version]
- Fibigr, J.; Šatínský, D.; Solich, P. Current trends in the analysis and quality control of food supplements based on plant extracts. Anal. Chim. Acta 2018, 1036, 1–15. [Google Scholar] [CrossRef]
- Perestrelo, R.; Silva, P.; Porto-Figueira, P.; Pereira, J.A.M.; Silva, C.; Medina, S.; Câmara, J.S. QuEChERS—Fundamentals, relevant improvements, applications and future trends. Anal. Chim. Acta 2019, 1070, 1–28. [Google Scholar] [CrossRef]
- Nan, J.; Wang, J.; Piao, X.; Yang, C.; Wu, X.; Quinto, M.; Li, D. Novel and rapid method for determination of organophosphorus pesticide residues in edible fungus using direct gas purge microsyringe extraction coupled on-line with gas chromatography-mass spectrometry. Talanta 2015, 142, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Manousi, N.; Kabir, A.; Zachariadis, G.A. Recent advances in the extraction of triazine herbicides from water samples. J. Sep. Sci. 2021. [Google Scholar] [CrossRef]
- Kalogiouri, N.P.; Samanidou, V.F. Recent Trends in the Development of Green Microextraction Techniques for the Determination of Hazardous Organic Compounds in Wine. Curr. Anal. Chem. 2019, 15, 788–800. [Google Scholar] [CrossRef]
- Mastellone, G.; Marengo, A.; Sgorbini, B.; Rubiolo, P.; Cagliero, C. New phases for analytical scale extraction from plants: Current and future trends. TrAC—Trends Anal. Chem. 2021, 141, 116288. [Google Scholar] [CrossRef]
- Ihnat, M. Sample Preparation for Food Analysis; Elsevier: Amsterdam, The Netherlands, 2003; Volume 41, ISBN 9780444511010. [Google Scholar]
- Pallaroni, L.; Von Holst, C.; Eskilsson, C.S.; Björklund, E. Microwave-assisted extraction of zearalenone from wheat and corn. Anal. Bioanal. Chem. 2002, 374, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Liazid, A.; Palma, M.; Brigui, J.; Barroso, C.G. Investigation on Ochratoxin A stability using different extraction techniques. Talanta 2007, 71, 976–980. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, H. Development of a microwave-assisted-extraction-based method for the determination of aflatoxins B1, G1, B2, and G2 in grains and grain products. Anal. Bioanal. Chem. 2013, 405, 1623–1630. [Google Scholar] [CrossRef]
- Anastassiades, M.; Lehotay, S.J.; Štajnbaher, D.; Schenck, F.J. Fast and easy multiresidue method employing acetonitrile extraction/partitioning and. J. AOAC Int. 2003, 86, 412–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Commision. Guidance Document on Analytical Quality Control and Method Validation for Pesticide Residues Analysis in Food and Feed. 2019. Document No. SANTE/12682/2019. Available online: https://www.eurl-pesticides.eu/docs/public/tmplt_article.asp?CntID=727 (accessed on 16 November 2021).
- Satpathy, G.; Tyagi, Y.K.; Gupta, R.K. A novel optimised and validated method for analysis of multi-residues of pesticides in fruits and vegetables by microwave-assisted extraction (MAE)-dispersive solid-phase extraction (d-SPE)-retention time locked (RTL)-gas chromatography-mass spectrometry. Food Chem. 2011, 127, 1300–1308. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, E.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Determination of patulin in apple and derived products by uhplc-ms/ms. Study of matrix effects with atmospheric pressure ionisation sources. Food Chem. 2014, 142, 400–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadok, I.; Stachniuk, A.; Staniszewska, M. Developments in the Monitoring of Patulin in Fruits Using Liquid Chromatography: An Overview. Food Anal. Methods 2019, 12, 76–93. [Google Scholar] [CrossRef]
- Eskilsson, C.S.; Bjorklund, E. Analytical-scale microwave-assisted extraction. J. Chromatogr. A 2000, 902, 227–250. [Google Scholar] [CrossRef]
- Rizzetti, T.M.; Kemmerich, M.; Martins, M.L.; Prestes, O.D.; Adaime, M.B.; Zanella, R. Optimization of a QuEChERS based method by means of central composite design for pesticide multiresidue determination in orange juice by UHPLC-MS/MS. Food Chem. 2016, 196, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Jiang, N.; Xian, H.; Wei, D.; Shi, L.; Feng, X. A single-step solid phase extraction for the simultaneous determination of 8 mycotoxins in fruits by ultra-high performance liquid chromatography tandem mass spectrometry. J. Chromatogr. A 2016, 1429, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Bester, K.; Bordin, G.; Rodriguez, A.; Schimmel, H.; Pauwels, J.; VanVyncht, G. How to overcome matrix effects in the determination of pesticides in fruit by HPLC-ESI-MS-MS. Anal. Bioanal. Chem. 2001, 371, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Lagunas-Allué, L.; Sanz-Asensio, J.; Martínez-Soria, M.T. Comparison of four extraction methods for the determination of fungicide residues in grapes through gas chromatography-mass spectrometry. J. Chromatogr. A 2012, 1270, 62–71. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Accuracy | Repeatability | Reproducibility | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Recoveries (%) | RSD (%) | Recoveries (%) | RSD (%) | LOQ | LOD | |||||
| 0.01 μg/g | 0.03 μg/g | 0.1 μg/g | 1 μg/g | 0.03 μg/g | 0.1 μg/g | 0.1 μg/g | 0.1 μg/g | (μg/g) | (μg/g) | |
| abamectin (B1a) | - | 100 | 98 | 82 | 7 | 19 | 88 | 18 | 0.030 | 0.005 |
| acetamiprid | 107 | 92 | 87 | 88 | 7 | 9 | 87 | 10 | 0.010 | 0.006 |
| acrinathrin | - | 80 | 87 | 70 | 18 | 15 | 98 | 20 | 0.030 | 0.011 |
| alternariol | 120 | 106 | 80 | 81 | 9 | 4 | 77 | 16 | 0.010 | 0.008 |
| alternariol monomethyl ether | 116 | 105 | 82 | 89 | 10 | 6 | 88 | 11 | 0.010 | 0.009 |
| boscalid | 107 | 97 | 82 | 86 | 6 | 6 | 94 | 14 | 0.010 | 0.007 |
| bupirimate | - | 89 | 90 | 81 | 13 | 8 | 91 | 17 | 0.030 | 0.007 |
| carbendazim | 108 | 104 | 91 | 99 | 19 | 12 | 91 | 15 | 0.010 | 0.005 |
| chlorpyrifos | 86 | 53 | 61 | 59 | 7 | 10 | 51 | 18 | 0.010 | 0.007 |
| clofentezine | - | 57 | 64 | 52 | 11 | 5 | 60 | 22 | 0.010 | 0.009 |
| clothianidin | 101 | 92 | 86 | 88 | 9 | 4 | 88 | 12 | 0.010 | 0.006 |
| cyproconazole | 96 | 88 | 83 | 83 | 18 | 12 | 86 | 14 | 0.010 | 0.007 |
| cyprodinil | 109 | 84 | 83 | 73 | 13 | 7 | 89 | 19 | 0.010 | 0.006 |
| deltamethrin | - | 100 | 88 | 73 | 17 | 20 | 64 | 12 | 0.030 | 0.011 |
| difenoconazole | - | 90 | 75 | 80 | 10 | 7 | 81 | 11 | 0.030 | 0.009 |
| etofenprox | - | 97 | 82 | 85 | 19 | 15 | 74 | 9 | 0.030 | 0.011 |
| fenbuconazole | 115 | 95 | 84 | 81 | 11 | 12 | 85 | 16 | 0.010 | 0.007 |
| fenpyroximate | - | 101 | 90 | 86 | 11 | 5 | 76 | 7 | 0.030 | 0.014 |
| flonicamid | 102 | 93 | 84 | 83 | 9 | 8 | 88 | 14 | 0.010 | 0.009 |
| fludioxonil | 78 | 88 | 76 | 70 | 7 | 4 | 81 | 7 | 0.010 | 0.007 |
| fluopyram | 108 | 94 | 86 | 86 | 17 | 13 | 92 | 13 | 0.010 | 0.008 |
| fluquinconazole | 97 | 88 | 83 | 87 | 14 | 9 | 96 | 18 | 0.010 | 0.006 |
| flutriafol | 100 | 100 | 84 | 86 | 17 | 15 | 86 | 14 | 0.010 | 0.005 |
| hexythiazox | - | 100 | 82 | 90 | 18 | 6 | 80 | 14 | 0.030 | 0.014 |
| imazalil | 120 | 80 | 89 | 75 | 17 | 19 | 90 | 17 | 0.010 | 0.008 |
| indoxacarb | - | 100 | 80 | 86 | 16 | 5 | 87 | 25 | 0.03 | 0.012 |
| kresoxym-methyl | 114 | 89 | 85 | 87 | 11 | 4 | 86 | 20 | 0.010 | 0.011 |
| lambda-cyhalothrin | - | 107 | 78 | 74 | 10 | 5 | 67 | 16 | 0.030 | 0.015 |
| methoxyfenozide | 106 | 93 | 91 | 94 | 11 | 4 | 95 | 11 | 0.010 | 0.011 |
| myclobutanil | 101 | 93 | 85 | 83 | 12 | 11 | 90 | 15 | 0.010 | 0.007 |
| penconazole | 114 | 89 | 84 | 80 | 10 | 3 | 85 | 13 | 0.010 | 0.006 |
| pirimicarb | 105 | 85 | 86 | 86 | 17 | 12 | 83 | 13 | 0.010 | 0.008 |
| pyraclostrobin | - | 92 | 83 | 85 | 16 | 14 | 90 | 19 | 0.030 | 0.009 |
| pyrimethanil | 94 | 82 | 81 | 79 | 20 | 14 | 86 | 16 | 0.010 | 0.007 |
| pyriproxyfen | - | 101 | 90 | 92 | 19 | 15 | 81 | 11 | 0.030 | 0.013 |
| spinosyn A | - | 96 | 91 | 74 | 18 | 20 | 79 | 18 | 0.030 | 0.011 |
| spinosyn D | - | 97 | 90 | 70 | 9 | 5 | 72 | 17 | 0.030 | 0.014 |
| spirodiclofen | - | 86 | 83 | 85 | 15 | 11 | 75 | 11 | 0.030 | 0.014 |
| tau-fluvalinate | - | 91 | 89 | 70 | 14 | 9 | 89 | 24 | 0.03 | 0.011 |
| tebuconazole | 116 | 92 | 81 | 82 | 16 | 3 | 87 | 17 | 0.010 | 0.007 |
| tebufenozide | 115 | 93 | 93 | 94 | 9 | 7 | 96 | 13 | 0.010 | 0.011 |
| tebufenpyrad | - | 100 | 85 | 86 | 10 | 13 | 83 | 16 | 0.030 | 0.011 |
| thiabendazole | 97 | 88 | 89 | 78 | 16 | 11 | 92 | 17 | 0.010 | 0.010 |
| thiacloprid | 102 | 92 | 87 | 89 | 11 | 4 | 88 | 11 | 0.010 | 0.007 |
| thiamethoxam | 104 | 87 | 91 | 90 | 13 | 18 | 94 | 5 | 0.010 | 0.008 |
| trifloxystrobin | - | 96 | 80 | 86 | 10 | 16 | 88 | 16 | 0.030 | 0.011 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kalogiouri, N.P.; Papadakis, E.-N.; Maggalou, M.G.; Karaoglanidis, G.S.; Samanidou, V.F.; Menkissoglu-Spiroudi, U. Development of a Microwave-Assisted Extraction Protocol for the Simultaneous Determination of Mycotoxins and Pesticide Residues in Apples by LC-MS/MS. Appl. Sci. 2021, 11, 10931. https://doi.org/10.3390/app112210931
Kalogiouri NP, Papadakis E-N, Maggalou MG, Karaoglanidis GS, Samanidou VF, Menkissoglu-Spiroudi U. Development of a Microwave-Assisted Extraction Protocol for the Simultaneous Determination of Mycotoxins and Pesticide Residues in Apples by LC-MS/MS. Applied Sciences. 2021; 11(22):10931. https://doi.org/10.3390/app112210931
Chicago/Turabian StyleKalogiouri, Natasa P., Emmanouil-Nikolaos Papadakis, Maria G. Maggalou, George S. Karaoglanidis, Victoria F. Samanidou, and Urania Menkissoglu-Spiroudi. 2021. "Development of a Microwave-Assisted Extraction Protocol for the Simultaneous Determination of Mycotoxins and Pesticide Residues in Apples by LC-MS/MS" Applied Sciences 11, no. 22: 10931. https://doi.org/10.3390/app112210931
APA StyleKalogiouri, N. P., Papadakis, E.-N., Maggalou, M. G., Karaoglanidis, G. S., Samanidou, V. F., & Menkissoglu-Spiroudi, U. (2021). Development of a Microwave-Assisted Extraction Protocol for the Simultaneous Determination of Mycotoxins and Pesticide Residues in Apples by LC-MS/MS. Applied Sciences, 11(22), 10931. https://doi.org/10.3390/app112210931