
2D Solid-State HETCOR 1H-13C NMR Experiments with Variable Cross Polarization Times as a Tool for a Better Understanding of the Chemistry of Cellulose-Based Pyrochars—A Tutorial



Abstract
:Featured Application
Abstract
1. Introduction
2. Materials and Methods
2.1. Production of the Pyrochar
2.2. Elemental Composition and pH
2.3. Solid-State 13C NMR Spectroscopy
3. Results
3.1. Elemental Composition and pH of the Cellulose Pyrochar
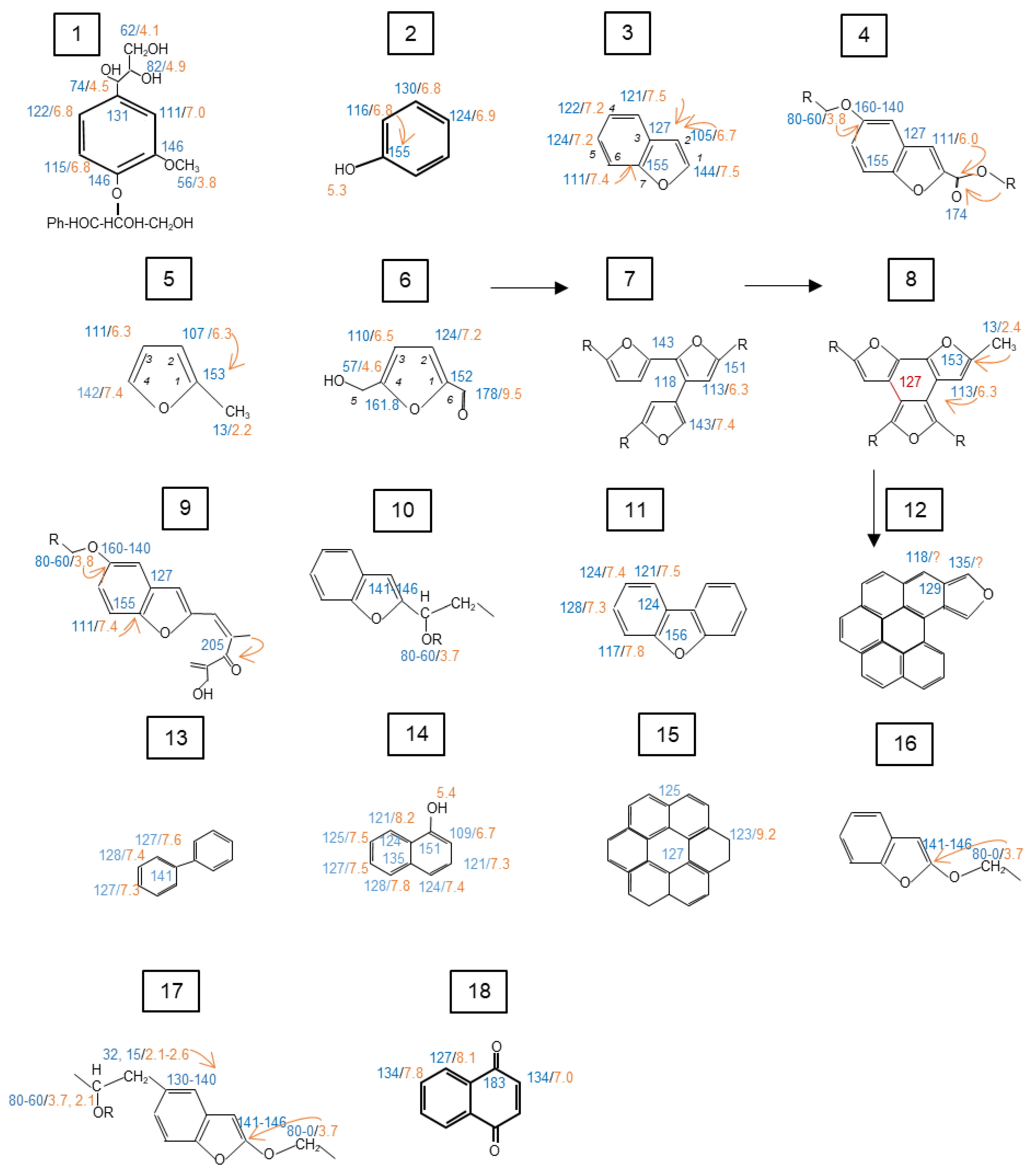
3.2. Characterization of Cellulose by 1D and 2D NMR Spectroscopy
3.2.1. Some Theoretical Background of the Cross-Polarization Experiment
3.2.2. Solid-State 1D CPMAS 13C NMR Spectrum of α–Cellulose
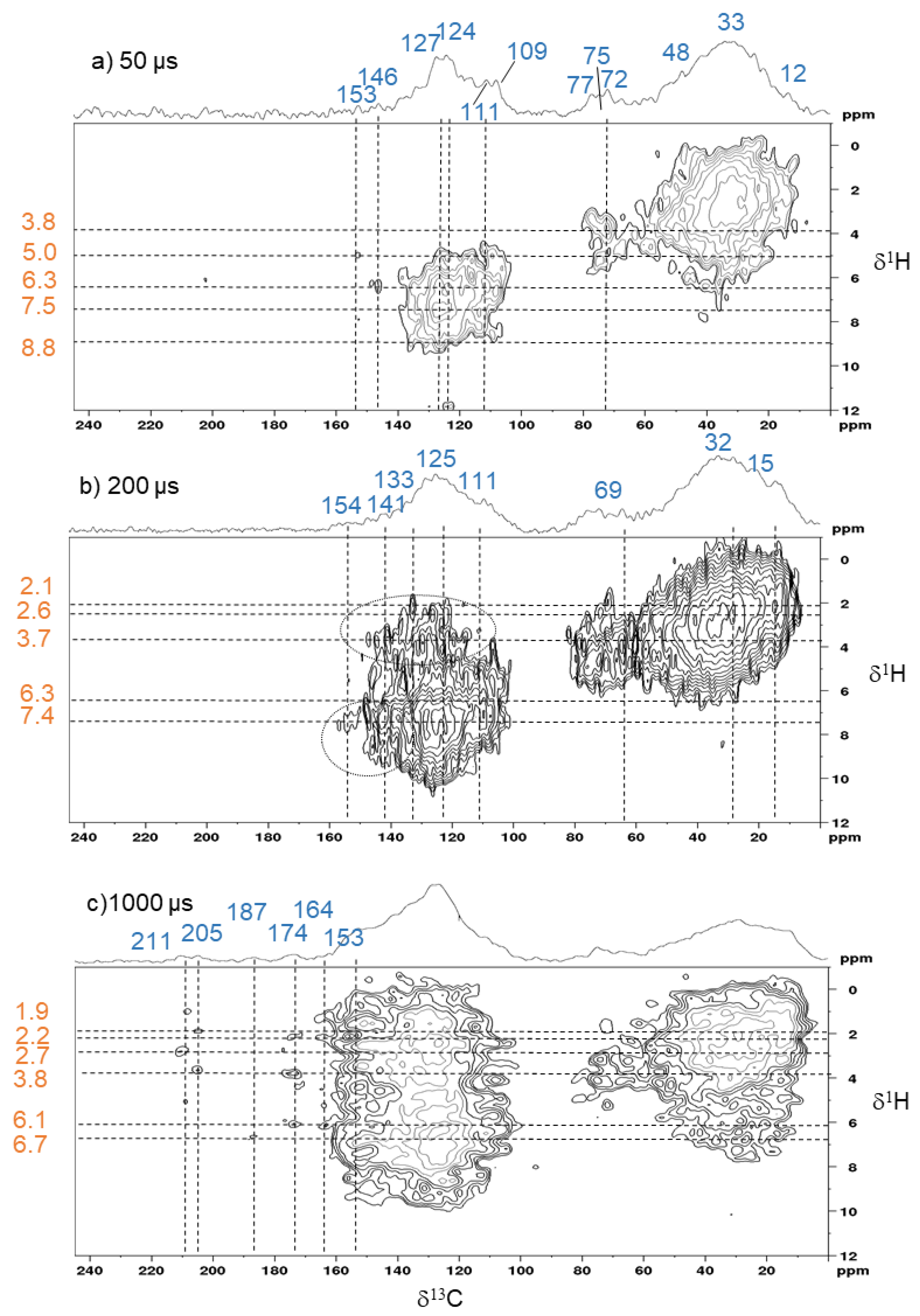
3.2.3. D Solid-State 1H-13C HETCOR NMR Spectra of α-Cellulose and Its T1H
3.3. Solid-State NMR Spectroscopic Characterization of Cellulose Pyrochar Pyrolzed at 350 °C
3.3.1. General Information Obtained by 1D Solid-State CPMAS 13C NMR Spectroscopy
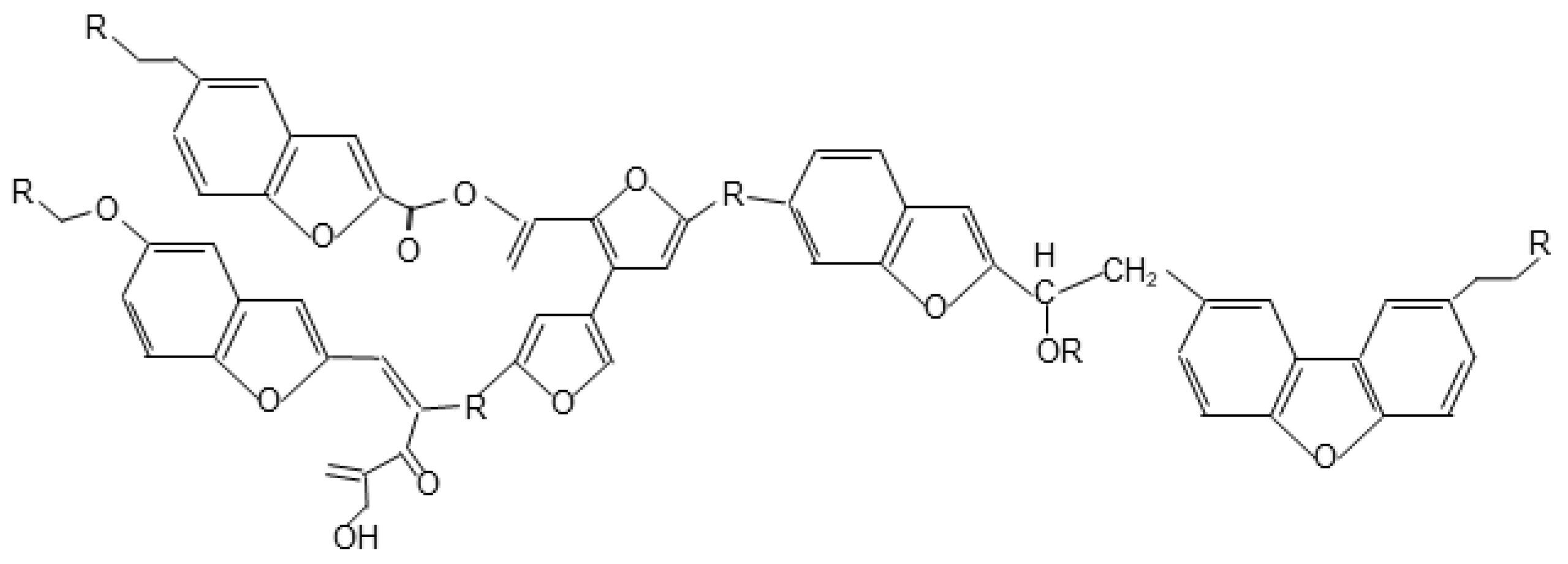
3.3.2. Solid-State 2D 1H-13C HETCOR NMR Spectra of the Cellulose Derived Pyrochar
4. Implications of the Findings for a Better Understanding of the Nature of Biochar
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- You, K.; Zhou, Z.; Gao, C.; Yang, Q. One-Step Preparation of Biochar Electrodes and Their Applications in Sediment Microbial Electrochemical Systems. Catalysts 2021, 11, 508. [Google Scholar] [CrossRef]
- Schmidt, H.P.; Hagemann, N.; Draper, K.; Kammann, C. The use of biochar in animal feeding. PeerJ 2019, 7, e7373. [Google Scholar] [CrossRef] [Green Version]
- Mendez, A.; Terradillos, M.; Gasco, G. Physicochemical and agronomic properties of biochar from sewage sludge pyrolysed at different temperatures. J. Anal. Appl. Pyrolysis 2013, 102, 124–130. [Google Scholar] [CrossRef]
- Nocentini, M.; Panettieri, M.; García de Castro Barragán, J.M.; Mastrolonardo, G.; Knicker, H. Recycling pyrolyzed organic waste from plant nurseries, rice production and shrimp industry as peat substitute in potting substrates. J. Environ. Manag. 2021, 277, 111436. [Google Scholar] [CrossRef]
- Campos, P.; De la Rosa, J.M. Assessing the Effects of Biochar on the Immobilization of Trace Elements and Plant Development in a Naturally Contaminated Soil. Sustainability 2020, 12, 6025. [Google Scholar] [CrossRef]
- Campos, P.; Miller, A.Z.; Prats, S.A.; Knicker, H.; Hagemann, N.; De la Rosa, J.M. Biochar amendment increases bacterial diversity and vegetation cover in trace element-polluted soils: A long-term field experiment. Soil Biol. Biochem. 2020, 150, 108014. [Google Scholar] [CrossRef]
- Medina, J.; Monreal, C.M.; Antilén, M.; Calabi-Floody, M.; Velasco-Molina, M.; Meier, S.; Borie, F.; Cornejo, P.; Knicker, H. Influence of inorganic additives on wheat straw composting: Characterization and structural composition of organic matter derived from the process. J. Environ. Manag. 2020, 260, 110137. [Google Scholar] [CrossRef]
- Antonangelo, J.A.; Sun, X.; Zhang, H. The roles of co-composted biochar (COMBI) in improving soil quality, crop productivity, and toxic metal amelioration. J. Environ. Manag. 2021, 277, 111443. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; He, L.; Lu, K.; Sarmah, A.; Li, J.; Bolan, N.S.; Pei, J.; Huang, H. Using biochar for remediation of soils contaminated with heavy metals and organic pollutants. Environ. Sci. Pollut. Res. 2013, 20, 8472–8483. [Google Scholar] [CrossRef]
- Wiedemeier, D.B.; Abiven, S.; Hockaday, W.C.; Keiluweit, M.; Kleber, M.; Masiello, C.A.; McBeath, A.V.; Nico, P.S.; Pyle, L.A.; Schneider, M.P.W.; et al. Aromaticity and degree of aromatic condensation of char. Org. Geochem. 2015, 78, 135–143. [Google Scholar] [CrossRef] [Green Version]
- Novak, J.M.; Busscher, W.J. Selection and Use of Designer Biochars to Improve Characteristics of Southeastern USA Coastal Plain Degraded Soils. In Advanced Biofuels and Bioproducts; Lee, J.W., Ed.; Springer: New York, NY, USA, 2013; pp. 69–96. ISBN 978-1-4614-3348-4. [Google Scholar]
- Zhao, S.-X.; Ta, N.; Wang, X.-D. Effect of Temperature on the Structural and Physicochemical Properties of Biochar with Apple Tree Branches as Feedstock Material. Energies 2017, 10, 1293. [Google Scholar] [CrossRef] [Green Version]
- Campos, P.; Miller, A.Z.; Knicker, H.; Costa-Pereira, M.F.; Merino, A.; De la Rosa, J.M. Chemical, physical and morphological properties of biochars produced from agricultural residues: Implications for their use as soil amendment. Waste Manag. 2020, 105, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Song, H.; Pan, S.; Dong, N.; Wang, X.; Sun, S. Initial pyrolysis mechanism and product formation of cellulose: An Experimental and Density functional theory(DFT) study. Sci. Rep. 2020, 10, 3626. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yan, R.; Chen, H.; Lee, D.H.; Zheng, C. Characteristics of hemicellulose, cellulose and lignin pyrolysis. Fuel 2007, 86, 1781–1788. [Google Scholar] [CrossRef]
- Lédé, J. Cellulose pyrolysis kinetics: An historical review on the existence and role of intermediate active cellulose. J. Anal. Appl. Pyrolysis 2012, 94, 17–32. [Google Scholar] [CrossRef]
- Lehmann, J. A handful of carbon. Nature 2007, 447, 143–144. [Google Scholar] [CrossRef] [PubMed]
- van Rossum, B.J.; Förster, H.; de Groot, H.J.M. High-Field and High-Speed CP-MAS13C NMR Heteronuclear Dipolar-Correlation Spectroscopy of Solids with Frequency-Switched Lee–Goldburg Homonuclear Decoupling. J. Magn. Reson. 1997, 124, 516–519. [Google Scholar] [CrossRef]
- Bronnimann, C.E.; Ridenour, C.F.; Kinney, D.R.; Maciel, G.E. 2D 1H-133C heteronuclear correlation spectra of representative organic solids. J. Magn. Reson. 1992, 97, 522–534. [Google Scholar] [CrossRef]
- Lattao, C.; Birdwell, J.; Wang, J.J.; Cook, R.L. Studying organic matter molecular assemblage within a whole organic soil by nuclear magnetic resonance. J. Environ. Qual. 2008, 37, 1501–1509. [Google Scholar] [CrossRef]
- Mao, J.D.; Xing, B.; Schmidt-Rohr, K. New structural information on a humic acid from two-dimensional 1H-13C correlation solid-state nuclear magnetic resonance. Environ. Sci. Technol. 2001, 35, 1928–1934. [Google Scholar] [CrossRef]
- Chu, W.; Cao, X.; Schmidt-Rohr, K.; Birdwell, J.E.; Mao, J. Investigation into the Effect of Heteroatom Content on Kerogen Structure Using Advanced 13 C Solid-State Nuclear Magnetic Resonance Spectroscopy. Energy Fuels 2019, 33, 645–653. [Google Scholar] [CrossRef]
- Le Brech, Y.; Delmotte, L.; Raya, J.; Brosse, N.; Gadiou, R.; Dufour, A. High resolution solid state 2D NMR analysis of biomass and biochar. Anal. Chem. 2015, 87, 843–847. [Google Scholar] [CrossRef] [PubMed]
- Fengel, D.; Wegener, G. Wood: Chemistry, Ultrastructure, Reaction; De Gruyter: Berlin, Germany, 1984. [Google Scholar]
- Haider, K. Problems related to the humification processes in soils of temperate climate. In Soil Biochemistry; Bollag, J.-M., Stotzky, G., Eds.; Marcel Dekker: New York, NY, USA, 1991; Volume 7, pp. 55–94. [Google Scholar]
- Sun, H.; Hockaday, W.C.; Masiello, C.A.; Zygourakis, K. Multiple Controls on the Chemical and Physical Structure of Biochars. Ind. Eng. Chem. Res. 2012, 51, 3587–3597. [Google Scholar] [CrossRef]
- Mehring, M. High Resolution NMR in Solids, 2nd ed.; Springer: Berlin, Germany, 1983. [Google Scholar]
- Abelmann, K.; Totsche, K.U.; Knicker, H.; Kögel-Knabner, I. CP dynamics of heterogeneous organic material: Characterization of molecular domains in coals. Solid State Nucl. Magn. Reson. 2004, 25, 252–266. [Google Scholar] [CrossRef] [PubMed]
- Knicker, H.; Hilscher, A.; González-Vila, F.J.; Almendros, G. A new conceptual model for the structural properties of char produced during vegetation fires. Org. Geochem. 2008, 39, 935–939. [Google Scholar] [CrossRef] [Green Version]
- Knicker, H. “Black nitrogen”—An important fraction in determining the recalcitrance of charcoal. Org. Geochem. 2010, 41, 947–950. [Google Scholar] [CrossRef]
- Schmidt, H.P.; Bucheli, T.; Kammann, C.; Glaser, B.; Abiven, S.; Leifeld, J. EBC European Biochar Certificate Guidelines for a Sustainable Production of Biochar. European Biochar Certificate. 2021. Available online: https://www.european-biochar.org/en/ct/2-EBC-guidelines-documents-for-the-certification (accessed on 6 August 2021).
- Teeäär, R.; Lippmaa, E. Solid state 13C NMR of cellulose. A relaxation study. Polym. Bull. 1984, 12, 315–318. [Google Scholar] [CrossRef]
- Pines, A.; Gibby, G.G.; Waugh, J.S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973, 59, 569–590. [Google Scholar] [CrossRef]
- Knicker, H. Solid state CPMAS 13C and 15N NMR spectroscopy in organic geochemistry and how spin dynamics can either aggravate or improve spectra interpretation. Org. Geochem. 2011, 42, 867–890. [Google Scholar] [CrossRef]
- Wilson, M.A. NMR Techniques and Applications in Geochemistry and Soil Chemistry; Pergamon Press: Oxford, UK, 1987. [Google Scholar]
- Kalinowski, H.-O.; Berger, S.; Braun, S. 13C NMR Spektroskopie; Thieme: Stuttgart, Germany, 1984. [Google Scholar]
- Earl, W.L.; VanderHart, D.L. Observations by high-resolution carbon-13 nuclear magnetic resonance of cellulose I related to morphology and crystal structure. Macromolecules 1981, 14, 570–574. [Google Scholar] [CrossRef]
- Atalla, R.H.; VanderHart, D.L. The role of solid state 13C NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl. Magn. Reson. 1999, 15, 1–19. [Google Scholar] [CrossRef]
- Wooten, J.B.; Seeman, J.I.; Hajaligol, M.R. Observation and Characterization of Cellulose Pyrolysis Intermediates by 13C CPMAS NMR. A New Mechanistic Model. Energy Fuels 2004, 18, 1–15. [Google Scholar] [CrossRef]
- McBeath, A.V.; Smernik, R.J.; Krull, E.S.; Lehmann, J. The influence of feedstock and production temperature on biochar carbon chemistry: A solid-state 13C NMR study. Biomass Bioenergy 2014, 60, 121–129. [Google Scholar] [CrossRef]
- Bourke, J.; Manley-Harris, M.; Fushimi, C.; Dowaki, K.; Nunoura, T.; Antal, M.J. Do All Carbonized Charcoals Have the Same Chemical Structure? 2. A Model of the Chemical Structure of Carbonized Charcoal. Ind. Eng. Chem. Res. 2007, 46, 5954–5967. [Google Scholar] [CrossRef]
- Ben, H.; Hao, N.; Liu, Q.; Ragauskas, A.J. Solid-State NMR Investigation of Bio-chars Produced from Biomass Components and Whole Biomasses. BioEnergy Res. 2017, 10, 1036–1044. [Google Scholar] [CrossRef]
- Falco, C.; Perez Caballero, F.; Babonneau, F.; Gervais, C.; Laurent, G.; Titirici, M.-M.; Baccile, N. Hydrothermal Carbon from Biomass: Structural Differences between Hydrothermal and Pyrolyzed Carbons via 13C Solid State NMR. Langmuir 2011, 27, 14460–14471. [Google Scholar] [CrossRef] [Green Version]
- Mettler, M.S.; Vlachos, D.G.; Dauenhauer, P.J. Top ten fundamental challenges of biomass pyrolysis for biofuels. Energy Environ. Sci. 2012, 5, 7797–7809. [Google Scholar] [CrossRef]
- Falco, C.; Baccile, N.; Titirici, M.-M. Morphological and structural differences between glucose, cellulose and lignocellulosic biomass derived hydrothermal carbons. Green Chem. 2011, 13, 3273. [Google Scholar] [CrossRef] [Green Version]
- Schulman, J.M.; Disch, R.L. Aromatic Character of [n]Helicenes and [n]Phenacenes. J. Phys. Chem. A 1999, 103, 6669–6672. [Google Scholar] [CrossRef]
- Pu, Y.; Hallac, B.; Ragauskas, A.J. Plant Biomass Characterization: Application of Solution- and Solid-State NMR Spectroscopy. In Aqueous Pretreatment of Plant Biomass for Biological and Chemical Conversion to Fuels and Chemicals; John Wiley & Sons, Ltd.: Chichester, UK, 2013; pp. 369–390. ISBN 9780470975831. [Google Scholar]
- Reich, H. Hans Reich’s Collection. NMR Spectroscopy. Available online: https://organicchemistrydata.org/hansreich/resources/nmr/?page=nmr-content%2F (accessed on 20 January 2021).
- Thonhauser, T.; Ceresoli, D.; Marzari, N. NMR shifts for polycyclic aromatic hydrocarbons from first-principles. Int. J. Quantum Chem. 2009, 109, 3336–3342. [Google Scholar] [CrossRef] [Green Version]
- Sekiguchi, Y.; Frye, J.S.; Shafizadeh, F. Structure and formation of cellulosic chars. J. Appl. Polym. Sci. 1983, 28, 3513–3525. [Google Scholar] [CrossRef]
- Baldock, J.A.; Smernik, R.J. Chemical composition and bioavailability of thermally altered Pinus resinosa (Red pine) wood. Org. Geochem. 2002, 33, 1093–1109. [Google Scholar] [CrossRef]
- Xin, S.; Yang, H.; Chen, Y.; Yang, M.; Chen, L.; Wang, X.; Chen, H. Chemical structure evolution of char during the pyrolysis of cellulose. J. Anal. Appl. Pyrolysis 2015, 116, 263–271. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Weight-Loss (%) | C (g/kg) | H (g/kg) | O 1 (g/kg) | H/Catm | O/Catm | pH | |||
|---|---|---|---|---|---|---|---|---|---|
| Ce25 | 0 | 419.1 | ±1.3 | 59.5 | ±0.5 | 521.4 | 1.7 | 0.9 | 7.2 |
| Ce350 | 64 | 701.1 | ±3.4 | 42.8 | ±1.0 | 256.1 | 0.7 | 0.3 | 5.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Knicker, H.; Velasco-Molina, M.; Knicker, M. 2D Solid-State HETCOR 1H-13C NMR Experiments with Variable Cross Polarization Times as a Tool for a Better Understanding of the Chemistry of Cellulose-Based Pyrochars—A Tutorial. Appl. Sci. 2021, 11, 8569. https://doi.org/10.3390/app11188569
Knicker H, Velasco-Molina M, Knicker M. 2D Solid-State HETCOR 1H-13C NMR Experiments with Variable Cross Polarization Times as a Tool for a Better Understanding of the Chemistry of Cellulose-Based Pyrochars—A Tutorial. Applied Sciences. 2021; 11(18):8569. https://doi.org/10.3390/app11188569
Chicago/Turabian StyleKnicker, Heike, Marta Velasco-Molina, and Michael Knicker. 2021. "2D Solid-State HETCOR 1H-13C NMR Experiments with Variable Cross Polarization Times as a Tool for a Better Understanding of the Chemistry of Cellulose-Based Pyrochars—A Tutorial" Applied Sciences 11, no. 18: 8569. https://doi.org/10.3390/app11188569
APA StyleKnicker, H., Velasco-Molina, M., & Knicker, M. (2021). 2D Solid-State HETCOR 1H-13C NMR Experiments with Variable Cross Polarization Times as a Tool for a Better Understanding of the Chemistry of Cellulose-Based Pyrochars—A Tutorial. Applied Sciences, 11(18), 8569. https://doi.org/10.3390/app11188569