
Inhibition of Breast Tumour Growth with Intravenously Administered PRKCA siRNA- and PTEN Tumour Suppressor Gene-Loaded Carbonate Apatite Nanoparticles


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Formulation of CA NPs
2.3. Isolation of Plasmid DNA for Gene Transfection
2.4. Reconstitution of PRKCA siRNA
2.5. Size and Zeta Potential Measurement
2.6. Cell Culture and Seeding
2.7. Cell Viability Assessment with MTT Assay
2.8. Western Blotting
2.9. Study of Tumor Regression Pattern in Breast Tumor-Induced Mouse Model (In Vivo)
2.10. Statistical Analysis
3. Results
3.1. Size and Zeta Potential of PRKCA Specific siRNA- and PTEN Plasmid-Loaded NPs
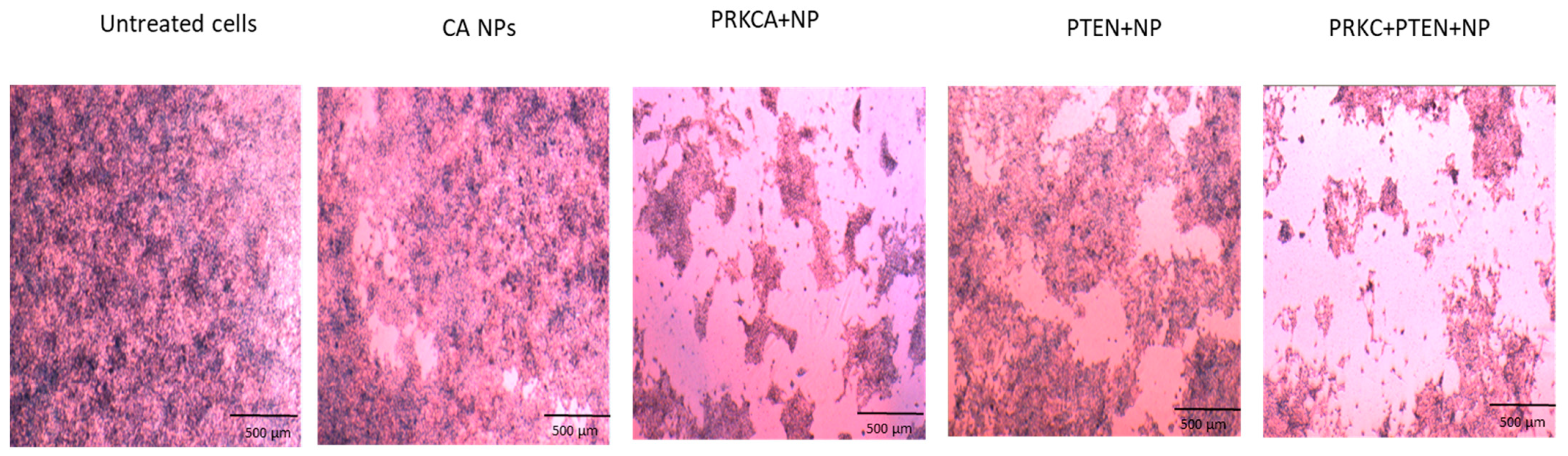
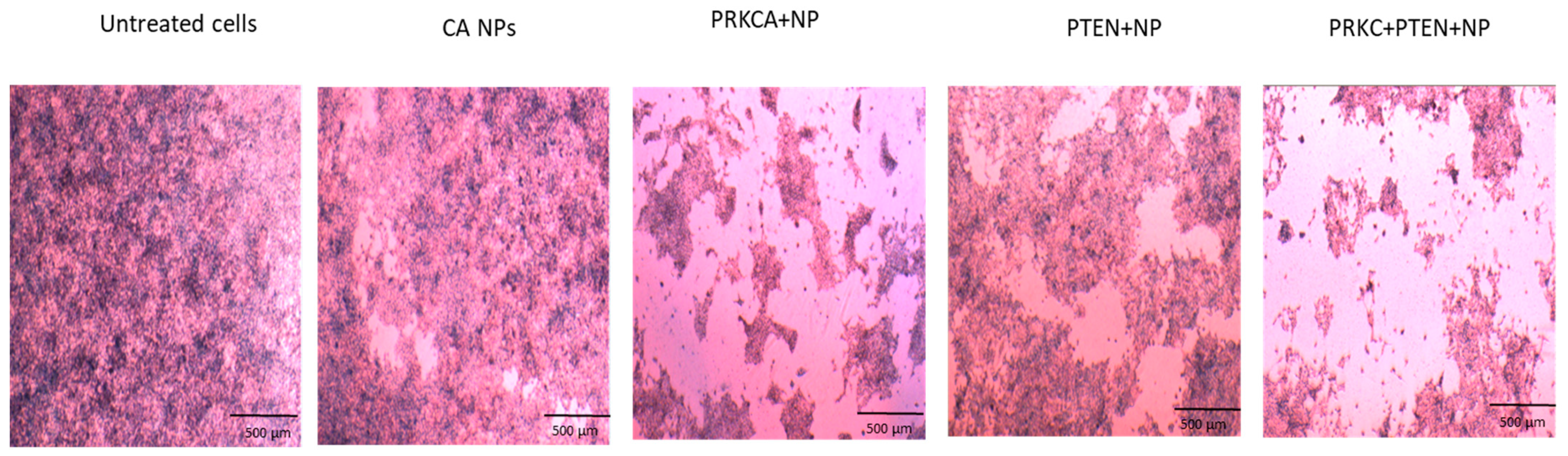
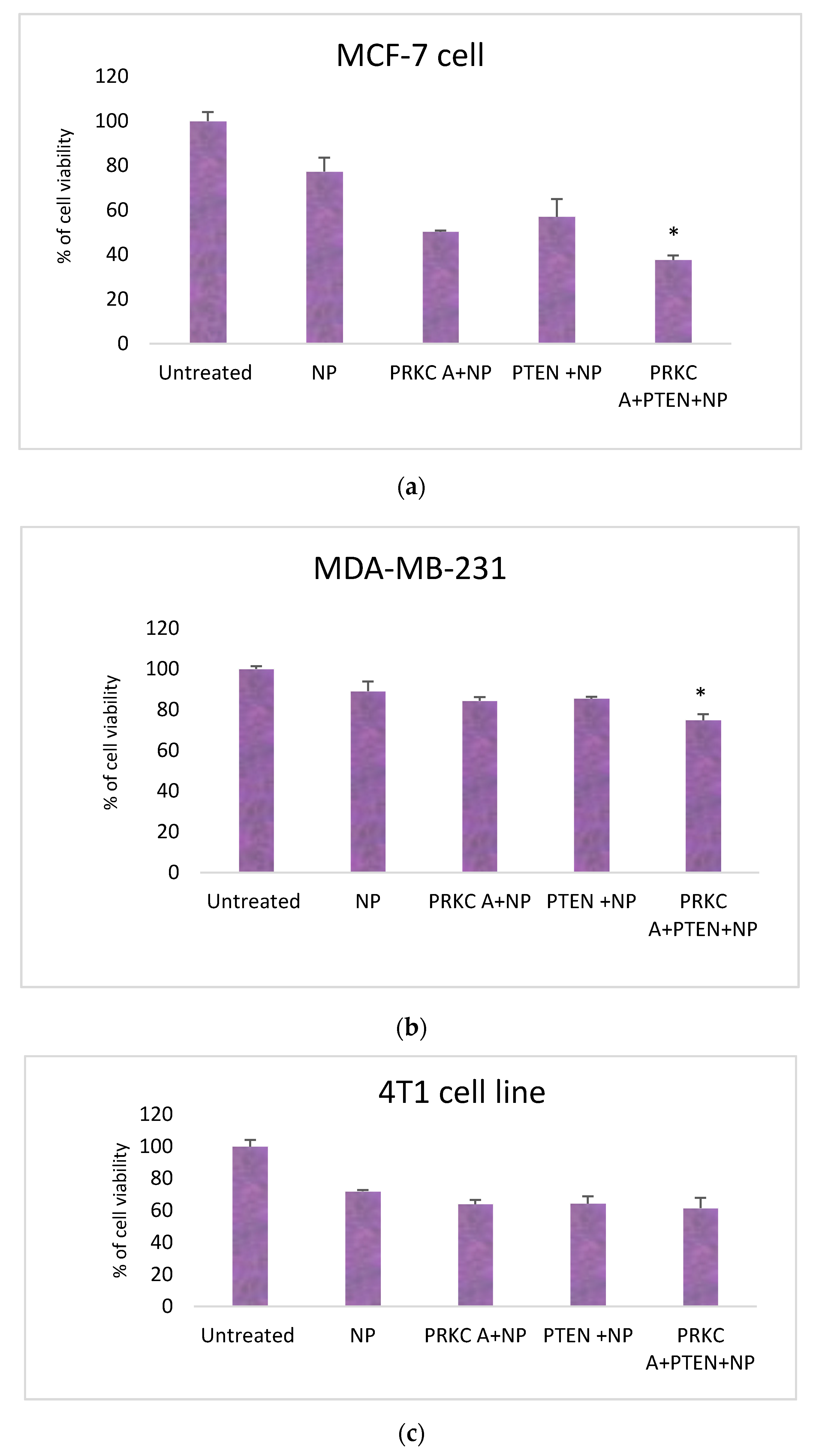
3.2. Co-Delivery of siRNA against PRKCA and PTEN Plasmid Reduced Growth of Breast Cancer Cell Lines
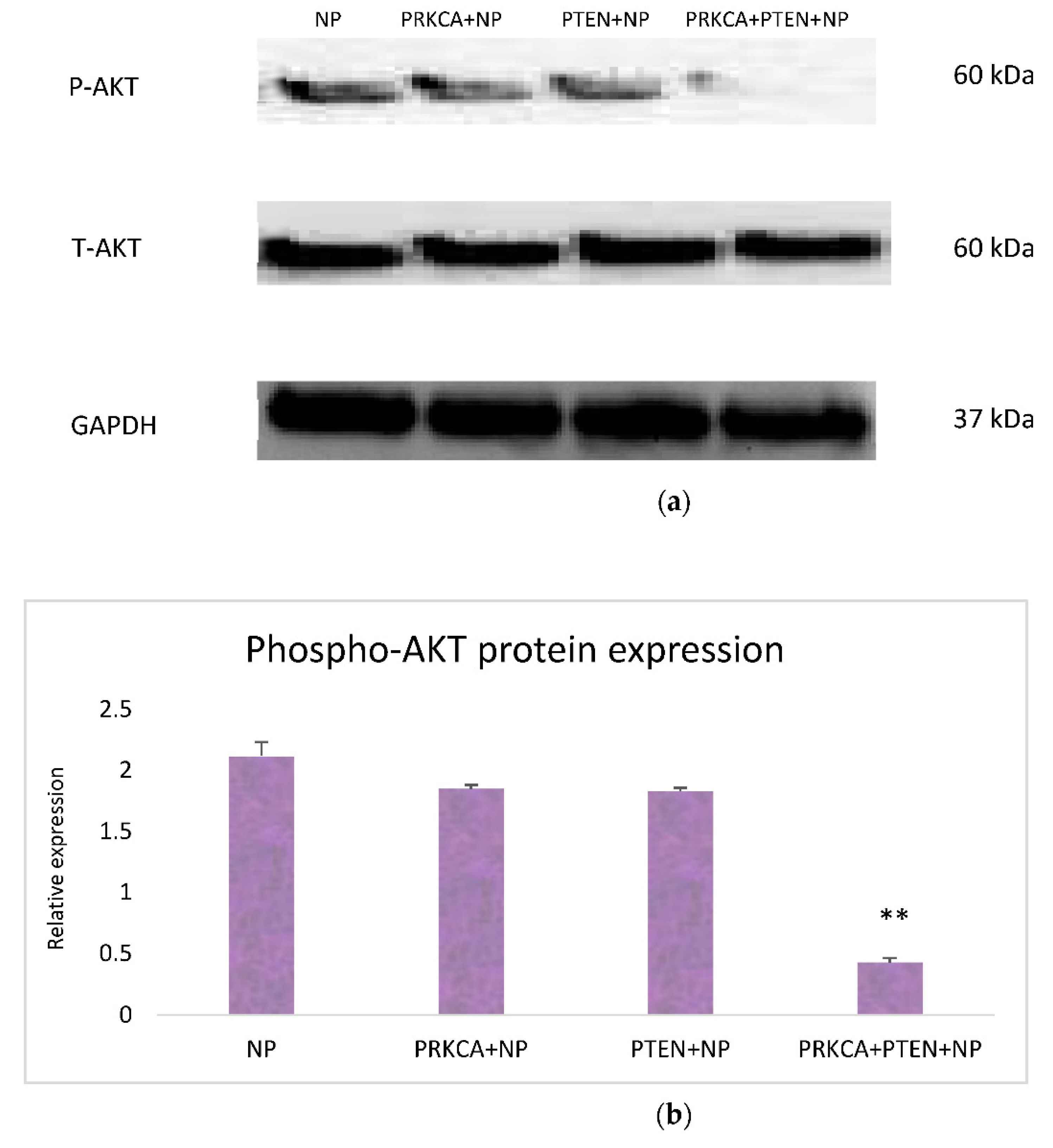
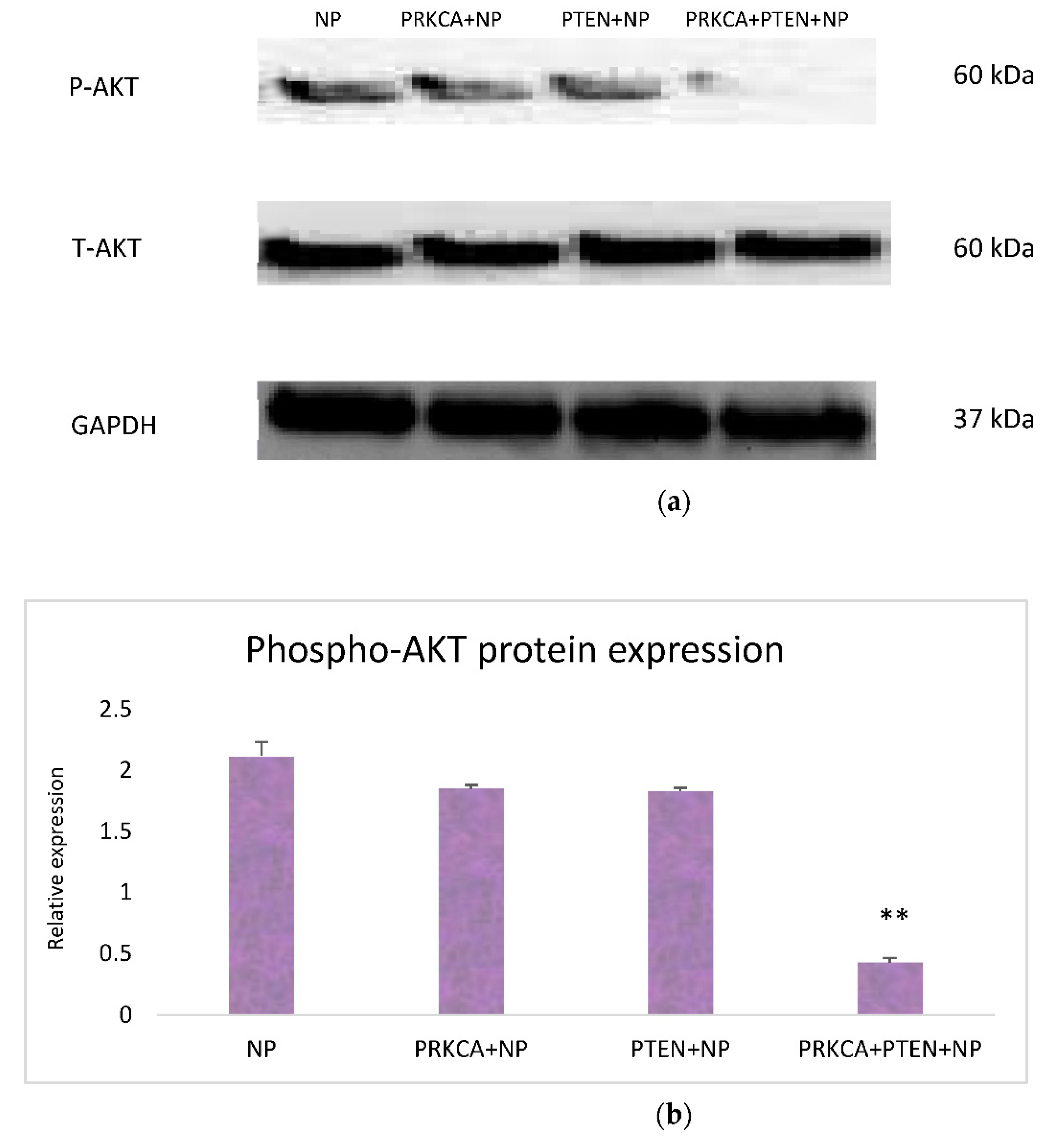
3.3. Effect of Co-Delivery of PRKCA siRNA and PTEN Gene on AKT Signaling Pathway: Western Blot Analysis
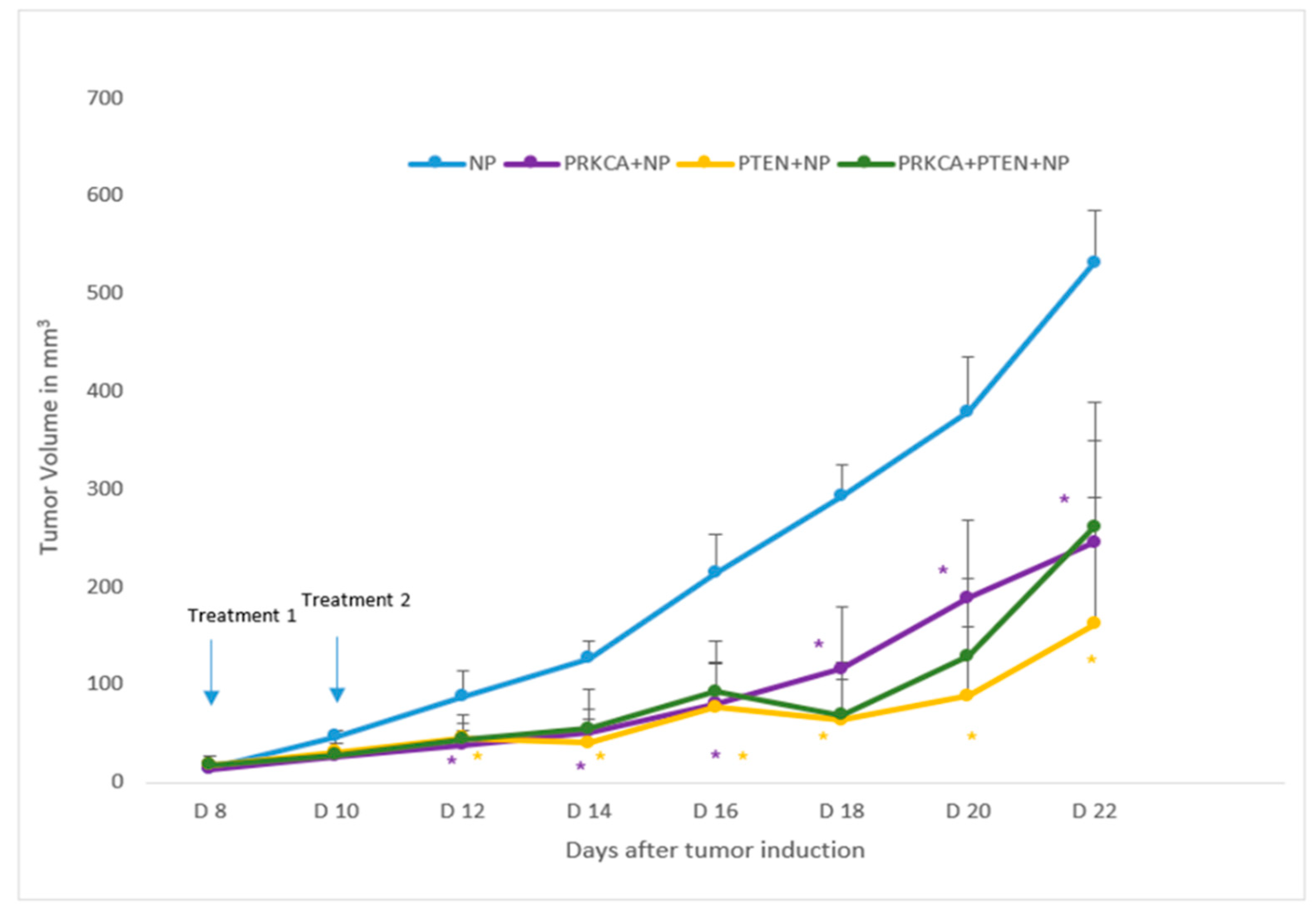
3.4. Influence of NPs-Facilitated PRKCA siRNA and PTEN Tumor Suppressor Gene Delivery on Tumor Regression
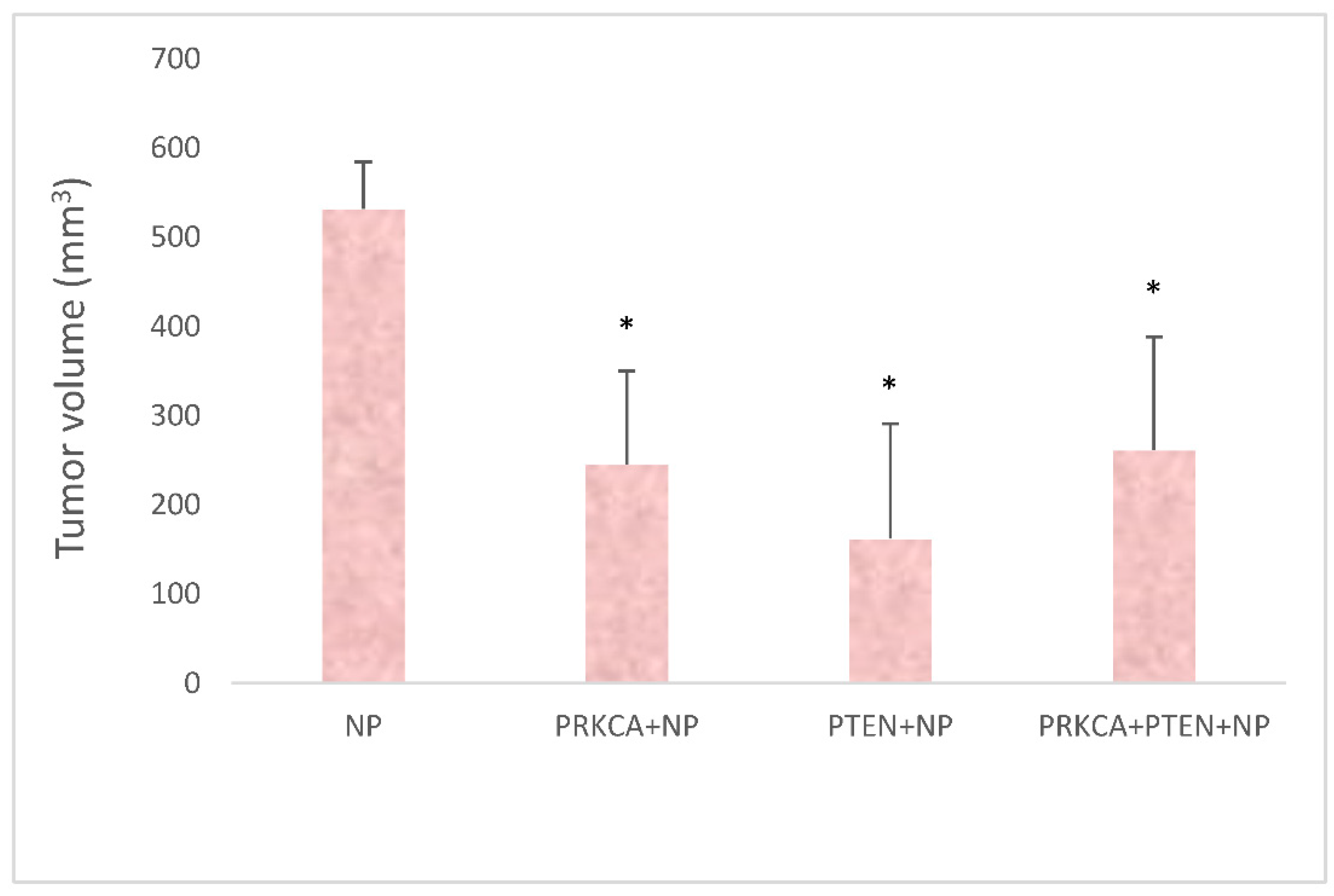
3.5. Quantitation of Tumor Volumes upon Treatments
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.B.; Ferlay, J.M.E.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global Cancer Statistics 2018: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. Cancer J. Clin. 2018, 68, 31. [Google Scholar] [CrossRef] [Green Version]
- Lønne, G.K.; Cornmark, L.; Omanovic Zahirovic, I.; Landberg, G.; Jirström, K.; Larsson, C. PKCalpha expression is a marker for breast cancer aggressiveness. Mol. Cancer 2010, 9, 76. [Google Scholar] [CrossRef] [Green Version]
- Cooke, M.; Magimaidas, A.; Casado-Medrano, V.; Kazanietz, M.G. Protein kinase C in cancer: The top five unanswered questions. Mol. Carcinog. 2017, 56, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Mellor, H.; Parker, P. The extended protein kinase C superfamily. Biochem. J. 1998, 332, 281–292. [Google Scholar] [CrossRef]
- Griner, E.M.; Kazanietz, M.G. Protein kinase C and other diacylglycerol effectors in cancer. Nat. Rev. Cancer 2007, 7, 281–294. [Google Scholar] [CrossRef]
- Webb, B.; Hirst, S.J.; Giembycz, M.A. Protein kinase C isoenzymes: A review of their structure, regulation and role in regulating airways smooth muscle tone and mitogenesis. Br. J. Pharmacol. 2000, 130, 1433–1452. [Google Scholar] [CrossRef] [Green Version]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2013, 33, 5225–5237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oster, H.; Leitges, M. Protein kinase C alpha but not PKCzeta suppresses intestinal tumor formation in ApcMin/+ mice. Cancer Res. 2006, 66, 6955–6963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haughian, J.M.; Reno, E.M.; Thorne, A.M.; Bradford, A.P. Protein kinase C alpha-dependent signaling mediates endometrial cancer cell growth and tumorigenesis. Int. J. Cancer 2009, 125, 2556–2564. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Fujii, T.; Yokoyama, G.; Kazanietz, M.G.; Yamana, H.; Shirouzu, K. Cell growth inhibition by all-trans retinoic acid in SKBR-3 breast cancer cells: Involvement of protein kinase C? And extracellular signal-regulated kinase mitogen-activated protein kinase. Mol. Carcinog. 2003, 38, 106–116. [Google Scholar] [CrossRef]
- Tonetti, D.A.; Szafran, A.; Gao, W.; Escarzaga, D.; Walters, K.; Coon, J.S. PKCα and ERβ Are Associated with Triple-Negative Breast Cancers in African American and Caucasian Patients. Int. J. Breast Cancer 2012, 2012, 740353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishizuka, Y. The molecular heterogeneity of protein kinase C and its implications for cellular regulation. Nature 1988, 334, 661–665. [Google Scholar] [CrossRef] [PubMed]
- Tonetti, D.A.; Morrow, M.; Kidwai, N.; Gupta, A.; Badve, S. Elevated protein kinase C alpha expression may be predictive of tamoxifen treatment failure. Br. J. Cancer 2003, 88, 1400–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, S.H.; Moon, B.I.; Suh, H.S.; Sung, S.H.; Han, W.S.; Cho, M.S.; Lee, N.S.; Choi, H.Y.; Koo, H.S. Loss of PTEN Expression in Breast Cancers. J. Pathol. Transl. Med. 2005, 39, 236–241. [Google Scholar]
- Li, J.; Yen, C.; Liaw, D.; Podsypanina, K.; Bose, S.; Wang, S.I.; Puc, J.; Miliaresis, C.; Rodgers, L.; McCombie, W.R.; et al. PTEN, a Putative Protein Tyrosine Phosphatase Gene Mutated in Human Brain, Breast, and Prostate Cancer. Science 1997, 275, 1943–1947. [Google Scholar] [CrossRef]
- Tamura, M.; Gu, J.; Matsumoto, K.; Aota, S.-I.; Parsons, R.; Yamada, K.M. Inhibition of Cell Migration, Spreading, and Focal Adhesions by Tumor Suppressor PTEN. Science 1998, 280, 1614–1617. [Google Scholar] [CrossRef]
- Carbognin, L.; Miglietta, F.; Paris, I.; Dieci, M.V. Prognostic and Predictive Implications of PTEN in Breast Cancer: Unfulfilled Promises but Intriguing Perspectives. Cancers 2019, 11, 1401. [Google Scholar] [CrossRef] [Green Version]
- Hollander, M.C.; Blumenthal, G.; Dennis, P.A. PTEN loss in the continuum of common cancers, rare syndromes and mouse models. Nat. Rev. Cancer 2011, 11, 289–301. [Google Scholar] [CrossRef]
- Planchon, S.M.; Waite, K.A.; Eng, C. The nuclear affairs of PTEN. J. Cell Sci. 2008, 121, 249–253. [Google Scholar] [CrossRef] [Green Version]
- Alimonti, A.; Carracedo, A.; Clohessy, J.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [Green Version]
- Papa, A.; Wan, L.; Bonora, M.; Salmena, L.; Song, M.S.; Hobbs, R.; Lunardi, A.; Webster, K.; Ng, C.; Newton, R.H.; et al. Cancer-Associated PTEN Mutants Act in a Dominant-Negative Manner to Suppress PTEN Protein Function. Cell 2014, 157, 595–610. [Google Scholar] [CrossRef] [Green Version]
- Kingston, B.; Bailleux, C.; Delaloge, S.; Schiavon, G.; Scott, V.; Lacroix-Triki, M.; Carr, T.H.; Kozarewa, I.; Gevensleben, H.; Kemp, Z.; et al. Exceptional Response to AKT Inhibition in Patients with Breast Cancer and Germline PTEN Mutations. JCO Precis. Oncol. 2019, 3, 1–7. [Google Scholar] [CrossRef]
- Waite, K.A.; Eng, C. Protean PTEN: Form and Function. Am. J. Hum. Genet. 2002, 70, 829–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mittendorf, E.A.; Philips, A.V.; Meric-Bernstam, F.; Qiao, N.; Wu, Y.; Harrington, S.; Su, X.; Wang, Y.; Gonzalez-Angulo, A.M.; Akcakanat, A.; et al. PD-L1 Expression in Triple-Negative Breast Cancer. Cancer Immunol. Res. 2014, 2, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, E.H.; Maruyama, A.; Nagaoka, K.M.; Kotaka, M.; Hirose, S.; Kunou, M.; Akaike, T. pH-sensing nano-crystals of carbonate apatite: Effects on intracellular delivery and release of DNA for efficient expression into mammalian cells. Gene 2006, 367, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, E.H. pH-responsive magnesium-and carbonate-substituted apatite nano-crystals for efficient and cell-targeted delivery of transgenes. Open J. Gen. 2013, 3, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Islam, R.A.; Al-Busaidi, H.; Zaman, R.; Abidin, S.A.Z.; Othman, I.; Chowdhury, E.H. Carbonate Apatite and Hydroxyapatite Formulated with Minimal Ingredients to Deliver SiRNA into Breast Cancer Cells In Vitro and In Vivo. J. Funct. Biomater. 2020, 11, 63. [Google Scholar] [CrossRef]
- Chowdhury, E.H.; Akaike, T. High performance DNA nano-carriers of carbonate apatite: Multiple factors in regulation of particle synthesis and transfection efficiency. Int. J. Nanomed. 2007, 2, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Hossain, S.; Stanislaus, A.; Chua, M.J.; Tada, S.; Tagawa, Y.-I.; Chowdhury, E.H.; Akaike, T. Carbonate apatite-facilitated intracellularly delivered siRNA for efficient knockdown of functional genes. J. Control. Release 2010, 147, 101–108. [Google Scholar] [CrossRef]
- Martinez, J.; Patkaniowska, A.; Urlaub, H.; Lührmann, R.; Tuschl, T. Single-Stranded Antisense siRNAs Guide Target RNA Cleavage in RNAi. Cell 2002, 110, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Tuschl, T.; Zamore, P.D.; Lehmann, R.; Bartel, D.P.; Sharp, P.A. Targeted mRNA degradation by double-stranded RNA in vitro. Genes Dev. 1999, 13, 3191–3197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vaughan, E.E.; Dean, D.A. Intracellular Trafficking of Plasmids during Transfection Is Mediated by Microtubules. Mol. Ther. J. Am. Soc. Gene Ther. 2006, 13, 422–428. [Google Scholar] [CrossRef] [PubMed]
- Honary, S.; Zahir, F. Effect of Zeta Potential on the Properties of Nano-Drug Delivery Systems—A Review (Part 2). Trop. J. Pharm. Res. 2013, 12, 9. [Google Scholar]
- Kass, G.E.; Orrenius, S. Calcium signaling and cytotoxicity. Environ. Health Perspect. 1999, 107, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nitulescu, G.M.; Van De Venter, M.; Nitulescu, G.; Ungurianu, A.; Juzenas, P.; Peng, Q.; Olaru, O.T.; Grădinaru, D.; Tsatsakis, A.; Tsoukalas, D.; et al. The Akt pathway in oncology therapy and beyond (Review). Int. J. Oncol. 2018, 53, 2319–2331. [Google Scholar]
- Xu, C.-F.; Wang, J. Delivery systems for siRNA drug development in cancer therapy. Asian J. Pharm. Sci. 2015, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kamaruzman, N.I.; Aziz, N.A.; Poh, C.L.; Chowdhury, E.H. Oncogenic Signaling in Tumorigenesis and Applications of siRNA Nanotherapeutics in Breast Cancer. Cancers 2019, 11, 632. [Google Scholar] [CrossRef] [Green Version]
- Torchilin, V.P. Passive and Active Drug Targeting: Drug Delivery to Tumors as an Example. Drug Deliv. 2009, 3–53. [Google Scholar] [CrossRef]
- Kamaruzman, N.I.; Tiash, S.; Ashaie, M.; Chowdhury, E.H. siRNAs Targeting Growth Factor Receptor and Anti-Apoptotic Genes Synergistically Kill Breast Cancer Cells through Inhibition of MAPK and PI-3 Kinase Pathways. Biomedicines 2018, 6, 73. [Google Scholar] [CrossRef] [Green Version]
- Chua, M.; Tiash, S.; Fatemian, T.; Noordin, M.I.; Keng, C.; Chowdhury, E. Carbonate apatite-facilitated intracellular delivery of c-ROS1 small interfering RNA sensitises MCF-7 breast cancer cells to cisplatin and paclitaxel. Cancer 2013, 1, 7. [Google Scholar] [CrossRef]
- Akimov, I.; Chernolovskaya, E.L.; Spitsyna, Y.E.; Ryabchikova, E.I.; Zenkova, M.A. Silencing of Her2, CCNB1 and pKC Genes by siRNA Results in Prolonged Retardation of Neuroblastoma Cell Division. Acta Nat. 2011, 3, 29–39. [Google Scholar] [CrossRef]
- Wu, T.-T.; Hsieh, Y.-H.; Hsieh, Y.-S.; Liu, J.-Y. Reduction of PKCα decreases cell proliferation, migration, and invasion of human malignant hepatocellular carcinoma. J. Cell. Biochem. 2008, 103, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Bonneau, D.; Longy, M. Mutations of the human PTEN gene. Hum. Mutat. 2000, 16, 109–122. [Google Scholar] [CrossRef]
- Li, X.; Lin, G.; Wu, B.; Zhou, X.; Zhou, K. Overexpression of PTEN Induces Cell Growth Arrest and Apoptosis in Human Breast Cancer ZR-75-1 Cells. Acta Biochim. Biophys. Sin. 2007, 39, 745–750. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Wang, C.; Jiang, C.; Chen, D. Exogenous PTEN gene induces apoptosis in breast carcinoma cell line MDA468. Acta Acad. Med. Wuhan 2007, 27, 61–64. [Google Scholar] [CrossRef]
- Shigeru, N. Protein Kinase Ca (PKCα): Regulation and Biological Function. J. Biochem. 2002, 132, 669–675. [Google Scholar]
- Li, W.; Zhang, J.; Flechner, L.; Hyun, T.; Yam, A.; Franke, T.F.; Pierce, J.H. Protein kinase C-α overexpression stimulates Akt activity and suppresses apoptosis induced by interleukin 3 withdrawal. Oncogene 1999, 18, 6564–6572. [Google Scholar] [CrossRef] [Green Version]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef] [Green Version]
- Holliday, D.L.; Speirs, V. Choosing the right cell line for breast cancer research. Breast Cancer Res. 2011, 13, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Sandra, R.; Susanne, S. Protein Corona of Nanoparticles: Distinct Proteins Regulate the Cellular Uptake. Biomacromolecules 2015, 16, 1311–1321. [Google Scholar]
- Chowdhury, E.H. Nanotherapeutics from Laboratory to Clinic; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Corbo, C.; Molinaro, R.; Tabatabaei, M.; Farokhzad, O.C.; Mahmoudi, M. Personalized protein corona on nano-particles and its clinical implications. Biomater. Sci. 2017, 5, 378–387. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ibnat, N.; Islam, R.A.; Chowdhury, E.H. Inhibition of Breast Tumour Growth with Intravenously Administered PRKCA siRNA- and PTEN Tumour Suppressor Gene-Loaded Carbonate Apatite Nanoparticles. Appl. Sci. 2021, 11, 8133. https://doi.org/10.3390/app11178133
Ibnat N, Islam RA, Chowdhury EH. Inhibition of Breast Tumour Growth with Intravenously Administered PRKCA siRNA- and PTEN Tumour Suppressor Gene-Loaded Carbonate Apatite Nanoparticles. Applied Sciences. 2021; 11(17):8133. https://doi.org/10.3390/app11178133
Chicago/Turabian StyleIbnat, Nabilah, Rowshan Ara Islam, and Ezharul Hoque Chowdhury. 2021. "Inhibition of Breast Tumour Growth with Intravenously Administered PRKCA siRNA- and PTEN Tumour Suppressor Gene-Loaded Carbonate Apatite Nanoparticles" Applied Sciences 11, no. 17: 8133. https://doi.org/10.3390/app11178133
APA StyleIbnat, N., Islam, R. A., & Chowdhury, E. H. (2021). Inhibition of Breast Tumour Growth with Intravenously Administered PRKCA siRNA- and PTEN Tumour Suppressor Gene-Loaded Carbonate Apatite Nanoparticles. Applied Sciences, 11(17), 8133. https://doi.org/10.3390/app11178133