Abstract
The intra-cage behaviour of guest H2 and D2 molecules in doubly occupied 51264 cages in structure-II (sII) clathrate hydrates were investigated using classical and path-integral molecular dynamics at 100 K. We probed the structure of tetrahedral sites, proton vibrations, localised molecular rattling timescales at sites, and the jump-diffusion travel of H2 and D2 molecules between sites. The site-diffusion model was correlated with experimental neutron scattering data, and the cage occupancies were then discussed in light of recent state-of-the-art experimental and theoretical findings in the literature.
1. Introduction
Hydrogen is considered to be the ultimate alternative clean fuel to supplant the use of hydrocarbons in many contexts. Given its very high energy density, it could also serve as a temporary medium for the storage and transport of energy that can seamlessly integrate into renewable energy applications. Despite its very high energy density, rendering it a tempting alternative, its relatively low volumetric density (in gaseous form) and associated hazards, makes finding a safe and affordable hosting/storage material with high capacity a ‘Holy Grail’. Although various candidate materials may appear to challenge these material requirements, most of the proposed solutions are bedevilled with a high manufacturing cost or insufficiently low capacity, thus ruling them out of contention [1].
Clathrate hydrates are non-stoichiometric inclusion compounds in which a host water lattice entraps gas molecules [2]. Clathrate hydrates offer a potentially interesting storage medium for hydrogen—a means for ’green mass storage’ [2]. The prospect of using gas hydrates as a cheap and green alternative to metal–organic frameworks, zeolites and hydrides for hydrogen storage to facilitate the green and hydrogen economies is certainly attractive [2]. Although pure hydrogen hydrates (consisting only of hydrogen in sII clathrate cages) have been realised experimentally, first by Dyadin et al. [3,4], they require very high pressures (around 1.5–2 kbar) to be structurally stable at near-ambient temperatures [2,3,4,5] and are less practical for engineering applications [2,5,6]. The maximum theoretical hydrogen capacity of the hydrogen hydrate is 5.3 wt.% (for sII hydrates) at significantly elevated pressures of the order of 2–3 kbar for this higher level of cage-filling, and temperatures of around 250 K [7,8,9,10]. However, this maximal storage capacity of circa 5 wt.% is reliant on double and quadruple occupation of small (512) and large (51264) sII-hydrate cages, respectively. This is a somewhat controversial interpretation of high-pressure neutron-diffraction data. More realistic (lower) cage occupancies have been proposed (e.g., closer to 1 for small cages and typically no more than 2 or sometimes 3 in the large cage [2,11]), leveraging sophisticated ab-initio molecular-dynamics simulation to assess cage occupancies systematically [11].
Of course, the high-pressure requirements to afford stability of pure hydrogen and realise a larger hydrogen-bearing capacity approaching United States Department of Energy (DOE) goals [5,6] makes it somewhat of an impractical hydrogen-storage medium [1] despite interesting ‘ice-coating’ proposals [12]. A pragmatic approach for engineering applications hinges on the use of a mixing gas for binary hydrates, in which a ‘helper’ molecule fills (most) large cavities and the smaller cages are free to host hydrogen. In this respect, both propane [13,14] and tetrahydrofuran (THF) hydrate have been advanced as potentially attractive propositions [15]. Such ‘doping’, however, does tend to undermine the hydrogen-bearing hydrates’ ‘green’ credentials, and must be considered in the appraisal of green hydrogen-storage strategies carefully.
In any event, having briefly discussed the ‘green-storage’ context and motivation for hydrogen storage in hydrates (especially the ‘pure’ case), together with some controversy over large-cage (and even small-cavity) occupancies giving rise to perhaps inflated H2-occupancy estimates [7,8,9,10], the question remains to be answered about the fundamentals of intra-cage guest (H2 and D2) phenomena; notably their structure, vibrations and ‘site-hopping’ behaviour in the large 51264 sII-lattice cages. Although there has certainly been a number of high-quality studies on guest inter-cage dynamics and diffusivity throughout the sII lattice [16,17,18], as well as inter-cage H2-transfer energy barriers [11,19,20,21], such intra-cage dynamics have been less studied. Futera et al. have carried out sophisticated Raman-type analysis of hydrogen hydrates with state-of-the-art ab initio molecular-dynamics (AIMD) simulation and Raman experiments—although the AIMD was propagated classically [22]. Very recently, Ranieri et al. carried out inelastic neutron scattering (INS) measurements of quantum dynamics of D2 and H2 molecules confined in pure- and binary-hydrate structures (helium in the latter case), determining [23] and assessing molecular quantal rotation and translations. Bacic and co-workers made substantial recent advances in more rigorous 6- and 8-D quantum treatments of singly and doubly occupied sII-hydrate cages [24,25]. In particular, the INS studies of reference [23] suggest that the large cage in sII hydrogen hydrates is rarely more than doubly occupied, with average occupancies of circa 1.5–1.8 at kilobar pressures (and 0.85-1 for 512 cages), while reference [11] predicted that singly occupied 512 and doubly or triply occupied 51264 cages are more energetically realistic across a range of temperatures and cavity occupancies. These findings more reasonably accord with the subsequently reported INS findings [23]. Given these careful intra-cage phenomena studies of references [11,19,20,21,22,23,24,25] and others, we concluded that the weight of most recent scientific evidence, from both experiment and theory, supports the view that we can (and probably should) use single- and double-H2 and D2 occupations of the small and large sII hydrate cavities, respectively, as being reasonable and ripe for further detailed study.
Keeping this mandate in mind (i.e., to study in further detail intra-cage phenomena for singly- and doubly occupied small and large sII-lattice cages, respectively), we noted that the tetrahedral structure of intra-cage ‘sites’ to accommodate guest molecules for various large-cage occupancies within sII-hydrate 51264 cages is less well characterised in the literature. However, reference [11] showed (from AIMD) a range of probable spatial distributions for likely H2 positions in large cages. Further analysis is warranted for the doubly occupied case, in particular the study of nuclear quantum effects impacting these positions. Moreover, although reference [26] reported some interesting quasielastic neutron scattering (QENS) measurements of classical “jump-diffusion” dynamics between these sites, reference [23] examined this from a quantum perspective for lower-temperature behaviour, where assumptions of classical site–site hopping are more questionable.
In this study, we sought to investigate further tetrahedral-site distributions for doubly occupied large cages using path-integral dynamics to incorporate nuclear quantum effects at 100 K. Unlike our previous work in references [11,19,27], in which occupancy of the 51264 cavities was an important consideration, in this study we emphasised again that the overwhelming (and very recent) consensus of the state-of-the art experiment and molecular simulation is definitively in favour of no more than double occupation of such cages as being routinely feasible. Keeping this in mind, we focused on doubly occupied cavities in the present contribution. Having studied such quantum site structural distributions for such doubly occupied 51264 cages, we then proceeded to assess further dynamical vibrations and intra-cage diffusive motion between these sites, as well as relaxation of residence time distributions thereat as a function of temperatures (80–125 K) where classical effects are dominant (i.e., above 50 K) [23,26].
2. Methodology
The empirical model used in this work was the force-matched model (FMM) of Burnham, Futera and English [27]. The model was parameterised against Born-Oppenheimer molecular dynamics (BOMD) trajectories of type II clathrate hydrates containing guest hydrogen molecules. The model was fit using a force-matching method, in which the forces on each particle during the course of the BOMD trajectory were fit. The results had good agreement with the density functional theory results for the free energy barriers for H2 molecules’ passage across large cages. This was true, in particular, along the path connecting pairs of large cages through their shared hexagonal face, through which the guest molecules hopped from one large cage to another.
Both classically propagated and 32-bead path-integral molecular dynamics (PIMD) were carried out under periodic boundary conditions for an sII unit cell with single and double occupation in small and large cages of both D2 and H2, respectively. The D2 and H2 were in respective D2O and H2O lattices. The molecular-dynamics methodology and other computational details are otherwise very similar to reference [27]. Canonical-ensemble dynamics (classical and 32-bead PI) were performed in an sII unit cell with a vanishingly small dipole moment [27] for 20 ps with a 0.2-fs time step. All cages in the unit-cell simulation box were occupied during molecular dynamics (MD) (single or double, as appropriate).
Initially, we wished to average the results over all cages (either 512 or 51264) and over all time steps. Doing so first required identifying all the cages of each type, and then placing those cages in a standard position/orientation. To do this required a method for producing an equivalent orthonormal x-, y-, z- axes set for each of the large (and small) cages, such that if the cages only differed by a rotation in the frame of the axes set, each cage would be brought into perfect alignment.
To place each of the 51264 large cages into a standard orientation, we first used an algorithm to identify the vertices of the four hexagonal faces. We then constructed four relative vectors from the cage-centre to the centre of each of the four hexagonal faces, which should, and does, form a tetrahedral vector set. The vectors were then normalised. It was then possible to form an orthonormal axis set from linear combinations of the tetrahedral vectors, which can be written in matrix form as , where are the four tetrahedral vectors, contain the orthonormal axis vectors (in ), and the orthogonal matrix is given by:
To place each of the 512 dodecahedral cages in a standard orientation, we first chose an arbitrary starting vertex on each dodecahedron. Then, starting from the initial vertex, we searched for the six second nearest neighbours. From these we constructed six unit vectors pointing from the starting vertex to the six second nearest neighbours. It can be shown that, for a regular dodecahedron, these six vectors comprised two sets of mutually orthonormal triplets, either of which were used to form an axis set. One set was found from the three left turns that were made at the connecting first nearest neighbour (to the second-nearest neighbour), and the other set was found from the three right turns. In fact, the two choices were not equivalent as they corresponded to two different orientations of the dodecahedron. For this reason, the equivalent axis set for each dodecahedron (choosing consistently either all-left or all-right turns) should be chosen carefully.
As noted above, and to mimic the quantum-INS experiments of reference [23], we were interested in the structure of the D2 guest molecules within the cages. A natural way to analyse the data is to generate a three-dimensional density of the centre of mass with respect to the large (or small) cages, averaged over all large (small) cages in the simulation cell and over all time steps in our (path-integral) molecular dynamics simulation. The resulting density can then be displayed through examination of isosurfaces of the spatial-density data (vide infra).
Turning to the matter of investigating the so-defined and resultant tetrahedral sites in terms of their hopping dynamics and distribution of dwell times therein, we identified every doubly occupied large cage in our simulation cell, recording the labels of the molecules forming the vertices of each cage. Using this information, we could then find the geometric centre of each large cage’s four hexagonal faces on each time-step of a molecular-dynamics simulation. Then, for each of the two guest molecules, we recorded which of the four hexagonal mid-points that the guest molecule was closest to on each step of the simulation. As the simulation progresses, the guest molecules gradually switched between the vicinities of different mid-points, which we characterised by taking a histogram of the hopping lifetimes. Each lifetime was an unbroken sequence of (classical) molecular-dynamics time steps in which a particular guest molecule was closest to a particular midpoint. Unfortunately, PIMD has well-known (in-principle) pathologies with respect to theoretically rigorous time-series representation of dynamical/vibrational properties, so we conducted this moderate towards ambient-temperature analysis (80, 100 and 125 K) for classical propagation only in a manner consistent with QENS studies [26], where quantum phenomena do not dominate [23].
Finally, we characterised the resulting histogram by fitting the long-time data points (t > 5 ps) to a double exponential decay profile for both guest types (a pair of D2 in the large cages of a D2O lattice, and a H2 duo in large cavities of H2O-lattice type), i.e.,:
where τ is the characteristic relaxation time for the guest molecules’ site-residence times, with τ2 giving the longer-time characteristic decay. From assessing a single to a double exponential fitting strategy [28], we found that the latter gave better-fidelity fits. This is reflective of the ‘bifurcated’ nature of caged/confined dynamics of guest molecules [28,29,30], which undergo local site-specific vibrational motion, and more diffusive inter-site translations [23,26]. Therefore, the initial decay, τ1, is more reflective of local site-specific rattling motion.
h(t) = A1 exp(−t/τ1) + A2 exp(−t/τ2)
3. Results and Discussion
After examining D2-guest structural distributions first [11,23], isosurface plots for both large and small cages and for both classical and path integral molecular dynamics simulations are plotted below. Because no single isosurface can capture the full extent of the three-dimensional density data, we chose to use two isosurfaces for each case: one at 5% the density maximum (in light transparent grey) and one at 10% the density maximum (in darker grey).
Turning to classical results, we observed that for guest D2 molecules in the singly occupied small 512 cages, their centre of masses (COMs) were distributed close to the cage-centres with no discernible anisotropy. The large 51264 cages, which were each occupied by two D2 guest molecules, showed much more structure in their COM positions. The guest molecule COMs were preferentially located in tetrahedrally-oriented sites, one behind the centre of each of the large cage’s four hexagonal faces. There was also significant population at off-tetrahedral sites, with the density isosurface taking on a somewhat cubic shape overall. Additionally, there was some population at the centre of each hexagonal face, which were shared by large cage pairs.
Turning to the path-integral results, we observed that the guest density in the small cages was practically identical to the classical results, where the guest centre of masses confined to the cage-centre region. Although similar to the classical results, there was a more observable difference for the large cage results (i.e., the off-tetrahedral locations appear more heavily populated, such that the density within the large cages appears almost symmetrically distributed along the eight vertices of a cube). For the reader’s closer reference, Gaussian-type ‘cube’ files are provided as part of Supplementary Material for both classical and PIMD spatial density results.
Further details on temperature and occupation effects on the nature of quantum and classical spatial distributions of such D2 and H2 guests in the 51264 cages have been outlined (in detail) in reference [11]. In brief, the present results for double occupation are, by and large, compatible with 100 K results in reference [11], sampled from high-fidelity Density-Functional Theory (DFT) calculations; indeed, the H2-water potential model, optimised for hydrogen hydrates was developed using such DFT calculations [27]. In essence, for H2 occupancies of 1 to 5 at 50, 100 and 200 K, the guests’ density isosurfaces showed how the density became more or less perfect during the quadruple occupation [11] (which is in accord with the findings of Figure 1 in the present work). When temperature increased, the extent of delocalisation increases (and not just for the guests), serving to ‘smear’ the intra-cage iso-densities more greatly [11]. This resulted in slightly less well-defined adherence to the tetrahedral sites and, to a greater extent, off-site spatial density [11].
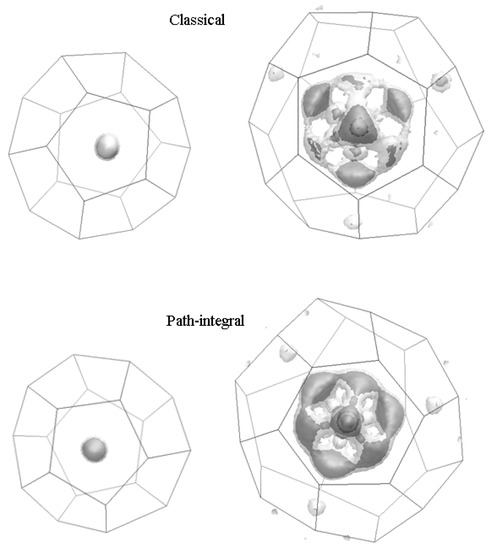
Figure 1.
Spatial density isosurface plots (0.1 Å) at 100 K in singly and doubly occupied small and large cages.
In terms of these singly occupied 512 cages, using the orthonormal tetrahedral-vector approach above to identify local-axis systems therein from both classical and path-integral MD, there was no real preferred orientations of the D-D axis in its angular histogram in either case, which indicates no preferred local orientational alignment and relatively free rotational motion.
In terms of free energies of the guests, and on related energy barriers (as mentioned previously), our previous work in references [11,19,27] established that H2 occupations of 2 or 3 in 51,264 cages were the most thermodynamically feasible. It was also found that such free energy terms were lower in the classical case than for the quantum case, owing to quantal delocalisation increasing these terms. More specifically, in reference [11], we calculated free energy profiles of the molecules over the volume of the 51264 cages’ interiors at 50, 100 and 200 K. We showed that the associated barrier reduced almost linearly for 1–3 molecules per large cage but grew larger than expected for quadruple occupancy, with this departure from linearity becoming more pronounced at lower temperatures [11]. The barriers tended to rise with rising temperature, with nuclear quantum effects (NQEs) raising further due to quantal delocalisation [11].
In terms of intra-cage dynamical motion (to study classical site-jump phenomena), we first studied the vibrational (proton) velocity-spectra of the guest and host species (i.e., H2 in H2O and D2 in D2O) at 80, 100 and 125 K for 200 ps. These are provided in the Supplementary Information (Figures S1 and S2), and they obey the expected isotope vibrational effect quite well, i.e., doubling the mass in going from H2 to D2 (and H2O to D2O) reduces the frequency by √2.
For double occupation of the 51,264 cages, the double exponential decay fit for the site-residence distribution h(t), from Equation (1), gives an expected relaxation time τ2 at 100 K of around 10 and 13 ps for H2 and D2, respectively, with r2 values of over 0.95. The shorter τ1 times, reflective of localised rattling, were about 2.0 and 3.1 ps for H2 and D2, respectively. The h(t) data is shown in Figure 2 at 100 K for both D2 and H2 guests, with the H2-fit as an example. QENS signals observed in the spectra of reference [23] indicate that increasing temperature in the H2O-H2 sII clathrate corresponds to a change in H2 dynamics from a strictly quantum to a more mixed quantum-classical regime around 10–15 K. At all investigated temperatures up to around 50 K, the quasielastic broadening width was Q-independent23, suggesting, as expected in cage/confined dynamics [26,28,29,30], a localised rattling motion (reflective of the shorter-time transient relaxation process τ1 in Equation (1)), as opposed to more jump-/hop-diffusion exploration of other tetrahedral sites. For temperatures between 25 and 45 K, it was found (using QENS) that the width of this hybrid quantum-classical process ranged between approximately 0.55 and 0.65 meV, which corresponded to between 8.2 and 6.4 ps [23]. At around 50 K, there was an onset of fully classical site-jump diffusional behaviour, i.e., with Q-dependence of the quasielastic broadening width emerging clearly [23], and so extrapolation of the QENS width to the fully classical hop-diffusion regime at 100 K (in the present work) was expected to yield localised rattling-timescales of no more than a few picoseconds. This is not inconsistent with our present τ1 estimates (i.e., 2.0 and 3.1 ps for H2 and D2, resp.). In addition, taking into account shorter time residences of the tetrahedral sites (<5 ps), the present work’s (time-weighted) means of the dwell time distributions at 100 K were 1.38 and 1.21 ps for H2 and D2, respectively. This is broadly in line with site-specific timescales in a classical jump-diffusion regime at 100 K from neutron scattering experiments [23,26].
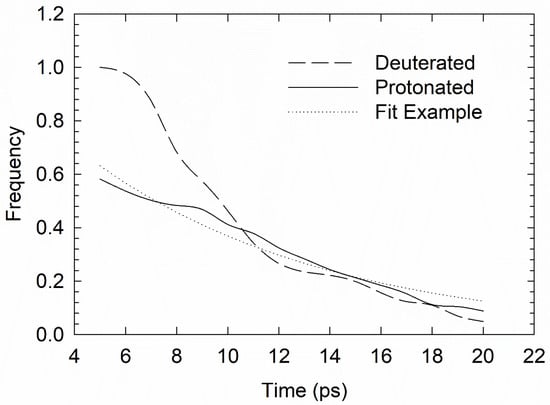
Figure 2.
Decay of spatial self-correlation, as measured by deuterated-normalised histogram of tetrahedral-site dwell times, in the large-cage tetrahedral sites at 100 K from classical molecular dynamics (MD) (in arbitrary units); the double exponential decay fit is shown for the H2-in-H2O case.
In terms of temperature dependence of τ1 and τ2 bi-exponential times, Arrhenius plots are provided in Figure S3 for 80, 100 and 125 K, revealing activation energies of 0.45, 0.50, 0.59 and 0.66 kJ/mol for τ1 (H2, D2) and τ2 (H2, D2), respectively. As expected, this is a deal smaller than for inter-cage phenomena [11,19,27]. The relative linearity of the Arrhenius fits shows that they are essentially undergoing a quasi-classical-type jump diffusion as the basic mechanism. The underlying τ1 (H2, D2) and τ2 (H2, D2) values are summarised below in Table 1, with r2 values well over 0.95. It can be further observed that the D2-inter-site hop relaxation dynamics was the most sluggish, with the largest activation energy barrier in its temperature dependence.

Table 1.
τ2 and τ2 intra-cage site-hop relaxation times in picoseconds.
On the importance of NQEs in the present work, it is clear that there is a stark effect at low temperature (of the order of 100 K) in terms of spatial distribution of the guests (Figure 1) together with their intra-cage, tetrahedral-site hopping dynamics. However, lest it be overlooked, one must also bear in mind NQEs in the context of the water lattice in the hydrate itself. Certainly, it is known that some NQEs are exhibited by protons in liquid water even at temperatures above 100 K, e.g., for auto-protolysis [31] and electric-field-induced proton conductivity [32], albeit with their magnitude becoming more decisive at lower temperatures. In the general ’water-NQE’ context, Conde et al. found that, for hydrate lattices, a reliable estimation of hydrate densities below 150 K, alongside sublimation energies, constant-pressure heat capacity and radial distribution functions, PI approaches are needed to take into account NQEs, while other properties were less affected below 150 K [33,34]. Indeed, our previous work [11,19,27] highlighted that quantal structural delocalisation (ever more important at lower temperatures) is a central feature in altering energy barriers. Naturally, the water lattice network itself also plays an important role in these spatial-delocalisation processes.
4. Conclusions
The intra-cage behaviour of guest H2 and D2 molecules in doubly occupied 51264 cages in sII clathrate at 100 K was investigated in the present study. We found that the singly occupied small cages did not evince any preferred orientation of the guest, while there was a greater population of off-tetrahedral guest occupation of large cages in the case of PIMD, with a more cubic-shaped density isosurface. Their greater population at the centre of each hexagonal face (shared by large-cage pairs) also reflected the stark effect of nuclear quantum effects in lowering inter-cage transition free energy barriers [11,19,21].
The tetrahedral sites found in Figure 1, featuring their ’musical chairs’ type of classical jump-diffusion phenomena (Figure 2), were found to be qualitatively consistent with the neutron scattering classical diffusion findings in reference [26]. This offered a semi-quantitative agreement in terms of rattling timescales with rough expected QENS width time equivalences [23]. However, it is possible that by reassessing our probability density boundaries of where the tetrahedral sites are located, and which are somewhat subjective and ‘fuzzy’, one may find slightly different quantitative values. For this reason, any direct comparison with neutron scattering data, while instructive, should also be treated with caution.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-3417/11/1/54/s1. Gaussian-cube isosurface density files for D2 in small and large cages, both classical and path-integral.
Author Contributions
N.J.E. conceptualised, performed selected simulations and analysis, and took charge of overall writing and project management. C.J.B. performed detailed mathematical and simulation programming, ran simulations themselves, as well as much analysis and technical writing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Science Foundation Ireland (SFI/15/ERC/I3142).
Acknowledgments
The authors acknowledge Science Foundation Ireland (SFI/15/ERC/I3142) for funding, as well as Umbertoluca Ranieri, Andrzej Falenty, Werner Kuhs and Livia Bove for interesting discussions.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Di Profio, P.; Arca, S.; Rossi, F.; Filipponi, M. Comparison of hydrogen hydrates with existing hydrogen storage technologies: Energetic and economic evaluations. Int. J. Hydrog. Energy 2009, 34, 9173–9180. [Google Scholar] [CrossRef]
- Hassanpouryouzband, A.; Joonaki, E.; Farahani, M.V.; Takeya, S.; Ruppel, C.; Yang, J.; English, N.J.; Schicks, J.M.; Edlmann, K.; Mehrabian, H.; et al. Gas hydrates in sustainable chemistry. Chem. Soc. Rev. 2020, 49, 5225–5309. [Google Scholar] [CrossRef] [PubMed]
- Dyadin, Y.A.; Larionov, E.G.; Manakov, A.Y.; Zhurko, F.V.; Aladko, E.Y.; Mikina, T.V.; Komarov, V.Y. Clathrate hydrates of hydrogen and neon. Mendeleev Commun. 1999, 9, 209–210. [Google Scholar] [CrossRef]
- Dyadin, Y.A.; Larionov, E.G.; Aladko, E.Y.; Manakov, A.Y.; Zhurko, F.V.; Mikina, T.V.; Komarov, V.Y.; Grachev, E.V. Clathrate formation in water-noble gas (hydrogen) systems at high pressures. J. Struct. Chem. 1999, 40, 790–795. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Kumar, R.; Linga, P. Hydrogen storage in clathrate hydrates: Current state of the art and future directions. Appl. Energy 2014, 122, 112–132. [Google Scholar] [CrossRef]
- Strobel, T.A.; Hester, K.C.; Koh, C.A.; Sum, A.K.; Sloan, E.D. Properties of the clathrates of hydrogen and developments in their applicability for hydrogen storage. Chem. Phys. Lett. 2009, 478, 97–109. [Google Scholar] [CrossRef]
- Mao, W.L.; Mao, H.K.; Goncharov, A.F.; Struzhkin, V.V.; Guo, Q.; Hu, J.; Shu, J.; Hemley, R.J.; Somayazulu, M.; Zhao, Y.; et al. Hydrogen clusters in clathrate hydrate. Science 2002, 297, 2247–2249. [Google Scholar] [CrossRef]
- Mao, W.L.; Mao, H.K. Hydrogen storage in molecular compounds. Proc. Natl. Acad. Sci. USA 2004, 101, 708–710. [Google Scholar] [CrossRef]
- Struzhkin, V.V.; Militzer, B.; Mao, W.L.; Mao, H.; Hemley, R.J. Hydrogen storage in molecular clathrates. Chem. Rev. 2007, 107, 4133–4151. [Google Scholar] [CrossRef]
- Lokshin, K.A.; Zhao, Y.; He, D.; Mao, W.L.; Mao, H.K.; Hemley, R.J.; Lobanov, M.V.; Greenblatt, M. Structure and dynamics of Hydrogen molecules in the novel clathrate hydrate by high pressure neutron diffraction. Phys. Rev. Lett. 2004, 93, 125503. [Google Scholar] [CrossRef]
- Burnham, C.J.; Futera, Z.; English, N.J. Quantum and classical inter-cage hopping of hydrogen molecules in clathrate hydrate: Temperature and cage-occupation effects. Phys. Chem. Chem. Phys. 2017, 19, 717–728. [Google Scholar] [CrossRef] [PubMed]
- Willow, S.Y.; Xantheas, S.S. Enhancement of hydrogen storage capacity in hydrate lattices. Chem. Phys. Lett. 2012, 525, 13–18. [Google Scholar] [CrossRef]
- Veluswamy, H.P.; Chen, J.Y.; Linga, P. Surfactant effect on the kinetics of mixed hydrogen/propane hydrate formation for hydrogen storage as clathrates. Chem. Eng. Sci. 2015, 126, 488–499. [Google Scholar] [CrossRef]
- Ghaani, M.R.; English, N.J. Hydrogen-/propane-hydrate decomposition: Thermodynamic and kinetic analysis. Mol. Phys. 2019, 117, 2434–2442. [Google Scholar] [CrossRef]
- Hester, K.C.; Strobel, T.A.; Sloan, E.D.; Koh, C.A.; Huq, A.; Schultz, A.J. Molecular hydrogen occupancy in binary THF−h2 clathrate hydrates by high resolution neutron diffraction. J. Phys. Chem. B 2006, 110, 14024–14027. [Google Scholar] [CrossRef]
- Okuchi, T.; Moudrakovski, I.; Ripmeester, J.A. Efficient storage of hydrogen fuel into leaky cages of clathrate hydrate. Appl. Phys. Lett. 2007, 91, 171903. [Google Scholar] [CrossRef]
- Frankcombe, T.J.; Kroes, G.J. Molecular dynamics simulations of Type-sII hydrogen clathrate hydrate close to equilibrium conditions. J. Phys. Chem. C 2007, 111, 13044–13052. [Google Scholar] [CrossRef]
- Cao, H.; English, N.J.; MacElroy, J.M.D. Diffusive hydrogen inter-cage migration in hydrogen and hydrogen-tetrahydrofuran clathrate hydrates. J. Chem. Phys. 2013, 138, 094507. [Google Scholar] [CrossRef]
- Burnham, C.J.; English, N.J. Free-energy calculations of the intercage hopping barriers of hydrogen molecules in clathrate hydrates. J. Phys. Chem. C 2016, 120, 16561–16567. [Google Scholar] [CrossRef]
- Alavi, S.; Ripmeester, J.A. Hydrogen-gas migration through clathrate hydrate cages. Angew. Chemie Int. Ed. 2007, 46, 6102–6105. [Google Scholar] [CrossRef]
- Cendagorta, J.R.; Powers, A.; Hele, T.J.H.; Marsalek, O.; Bačić, Z.; Tuckerman, M.E. Competing quantum effects in the free energy profiles and diffusion rates of hydrogen and deuterium molecules through clathrate hydrates. Phys. Chem. Chem. Phys. 2016, 18, 32169–32177. [Google Scholar] [CrossRef] [PubMed]
- Futera, Z.; Celli, M.; Del Rosso, L.; Burnham, C.J.; Ulivi, L.; English, N.J. Vibrational modes of hydrogen hydrates: An ab-initio molecular-dynamics and Raman-spectra study. J. Phys. Chem. C 2017, 121, 3690–3696. [Google Scholar] [CrossRef]
- Ranieri, U.; Koza, M.M.; Kuhs, W.F.; Gaal, R.; Klotz, S.; Falenty, A.; Wallacher, D.; Ollivier, J.; Gillet, P.; Bove, L.E. Quantum dynamics of H2 and D2 confined in hydrate structures as a function of pressure and temperature. J. Phys. Chem. C 2018, 123, 1888–1903. [Google Scholar] [CrossRef]
- Lauvergnat, D.; Felker, P.; Scribano, Y.; Benoit, D.M.; Bačić, Z. H2, HD, and D2 in the small cage of structure II clathrate hydrate: Vibrational frequency shifts from fully coupled quantum six-dimensional calculations of the vibration-translation-rotation eigenstates. J. Chem. Phys. 2019, 150, 154303. [Google Scholar] [CrossRef] [PubMed]
- Felker, P.M.; Lauvergnat, D.; Scribano, Y.; Benoit, D.M.; Bačić, Z. Intramolecular stretching vibrational states and frequency shifts of (H2)2 confined inside the large cage of clathrate hydrate from an eight-dimensional quantum treatment using small basis sets. J. Chem. Phys. 2019, 151, 124311. [Google Scholar] [CrossRef]
- Russina, M.; Kemner, E.; Mezei, F. Intra-cage dynamics of molecular hydrogen confined in cages of two different dimensions of clathrate hydrates. Sci. Rep. 2016, 6, 27417. [Google Scholar] [CrossRef]
- Burnham, C.J.; Futera, Z.; English, N.J. Study of hydrogen-molecule guests in type II clathrate hydrates using a force-matched potential model parameterised from ab initio molecular dynamics. J. Chem. Phys. 2018, 148, 102323. [Google Scholar] [CrossRef]
- English, N.J.; Tse, J.S.; Carey, D.J. Mechanisms for thermal conduction in various polymorphs of methane hydrate. Phys. Rev. B 2009, 80, 134306. [Google Scholar] [CrossRef]
- Garate, J.A.; English, N.J.; MacElroy, J.M.D. Human aquaporin 4 gating dynamics in dc and ac electric fields: A molecular dynamics study. J. Chem. Phys. 2011, 134, 055110. [Google Scholar] [CrossRef]
- Reale, R.; English, N.J.; Garate, J.A.; Marracino, P.; Liberti, M.; Apollonio, F. Human aquaporin 4 gating dynamics under and after nanosecond-scale static and alternating electric-field impulses: A molecular dynamics study of field effects and relaxation. J. Chem. Phys. 2013, 139, 205101. [Google Scholar] [CrossRef]
- Ceriotti, M.; Cuny, J.; Parrinello, M.; Manolopoulos, D.E. Nuclear quantum effects and hydrogen bond fluctuations in water. Proc. Natl. Acad. Sci. USA 2013, 110, 15591–15596. [Google Scholar] [CrossRef] [PubMed]
- Cassone, G. Nuclear quantum effects largely influence molecular dissociation and proton transfer in liquid water under an electric field. J. Phys. Chem. Lett. 2020, 11, 8983–8988. [Google Scholar] [CrossRef] [PubMed]
- Conde, M.M.; Vega, C.; McBride, C.; Noya, E.G.; Ramírez, R.; Sesé, L.M. Can gas hydrate structures be described using classical simulations? J. Chem. Phys. 2010, 132, 114503. [Google Scholar] [CrossRef] [PubMed]
- English, N.J.; El-Hendawy, M.M.; Mooney, D.A.; MacElroy, J.M.D. Perspectives on Atmospheric CO2 Fixation in Inorganic and Biomimetic Structures. Coord. Chem. Rev. 2014, 269, 85–95. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).