1. Introduction
The development of high-throughput sequencing technologies and computational capabilities has revolutionized genetics and genomics, resulting in a tremendous reduction in the cost of genome sequencing and the exponential growth of genomes in public repositories. Unfortunately, the annotation of both genes and proteins has not kept pace with genome sequencing, and this has led to a problem with misannotations. Recent research has shown that not only are protein annotations incorrect or confusing, but many protein sequences are missing annotations [
1,
2,
3,
4,
5]. For example, in [
5], protein sequences downloaded in 2016 using the National Center for Biotechnology Information (NCBI) GenBank ftp service [
6] were clustered, and it was found that of 2826 proteobacterial protein sequences that clustered into a single homologous group, 82% included GroEL or GroL in their annotation while the remaining sequences had a variety of annotations including “not yet annotated”,
mopA (obsolete gene name), thermosome, and 60 kDa chaperonin (11.78%). Moreover, 44 non-GroEL protein sequences were incorrectly annotated as chaperonin GroEL by the original annotators. The preferred annotation for this sequence listed in the UniProtKB/Swiss-Prot database is 60 kDa chaperonin while NCBI RefSeq annotates it as molecular chaperone GroEL [
5]. With a lack of consensus between these two mainstream databases for such a universally conserved protein, it is not surprising that major issues exist with protein annotation.
How can we reduce the problems with protein annotation? Manual curation would be ideal, but it demands substantial time and effort and, thus, the exponential growth in sequences has only been matched by a linear increase, with a relatively small slope, in the number of annotated entries with experimental validation [
7]. In the following paragraphs, we discuss our approach to this problem.
Unlike gene misannotation, which often occurs because of sequence discrepancies, protein misannotation arises from confusion whereby different names are used by the original annotators, and these names are propagated, sometimes appropriately and sometimes not; from lack of domain knowledge; and by error propagation. To improve protein annotation, we decided to use peer-reviewed biomedical papers that provide experimental evidence for manually curated protein function for a given protein annotation. Unfortunately, classifying whether a biomedical paper contains such evidence poses numerous challenges when determining the correct feature representation for the necessary relevant biological relationships. In [
8], protein features (protein lexicons) and assertion features (sentences expressing the main point of a paper) are extracted from publications, and a document classification corpus is constructed using evidence from the Function category of UniProtKB/Swiss-Prot. However, the set of target lexicons they use to indicate experimental confirmation, for example, “requir(e)”, “function”, “promot(e)”, and “control”, is very limited.
Compared with traditional techniques for language modeling such as hidden Markov models and handcrafted features as used in [
8], the simple yet useful bag-of-n-grams representation [
9] relies on a predetermined vocabulary of known words and measurement of the occurrence of each word. For n-grams (sequences of
n successive words), the probability of a word is computed from the
n-1 previous words using maximum likelihood estimation based on word frequency in a corpus and count normalization. Although a bag-of-n-grams contains some information on word order within a context size of
n, it is subject to the problem of sparsity because it underestimates the probability of words rarely occurring in a training corpus, and a zero or near-zero probability of the existence of such words makes it impossible to compute the probability of neighboring words in the test set [
10]. Most importantly, it is impractical to manually design a comprehensive vocabulary that contains the essential words from biomedical papers given the vast number of biomedical terms that exist.
More complex features such as part-of-speech (POS) tags, noun phrases [
11], and tree kernels [
12] are also widely used in feature representation. However, it is difficult to evaluate when POS tags and noun phrases are discriminatory features for a specific classification task [
13]. Tree kernels are able to generate many useful and relevant syntactic features, but their computational time complexity is superlinear with the number of tree nodes (syntactic features), and the accuracy using features generated by tree kernels is lower than that of linear models using manually crafted features [
14].
With recent improvements in deep learning, embedding methods such as word2vec [
15] and doc2vec [
10], which transform a word or a paragraph into a numeric vector representing its semantics, are widely used in feature representation. By using the densely distributed feature representation of word embeddings for each word as learned from its usage, words used in a similar context have similar representations to denote their meaning. Although word embedding uses a distinct numeric vector to represent a word, it overlooks the internal structure of words and is poor at learning words that do not appear in the training data.
In our work, we use BioWordVec [
16], a word embedding method that captures subword information, i.e., each word is further represented as a bag of n-gram characters [
17], and exploits the internal structure of words to enrich feature representation in the numeric vector. Once such an effective feature representation of a text input is available, it can be widely used for classification in deep learning models such as recurrent neural networks (RNNs) [
18] and convolutional neural networks (CNNs) [
19]. In RNNs, a sequence is broken into multiple words and the output is passed from the previous layer of the network to the current layer; in CNNs, a matrix is formed using word embeddings, and then convolutions using filters are performed on the matrix to generate feature maps. For our work, we chose to use a recurrent convolutional neural network (RCNN) to better capture contextual information and discriminative phrases using max pooling (a discretization process) and filtering [
20]. Given that ensemble learning works well when errors for the individual models are uncorrelated, we also use two discriminative classifiers, logistic regression and a support vector machine (SVM) with a linear kernel. Logistic regression is efficient for high-dimensional inputs, and regularization can be used to avoid overfitting. Similarly, regularization can be used with the SVM model. For the logistic regression and SVM models, we use BioSentVec [
21] rather than BioWordVec. The former is trained on large-scale biomedical texts to generate sentence embeddings and is more efficient for the traditional models.
To demonstrate the effectiveness of our ensemble learning method, we compare its performance with the state-of-the-art Bidirectional Encoder Representations from Transformers for Biomedical Text Mining (BioBERT) fine-tuned using the same data for the same task. The Bidirectional Encoder Representations from Transformers (BERT) model is a contextualized word representation model based on a masked language model, which predicts masked words randomly in a sequence [
22]. It learns information from both the left and right contexts of an input token during training and has been proven to achieve state-of-the-art performance for most NLP tasks. BioBERT [
23] is a biomedical-domain language representation model pre-trained on large-scale biomedical corpora (PubMed abstracts and PMC full-text articles), which extends BERT to the biomedical domain.
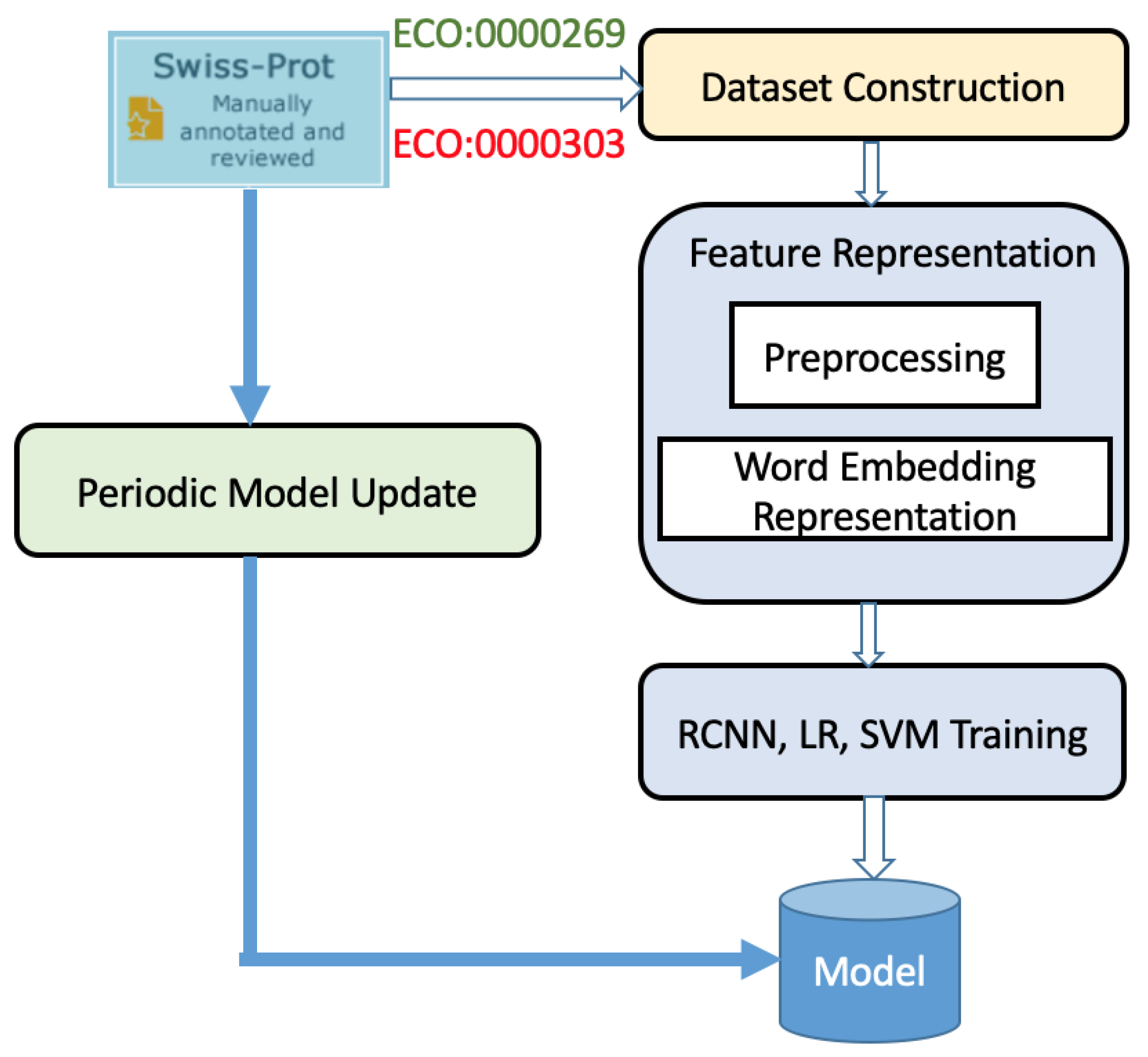
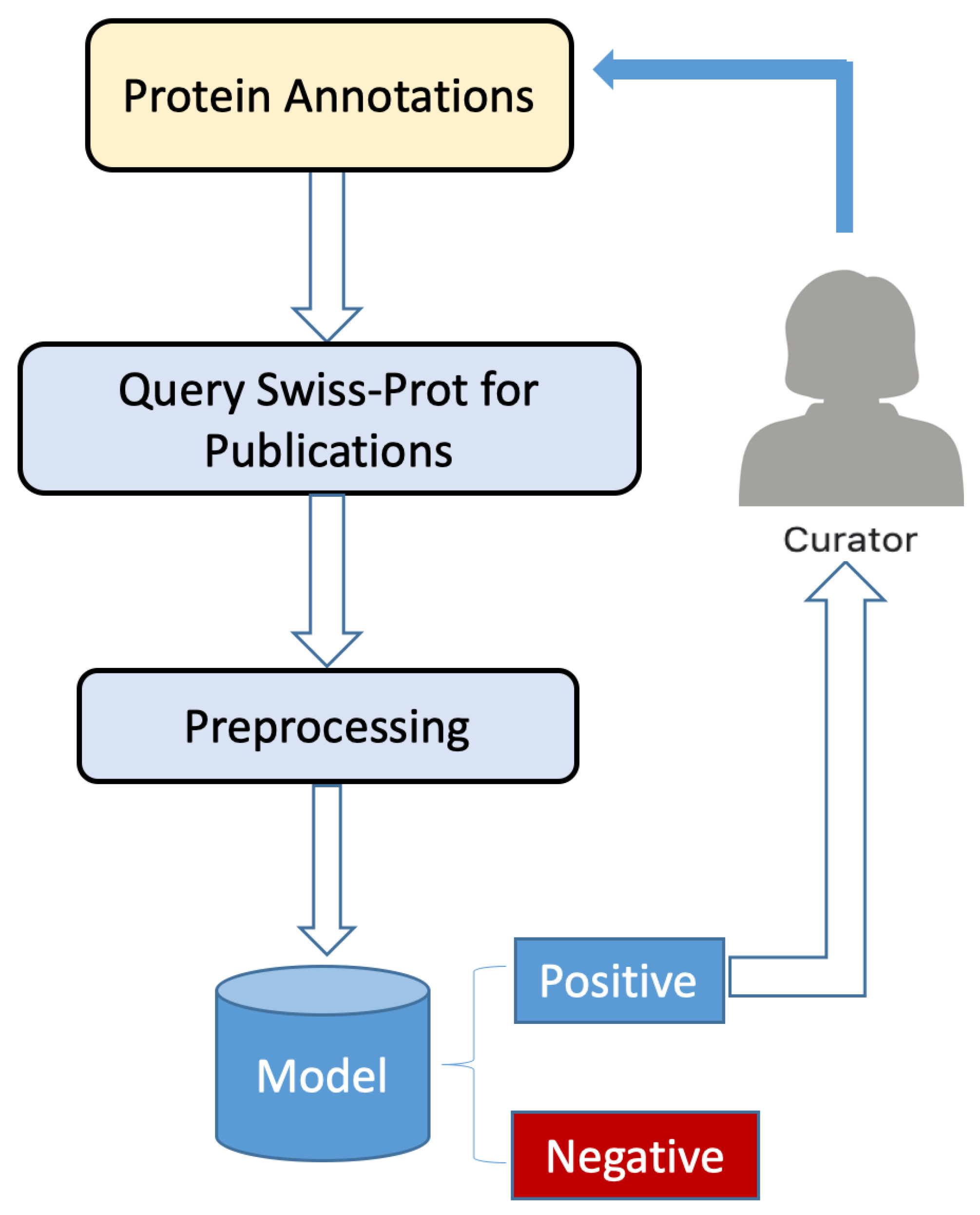
In an effort to ameliorate protein misannotation, we developed a smart software program based on natural language processing (NLP), which periodically queries the UniProtKB database for peer-reviewed articles on a given protein annotation. Word embedding using BioWordVec or BioSentVec is used on all pertinent publication titles retrieved, and an ensemble model is used to determine whether the publications demonstrate laboratory validation of protein function for a given protein annotation. Each of the three models used for ensemble learning—RCNN, logistic regression, and SVM—is individually trained using positive and negative data, and voting is used to classify whether a publication provides the necessary experimental confirmation. Annotations of proteins of which functions have been confirmed experimentally in peer reviewed publications are considered validated. Importantly, because we want to ensure the maximum number of verifications and because the results are reported to a human curator for confirmation, we use recall as our foremost metric.
In this paper, we describe a smart program used to verify protein annotation based on publications in the UniProtKB database and, in particular, classifying publications without any evidence tag for experimental validation, the majority of which are in the uncurated UniProtKB/TrEMBL database. While this smart program is the key to the success of our project, it is only a part of it. In the near future, we will present a system for individual researchers to use to assist them with annotating protein sequences of interest to them. More details on this system are presented in the Conclusion and Future Work section.
3. Results and Discussion
All data, both positive and negative examples, discussed in this paper were procured from the UniProtKB/Swiss-Prot database as of 10 April 2019. The dataset was randomly shuffled and divided into training (60%), validation (20%), and test (20%) sets. These consisted of 28202, 9398, and 9398 publications, respectively, with positive and negative examples equal in number. As mentioned previously, we tried to include word stemming in our pre-processing step, but non-stemmed words performed better with our pre-trained word embedding model. Thus, all results are based on the use of non-stemmed words. To test the effectiveness of our approach, we compared its performance with the state-of-the-art BioBERT model fine-tuned appropriately.
To evaluate the performance of our model, we used four metrics: accuracy, precision, recall, and the F1 score, as calculated using the following formulas:
where
TP (True Positive) correctly indicates publications have experimental evidence of protein function,
TN (True Negative) correctly indicates publications have no experimental evidence of protein function,
FP (False Positive) incorrectly indicates publications have experimental evidence of protein function, and
FN (False Negative) incorrectly indicates publications have no experimental confirmation of protein function.
Of the four evaluation metrics, recall is the most significant for our application. This is because our goal is to identify as many protein annotations as possible with experimental evidence. Thus, we would rather check publications that may not have experimental verification than to miss any possibilities of those that do. Because all results are sent to a human curator for manual review and curation after each periodic query has been performed, 100% precision can be guaranteed in the end. However, we did not ignore the other metrics. We wanted to achieve an overall balance, and in particular, we ensured a good F1 score, which is the harmonic mean of precision and recall. We did not want to completely sacrifice precision for recall. We could have, in fact, striven for a recall of one, but this would result in wasted time on the part of the curator.
As shown in
Table 1, the logistic regression (threshold = 0.5) and SVM models give similar results. This might be because the two classification categories are not well separated. The RCNN model outperforms both the logistic regression (threshold = 0.5) and SVM models by more than 10% for recall. This is most likely because the RCNN model is better at including contextual information and keywords. Recall that feature representation for the traditional logistic regression and SVM models differs from feature representation for the deep-learning RCNN model. For the traditional methods, conversion of a complete sentence into a numeric vector is accomplished using the BioSentVec model while for the deep learning model, key words are better captured by max pooling after we apply a non-linear activation function and a linear transformation on the word representation.
While the RCNN model achieves higher recall than the logistic regression (threshold = 0.5) and SVM models, the accuracy and precision metrics are not as good. Because recall is our most important metric, we further relaxed our logistic regression model with the use of a threshold of 0.25 and after cross-validation achieved an F1 score of 70% and a recall of 94.64%. With ensemble learning and for our objective, it is reasonable to use a weaker classifier with relatively low precision but high recall. In fact, when the three models are combined to create our ensemble learning model, the metrics generally improve. Importantly, our model achieves a recall of 91.25%. In addition, the F1 score of 76.05% is a marked improvement. Ensemble learning models work well when the error for the individual models is uncorrelated. Given that logistic regression, SVM, and RCNN models differ substantially, it is not surprising that combining them provides considerable improvement.
For comparison, we fine-tuned BioBERT on the same training set for the same classification task. Based on the performance of the validation set, we stopped at six epochs. We found that BioBERT performs better in terms of accuracy and precision mainly because of its deep bidirectionality, which masks random input words and conditions each word bidirectionally to predict the masked words. However, its recall value is much lower than that of our ensemble learning model, and its F1 score is lower as well. In addition, fine-tuning and inference were very slow due to BERT’s huge parameter size.
Table 2 presents results obtained using our ensemble learning model for a number of candidate protein annotations. The entry identifiers are unique and stable accession numbers in the UniProtKB database, which can be easily cross-referenced with accession numbers in the NCBI database. With the first two examples, we demonstrate that our ensemble learning model can identify peer-reviewed biomedical publications indicating experimental evidence of functional annotation for a target protein. We used our smart program with the UniProtKB/Swiss-Prot publications associated with glucarate dehydratase and methylaspartate ammonia-lyase, which are among a “gold standard” set of known enzyme superfamilies rigorously studied by domain experts [
32]. Our model successfully identified glucarate dehydratase (entry identifier P0AES2) and methylaspartate ammonia-lyase (entry identifier Q05514) as correct annotations based on the available publications. More specifically, of the six publications associated with glucarate dehydratase, our model identified “Evolution of enzymatic activities in the enolase superfamily: characterization of the (D)-glucarate/galactarate catabolic pathway in
Escherichia coli” and “Evolution of enzymatic activities in the enolase superfamily: crystallographic and mutagenesis studies of the reaction catalyzed by D-glucarate dehydratase from
Escherichia coli” as positive. Our classification results match exactly with UniProtKB/Swiss-Prot, which classifies only these two publications in the Function category. Of the eight publications associated with methylaspartate ammonia-lyase in UniProtKB/Swiss-Prot, four of them, “The purification and properties of beta-methylaspartase”, “Alteration of the diastereoselectivity of 3-methylaspartate ammonia lyase by using structure-based mutagenesis”, “The structure of 3-methylaspartase from
Clostridium tetanomorphum functions via the common enolase chemical step”, and “Engineering methylaspartate ammonia lyase for the asymmetric synthesis of unnatural amino acids”, were correctly classified as positive by our program. Our model only misclassified “Cloning, sequencing, and expression in
Escherichia coli of the
Clostridium tetanomorphum gene encoding beta-methylaspartase and characterization of the recombinant protein” as negative. Overall, however, our smart program identified this protein sequence as having the correct functional annotation.
With the remaining examples, we show that our smart program can also be applied to protein sequences from UniProtKB/Swiss-Prot without publications tagged in the Function category as well as to protein sequences in the unreviewed UniProtKB/TrEMBL database. For the UniProtKB/Swiss-Prot protein sequences 60S ribosomal protein L5-2 (entry identifier Q8L4L4) and Protein PsiE homolog (entry identifier Q81XB6), our model correctly classified both as negative and all their associated publications negative as well. For example, proteins from UniProtKB/TrEMBL, our model identified MEOX2 (entry identifier Q6FHY5) as positive by predicting 18 of 20 publications as positive. The exceptions were “Cloning of human full open reading frames in Gateway(TM) system entry vector (pDONR201)” and “Two candidate tumor suppressor genes, MEOX2 and SOSTDC1, identified in a 7p21 homozygous deletion region in a Wilms tumor”. Some publications in UniProtKB/TrEMBL are assigned to categories, but they are not manually reviewed; only the two publications listed above were assigned to the Sequences category, and our program did not think they contain function-related experimental evidence. Our model identified Beta catenin 1 (entry identifier B1MV73) as positive because it predicted both publications “Equine CTNNB1 and PECAM1 nucleotide structure and expression analyses in an experimental model of normal and pathological wound repair” and “Genome sequence, comparative analysis, and population genetics of the domestic horse” as positive although both these publications are tagged in the Sequences category. However, our human curator determined this to be a false positive case because the latter paper is a genome paper and the former paper looks at the expression of the gene and how it is regulated during wound repair rather than focusing on the target protein. The involvement of a human curator guarantees the accuracy of our final result after prediction by our smart program. Our model identified Maillard deglycase (entry identifier K7G4K8) as negative because it predicted both publications “The complete mitochondrial genome of the Korean soft-shelled turtle Pelodiscus sinensis (Testudines, Trionychidae)” and “The draft genomes of soft-shell turtle and green sea turtle yield insights into the development and evolution of the turtle-specific body plan” as negative. Our model identified Catenin (Cadherin-associated protein), beta 1, 88 kDa (entry identifier H2R2U1) as negative because it predicted both publications “Initial sequence of the chimpanzee genome and comparison with the human genome” and “De novo assembly of the reference chimpanzee transcriptome from NextGen mRNA sequences” as negative. Overall, our model correctly identified publications containing experimental evidence of protein function regardless of the publication category. As such, it can be used to identify sequences with experimental evidence of protein function. Publications associated with any annotations that have been predicted as positive are sent to a human curator for final review.
Our ensemble learning approach is able to effectively identify publications that are likely to provide experimental evidence of correct protein annotation and a partial solution to the protein annotation problem.





{kind=link}
{kind=link}