Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases


 ,
,  and
and 
Abstract
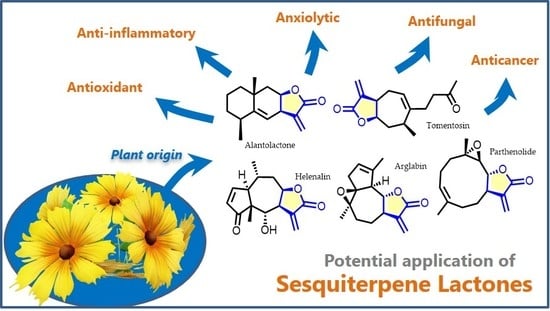
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Sesquiterpene Lactones with Significant In Vivo Activity
2.1. Alantolactone
2.2. Arglabin
2.3. Costunolide
2.4. Cynaropicrin
2.5. Helenalin
2.6. Inuviscolide
2.7. Lactucin and Its Derivatives Lactupicrin, and Lactucopicrin
2.8. Parthenolide
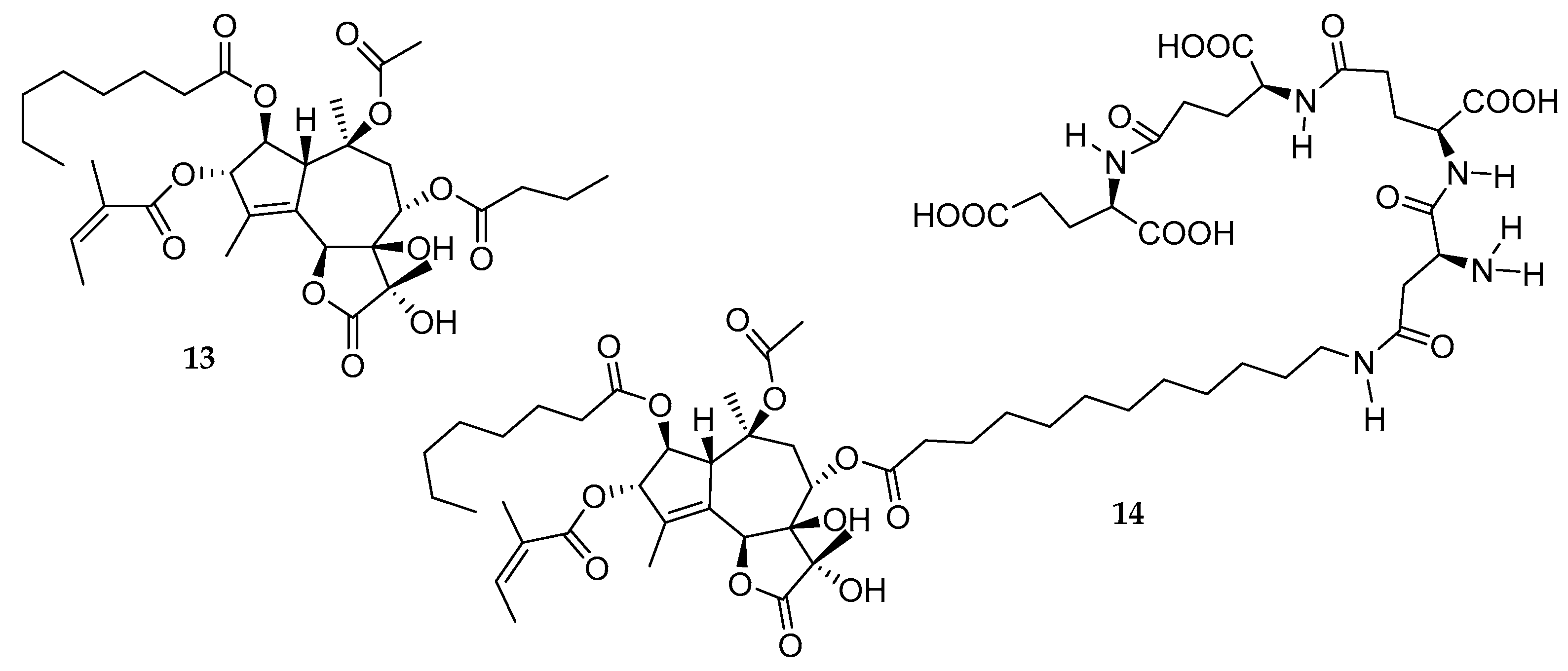
2.9. Thapsigargin
2.10. Tomentosin
3. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, Q.; Wang, Z.; Xie, Y.; Hu, H. Antitumor activity and mechanism of costunolide and dehydrocostus lactone: Two natural sesquiterpene lactones from the Asteraceae family. Biomed. Pharmacother. 2020, 125, 109955. [Google Scholar] [CrossRef] [PubMed]
- Stavrianidi, A. A classification of liquid chromatography mass spectrometry techniques for evaluation of chemical composition and quality control of traditional medicines. J. Chromatog. A 2020, 1609, 460501. [Google Scholar] [CrossRef] [PubMed]
- Gou, J.; Hao, F.; Huang, C.; Kwon, M.; Chen, F.; Li, C.; Liu, C.; Ro, D.-K.; Tang, H.; Zhang, Y. Discovery of a non-stereoselective cytochrome P450 catalyzing either 8α- or 8β-hydroxylation of germacrene A acid from the Chinese medicinal plant, Inula hupehensis. Plant J. 2018, 93, 92–106. [Google Scholar] [CrossRef] [PubMed]
- Perassolo, M.; Cardillo, A.B.; Busto, V.D.; Giulietti, A.M.; Talou, J.R. Biosynthesis of sesquiterpene lactones in plants and metabolic engineering for their biotechnological production. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V.P., Martino, V.S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 47–91. [Google Scholar] [CrossRef]
- Sülsen, V.P.; Martino, V.S. Overview. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V.P., Martino, V.S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 3–17. [Google Scholar] [CrossRef]
- Choodej, S.; Pudhom, K.; Yamauchi, K.; Mitsunaga, T. Inhibition of melanin production by sesquiterpene lactones from Saussurea lappa and their analogues. Med. Chem. Res. 2019, 28, 857–862. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Grigore, A.; Pinto, D.C.G.A.; Silva, A.M.S. The genus Inula and their metabolites: From ethnopharmacological to medicinal uses. J. Ethnopharmacol. 2014, 154, 286–310. [Google Scholar] [CrossRef]
- Bosco, A.; Golsteyn, R.M. Emerging anti-mitotic activities and other bioactivities of sesquiterpene compounds upon human cells. Molecules 2017, 22, 459. [Google Scholar] [CrossRef]
- Beer, M.F.; Bivona, A.E.; Sánchez Alberti, A.; Cerny, N.; Reta, G.F.; Martín, V.S.; Padrón, J.M.; Malchiodi, E.L.; Sülsen, V.P.; Donadel, O.J. Preparation of sesquiterpene lactone derivatives: Cytotoxic activity and selectivity of action. Molecules 2019, 24, 1113. [Google Scholar] [CrossRef]
- Sokovic, M.; Ciric, A.; Glamoclija, J.; Skaltsa, H. Biological activities of sesquiterpene lactones isolated from the genus Centaurea L. (Asteraceae). Curr. Pharm. Des. 2017, 23, 2767–2786. [Google Scholar] [CrossRef]
- Ma, C.; Meng, C.-W.; Zhou, Q.-M.; Peng, C.; Liu, F.; Zhang, J.-W.; Zhou, F.; Xiong, L. New sesquiterpenoids from the stems of Dendrobium nobile and their neuroprotective activities. Fitoterapia 2019, 138, 104351. [Google Scholar] [CrossRef]
- Ghantous, A.; Gali-Muhtasib, H.; Vuorela, H.; Saliba, N.A.; Darwiche, N. What made sesquiterpene lactones reach cancer clinical trials? Drug Discov. Today 2010, 15, 668–678. [Google Scholar] [CrossRef]
- Cheriti, A.; Belboukhari, N. Terpenoids of the Saharan medicinal plants Launaea Cass. genus (Asteraceae) and their biological activities. In Terpenoids and Squalene: Biosynthesis, Functions and Health Implications; Bates, A.R., Ed.; Nova Science Publishers, Inc.: New York, NY, USA, 2015; pp. 51–70. ISBN 978-1-63463-656-8. [Google Scholar]
- Wang, J.; Su, S.; Zhang, S.; Zhai, S.; Sheng, R.; Wu, W.; Guo, R. Structure-activity relationship and synthetic methodologies of α-santonin derivatives with diverse bioactivities: A mini-review. Eur. J. Med. Chem. 2019, 175, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J. Structure-activity and activity-activity relationships of sesquiterpene lactones. In Sesquiterpene Lactones: Advances in Their Chemistry and Biological Aspects; Sülsen, V., Martino, V., Eds.; Springer International Publishing AG: Cham, Switzerland, 2018; pp. 349–371. [Google Scholar] [CrossRef]
- Wang, G.-W.; Qin, J.-J.; Cheng, X.-R.; Shen, Y.-H.; Shan, L.; Jin, H.-Z.; Zhang, W.-D. Inula sesquiterpenoids: Structural diversity, cytotoxicity and anti-tumor activity. Expert Opin. Investig. Drugs 2014, 23, 317–345. [Google Scholar] [CrossRef] [PubMed]
- Reddy, D.M.; Qazi, N.A.; Sawant, S.D.; Bandey, A.H.; Srinivas, J.; Shankar, M.; Singh, S.K.; Verma, M.; Chashoo, G.; Saxena, A.; et al. Design and synthesis of spiro derivatives of parthenin as novel anti-cancer agents. Eur. J. Med. Chem. 2011, 46, 3210–3217. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Cao, J.; Huang, G.; Zhao, Q.; Shen, J. Biological activities of artemisinin derivatives beyond malaria. Curr. Top. Med. Chem. 2019, 19, 205–222. [Google Scholar] [CrossRef]
- Zhang, C.; Zhu, Y.; Yin, X.-P.; Wei, Q.-H.; Zhang, N.-N.; Li, C.-X.; Xie, T.; Chen, R. [Advances in synthesis of artemisinin based on plant genetic engineering]. Zhongguo Zhong Yao Za Zhi 2019, 44, 4285–4292. [Google Scholar] [CrossRef]
- Gao, F.; Sun, Z.; Kong, F.; Xiao, J. Artemisinin-derived hybrids and their anticancer activity. Eur. J. Med. Chem. 2020, 188, 112044. [Google Scholar] [CrossRef]
- Xu, R.; Peng, Y.; Wang, M.; Li, X. Intestinal absorption of isoalantolactone and alantolactone, two sesquiterpene lactones from radix inulae, using Caco-2 cells. Eur. J. Drug Metab. Pharmacokinet. 2019, 44, 295–303. [Google Scholar] [CrossRef]
- Circioban, D.; Pavel, I.Z.; Ledeti, A.; Ledeti, I.; Danciu, C.; Dehelean, C. Cytotoxic activity evaluation on breast cells of guest-host complexes containing artemisinin. Rev. Chim. 2019, 70, 2843–2846. [Google Scholar] [CrossRef]
- Shakeri, A.; Amini, E.; Asili, J.; Masullo, M.; Piacente, S.; Iranshahi, M. Screening of several biological activities induced by different sesquiterpene lactones isolated from Centaurea behen L. and Rhaponticum repens (L.) Hidalgo. Nat. Prod. Res. 2018, 32, 1436–1440. [Google Scholar] [CrossRef]
- Seca, A.M.L.; Pinto, D.C.G.A.; Silva, A.M.S. Metabolomic profile of the genus Inula. Chem. Biodivers. 2015, 12, 859–906. [Google Scholar] [CrossRef]
- Lim, H.S.; Jin, S.E.; Kim, O.S.; Shin, H.K.; Jeong, S.J. Alantolactone from Saussurea lappa exerts antiinflammatory effects by inhibiting chemokine production and STAT1 phosphorylation in TNF-α and IFN-γ-induced in HaCaT cells. Phytother. Res. 2015, 29, 1088–1096. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Choi, R.J.; Khan, S.; Lee, D.-S.; Kim, Y.-C.; Nam, Y.-J.; Lee, D.-U.; Kim, Y.S. Alantolactone suppresses inducible nitric oxide synthase and cyclooxygenase-2 expression by down-regulating NF-κB, MAPK and AP-1 via the MyD88 signaling pathway in LPS-activated RAW 264.7 cells. Int. Immunopharmacol. 2012, 14, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Cohen, N. The stereoselective total synthesis of alantolactone. J. Am. Chem. Soc. 1965, 87, 2773–2774. [Google Scholar] [CrossRef]
- Schultz, A.G.; Godfrey, J.D. An annelation approach to the synthesis of eudesmane and elemane sesquiterpene lactones. Total synthesis of dl-dihydrocallitrisin, dl-7,8-epialantolactone, dl-7,8-epiisoalantolactone, and dl-atractylon. J. Am. Chem. Soc. 1980, 102, 2414–2428. [Google Scholar] [CrossRef]
- Trendafilova, A.; Chanev, C.; Todorova, M. Ultrasound-assisted extraction of alantolactone and isoalantolactone from Inula helenium roots. Pharmacogn. Mag. 2010, 6, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.M.; Wang, J.; Liu, H.B.; Guo, C.Y.; Zhang, W.M. Microwave-assisted extraction of alantolactone and isoalantolactone from Inula helenium. Indian J. Pharm. Sci. 2015, 77, 116–120. [Google Scholar] [CrossRef]
- Chi, X.-F.; Yue, H.-L.; Zhao, X.-H.; Hu, F.-Z. Obtaining alantolactone and isoalantolactone from Inula racemose Hook.f. by optimized supercritical fluid extraction. Ind. Crops Prod. 2016, 79, 63–69. [Google Scholar] [CrossRef]
- Nikonova, L.P. Lactones of Inula magnifica. Chem. Nat. Comp. 1973, 9, 528. [Google Scholar] [CrossRef]
- Stampf, J.L.; Benezra, C.; Klecak, G.; Geleick, H.; Schulz, K.H.; Hausen, B. The sensitizing capacity of helenin and of two of its main constituents, the sesquiterpene lactones alantolactone and isoalantolactone: A comparison of epicutaneous and intradermal sensitizing methods in different strains of guinea pig. Contact Derm. 1982, 8, 16–24. [Google Scholar] [CrossRef]
- Kuno, Y.; Kawabe, Y.; Sakakibara, S. Allergic contact dermatitis associated with photosensitivity, from alantolactone in a chrysanthemum farmer. Contact Derm. 1999, 40, 224–225. [Google Scholar] [CrossRef]
- Marc, E.B.; Nelly, A.; Annick, D.-D.; Frederic, D. Plants used as remedies antirheumatic and antineuralgic in the traditional medicine of Lebanon. J. Ethnopharmacol. 2008, 120, 315–334. [Google Scholar] [CrossRef] [PubMed]
- Sekera, A.; Rahm, J. Natural antihelminthics. III. Helenin. Cesk. Farm. 1953, 2, 22–24. [Google Scholar] [PubMed]
- Kang, X.; Wang, H.; Li, Y.; Xiao, Y.; Zhao, L.; Zhang, T.; Zhou, S.; Zhou, X.; Li, Y.; Shou, Z.; et al. Alantolactone induces apoptosis through ROS-mediated AKT pathway and inhibition of PINK1-mediated mitophagy in human HepG2 cells. Artif. Cells Nanomed. Biotechnol. 2019, 47, 1961–1970. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Cao, X.; Kong, Y.; Wang, S.; Xia, Y.; Bi, R.; Liu, J. Apoptosis-promoting and migration-suppressing effect of alantolactone on gastric cancer cell lines BGC-823 and SGC-7901 via regulating p38 MAPK and NF-κB pathways. Hum. Exp. Toxicol. 2019, 38, 1132–1144. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Yang, Z.; Kong, Y.; He, Y.; Xu, Y.; Cao, X. Antitumor activity of alantolactone in lung cancer cell lines NCI-H1299 and Anip973. J. Food Biochem. 2019, 43, 12972. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Zhang, H.-M. Alantolactone induces gastric cancer BGC-823 cell apoptosis by regulating reactive oxygen species generation and the AKT signaling pathway. Oncol. Lett. 2019, 17, 4795–4802. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lan, Y.L.; Xing, J.S.; Lan, X.Q.; Wang, L.T.; Zhang, B. Alantolactone plays neuroprotective roles in traumatic brain injury in rats via anti-inflammatory, anti-oxidative and anti-apoptosis pathways. Am. J. Transl. Res. 2018, 10, 368–380. [Google Scholar]
- Tavares, W.R.; Seca, A.M.L. Inula L. secondary metabolites against oxidative stress-related human diseases. Antioxidants 2019, 8, 122. [Google Scholar] [CrossRef]
- Da Silva Castro, E.; Azeredo Alves Antunes, L.; Felipe Revoredo Lobo, J.; Arthur Ratcliffe, N.; Moreira Borges, R.; Rocha, L.; Burth, P.; Maria Fonte Amorim, L. Antileukemic properties of sesquiterpene lactones: A systematic review. Anti-Cancer Agents Med. Chem. 2018, 18, 323–334. [Google Scholar] [CrossRef]
- Xu, X.; Huang, L.; Zhang, Z.; Tong, J.; Mi, J.; Wu, Y.; Zhang, C.; Yan, H. Targeting non-oncogene ROS pathway by alantolactone in B cell acute lymphoblastic leukemia cells. Life Sci. 2019, 227, 153–165. [Google Scholar] [CrossRef]
- Chun, J.; Li, R.-J.; Cheng, M.-S.; Kim, Y.S. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015, 357, 393–403. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Cao, P.; Xia, Y.; Hong, L.; Zhang, T.; Shen, X.; Zheng, P.; Shen, H.; Zhao, Y.; Zou, P. Potent inhibition of gastric cancer cells by a natural compound via inhibiting TrxR1 activity and activating ROS-mediated p38 MAPK pathway. Free Radic. Res. 2019, 53, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Cao, P.; Xia, Y.; He, W.; Zhang, T.; Hong, L.; Zheng, P.; Shen, X.; Liang, G.; Cui, R.; Zou, P. Enhancement of oxaliplatin-induced colon cancer cell apoptosis by alantolactone, a natural product inducer of ROS. Int. J. Biol. Sci. 2019, 15, 1676–1684. [Google Scholar] [CrossRef] [PubMed]
- He, R.; Shi, X.; Zhou, M.; Zhao, Y.; Pan, S.; Zhao, C.; Guo, X.; Wang, M.; Li, X.; Qin, R. Alantolactone induces apoptosis and improves chemosensitivity of pancreatic cancer cells by impairment of autophagy-lysosome pathway via targeting TFEB. Toxicol. Appl. Pharmacol. 2018, 356, 159–171. [Google Scholar] [CrossRef]
- Rasul, A.; Khan, M.; Ali, M.; Li, J.; Li, X. Targeting apoptosis pathways in cancer with alantolactone and isoalantolactone. Sci. World J. 2013, 2013, 248532. [Google Scholar] [CrossRef]
- Quintana, J.; Estévez, F. Recent advances on cytotoxic sesquiterpene lactones. Curr. Pharm. Des. 2018, 24, 4355–4361. [Google Scholar] [CrossRef]
- Ren, Y.; Yue, B.; Ren, G.; Yu, Z.; Luo, X.; Sun, A.; Zhang, J.; Han, M.; Wang, Z.; Dou, W. Activation of PXR by alantolactone ameliorates DSS-induced experimental colitis via suppressing NF-κB signaling pathway. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef]
- Seo, J.Y.; Lim, S.S.; Kim, J.; Lee, K.W.; Kim, J.-S. Alantolactone and isoalantolactone prevent amyloid β25-35 -induced toxicity in mouse cortical neurons and scopolamine-induced cognitive impairment in mice. Phytother. Res. 2017, 31, 801–811. [Google Scholar] [CrossRef]
- Kumar, C.; Kumar, A.; Nalli, Y.; Lone, W.I.; Satti, N.K.; Verma, M.K.; Ahmed, Z.; Ali, A. Design, synthesis and biological evaluation of alantolactone derivatives as potential anti-inflammatory agents. Med. Chem. Res. 2019, 28, 849–856. [Google Scholar] [CrossRef]
- Li, X.; Lu, C.; Liu, S.; Liu, S.; Liu, S.; Su, C.; Xiao, T.; Bi, Z.; Sheng, P.; Huang, M.; et al. Synthesis and discovery of a drug candidate for treatment of idiopathic pulmonary fibrosis through inhibition of TGF-β1 pathway. Eur. J. Med. Chem. 2018, 157, 229–247. [Google Scholar] [CrossRef]
- Khan, M.; Yi, F.; Rasul, A.; Li, T.; Wang, N.; Gao, H.; Gao, R.; Ma, T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life 2012, 64, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Markowiak, T.; Kerner, N.; Neu, R.; Potzger, T.; Großer, C.; Zeman, F.; Hofmann, H.-S.; Ried, M. Adequate nephroprotection reduces renal complications after hyperthermic intrathoracic chemotherapy. J. Surg. Oncol. 2019, 120, 1220–1226. [Google Scholar] [CrossRef] [PubMed]
- Leach, C. Complications of systemic anti-cancer treatment. Medicine 2020, 48, 48–51. [Google Scholar] [CrossRef]
- Jain, K.K. An overview of drug delivery systems. Meth. Mol. Biol. 2020, 2059, 1–54. [Google Scholar] [CrossRef]
- Guo, C.; Zhang, S.; Teng, S.; Niu, K. Simultaneous determination of sesquiterpene lactones isoalantolactone and alantolactone isomers in rat plasma by liquid chromatography with tandem mass spectrometry: Application to a pharmacokinetic study. J. Sep. Sci. 2014, 37, 950–956. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Zhou, G.; Peng, Y.; Wang, M.; Li, X. Pharmacokinetics, tissue distribution and excretion of isoalantolactone and alantolactone in rats after oral administration of radix inulae extract. Molecules 2015, 20, 7719–7736. [Google Scholar] [CrossRef]
- Zhou, B.; Ye, J.; Yang, N.; Chen, L.; Zhuo, Z.; Mao, L.; Liu, Q.; Lan, G.; Ning, J.; Ge, G.; et al. Metabolism and pharmacokinetics of alantolactone and isoalantolactone in rats: Thiol conjugation as a potential metabolic pathway. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018, 1072, 370–378. [Google Scholar] [CrossRef]
- Bottex-Gauthier, C.; Vidal, D.; Picot, F.; Potier, P.; Menichini, F.; Appendino, G. In vitro biological activities of arglabin, a sesquiterpene lactone from the Chinese herb Artemisia myriantha Wall. (Asteraceae). Biotechnol. Ther. 1993, 4, 77–98. [Google Scholar]
- Zan, K.; Chen, X.Q.; Tu, P.F. Guaianolides from aerial parts of Artemisia myriantha. Zhongguo Zhongyao Zazhi. 2018, 43, 2295–2299. [Google Scholar] [CrossRef]
- Dylenova, E.P.; Randalova, T.E.; Tykheev, Z.A.; Zhigzhitzhapova, S.V.; Radnaeva, L.D. Artemisia jacutica Drob. as the source of terpenoids. IOP Conf. Ser. Earth Environ. Sci. 2019, 320, 012054. [Google Scholar] [CrossRef]
- Shaikenov, T.E.; Adekenov, S.M.; Williams, R.M.; Prashad, N.; Baker, F.L.; Madden, T.L.; Newman, R. Arglabin-DMA, a plant derived sesquiterpene, inhibits farnesyltransferase. Oncol. Rep. 2001, 8, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lone, S.H.; Bhat, K.A.; Khuroo, M.A. Arglabin: From isolation to antitumor evaluation. Chem.-Biol. Interact. 2015, 240, 180–198. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Reiser, O. Synthetic approaches towards structurally diverse gamma-butyrolactone natural-product-like compounds. Curr. Opin. Chem. Biol. 2005, 9, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kalidindi, S.; Jeong, S.W.B.; Schall, A.; Bandichhor, R.; Nosse, B.; Reiser, O. Enantioselective synthesis of arglabin. Angew. Chem. Int. Ed. 2007, 46, 6361–6363. [Google Scholar] [CrossRef] [PubMed]
- Gharpure, S.J.; Nanda, L.N. Application of oxygen/nitrogen substituted donor-acceptor cyclopropanes in the total synthesis of natural products. Tetrahedron Lett. 2017, 58, 711–720. [Google Scholar] [CrossRef]
- Lone, S.H.; Bhat, K.A. Hemisynthesis of a naturally occurring clinically significant antitumor arglabin from ludartin. Tetrahedron Lett. 2015, 56, 1908–1910. [Google Scholar] [CrossRef]
- Ganesan, A. The impact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef]
- Adekenov, S.M. Chemical modification of arglabin and biological activity of its new derivatives. Fitoterapia 2016, 110, 196–205. [Google Scholar] [CrossRef]
- Musulmanbekov, K.Z. Clinical use of the antitumor drug arglabin. In Proceedings of the International Scientific-Practical Confcrcnce, Karaganda, Kazakhstan, 16–18 October 2002; p. 46. [Google Scholar]
- Zhangabylov, N.S.; Dederer, L.Y.; Gorbacheva, L.B.; Vasil’eva, S.V.; Terekhov, A.S.; Adekenov, S.M. Sesquiterpene lactone arglabin influences DNA synthesis in P388 leukemia cells in vivo. Pharm. Chem. J. 2004, 38, 651–653. [Google Scholar] [CrossRef]
- Sirota, V.B.; Bochkova, N.V.; Kostrova, E.V.; Tselikova, N.L.; Kabildina, N.A. Application of the phytopreparate arglabin in combined treatment of patients with cancer. In Proceedings of the 1st Russian Phytotherapeutic Congress, Moscow, Russia, 14–16 March 2008; p. 148. [Google Scholar]
- Schepetkin, I.A.; Kirpotina, L.N.; Mitchell, P.T.; Kishkentaeva, A.C.; Shaimerdenova, Z.R.; Atazhanova, G.A.; Adekenov, S.M.; Quinn, M.T. The natural sesquiterpene lactones arglabin, grosheimin, agracin, parthenolide, and estafiatin inhibit T cell receptor (TCR) activation. Phytochemistry 2018, 146, 36–46. [Google Scholar] [CrossRef]
- He, W.; Lai, R.F.; Lin, Q.; Huang, Y.M.; Wang, L. Arglabin is a plant sesquiterpene lactone that exerts potent anticancer effects on human oral squamous cancer cells via mitochondrial apoptosis and downregulation of the mTOR/PI3K/Akt signaling pathway to inhibit tumor growth in vivo. J. Buon 2018, 23, 1679–1685. [Google Scholar] [PubMed]
- Tewari, D.; Rawat, P.; Singh, P.K. Adverse drug reactions of anticancer drugs derived from natural sources. Food Chem. Toxicol. 2019, 123, 522–535. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Jia, H.; Xia, H. Arglabin as a potential drug in the treatment of Freund’s complete adjuvant-induced arthritis in rats. Trop. J. Pharm. Res. 2018, 17, 1585–1590. [Google Scholar] [CrossRef]
- Abderrazak, A.; Couchie, D.; Mahmood, D.F.; Elhage, R.; Vindis, C.; Laffargue, M.; Mateo, V.; Buchele, B.; Ayala, M.R.; El Gaafary, M.; et al. Anti-inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high-fat diet. Circulation 2015, 131, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Baldrighi, M.; Mallat, Z.; Li, X. NLRP3 inflammasome pathways in atherosclerosis. Atherosclerosis 2017, 267, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.S.; Kelkar, G.R.; Bhattacharyya, S.C. Terpenoids—XXI: The structure of costunolide, a new sesquiterpene lactone from costus root oil. Tetrahedron 1960, 9, 275–283. [Google Scholar] [CrossRef]
- Ikezawa, N.; Göpfert, J.C.; Nguyen, D.T.; Kim, S.-U.; O’Maille, P.E.; Spring, O.; Ro, D.-K. Lettuce costunolide synthase (CYP71BL2) and its homolog (CYP71BL1) from sunflower catalyze distinct regio- and stereoselective hydroxylations in sesquiterpene lactone metabolism. J. Biol. Chem. 2011, 286, 21601–21611. [Google Scholar] [CrossRef]
- Kassuya, C.A.L.; Cremoneze, A.; Barros, L.F.L.; Simas, A.S.; Lapa, F.d.R.; Mello-Silva, R.; Stefanello, M.E.A.; Zampronio, A.R. Antipyretic and anti-inflammatory properties of the ethanolic extract, dichloromethane fraction and costunolide from Magnolia ovata (Magnoliaceae). J. Ethnopharmacol. 2009, 124, 369–376. [Google Scholar] [CrossRef]
- De Kraker, J.-W.; Franssen, M.C.R.; Joerink, M.; de Groot, A.; Bouwmeester, H.J. Biosynthesis of costunolide, dihydrocostunolide, and leucodin. Demonstration of cytochrome p450-catalyzed formation of the lactone ring present in sesquiterpene lactones of chicory. Plant Physiol. 2002, 129, 257–268. [Google Scholar] [CrossRef]
- Abdelwahab, S.I.; Taha, M.M.E.; Alhazmi, H.A.; Ahsan, W.; Rehman, Z.U.; Bratty, M.A.; Makeen, H. Phytochemical profiling of costus (Saussurea lappa Clarke) root essential oil, and its antimicrobial and toxicological effects. Trop. J. Pharm. Res. 2019, 18, 2155–2160. [Google Scholar] [CrossRef]
- Yang, Z.-J.; Ge, W.-Z.; Li, Q.-Y.; Lu, Y.; Gong, J.-M.; Kuang, B.-J.; Xi, X.; Wu, H.; Zhang, Q.; Chen, Y. Synthesis and biological evaluation of costunolide, parthenolide, and their fluorinated analogues. J. Med. Chem. 2015, 58, 7007–7020. [Google Scholar] [CrossRef] [PubMed]
- Pavan Kumar, C.; Devi, A.; Ashok Yadav, P.; Rao Vadaparthi, R.; Shankaraiah, G.; Sowjanya, P.; Jain, N.; Suresh Babu, K. “Click” reaction mediated synthesis of costunolide and dehydrocostuslactone derivatives and evaluation of their cytotoxic activity. Asian Nat. Prod. Res. 2016, 18, 1063–1078. [Google Scholar] [CrossRef] [PubMed]
- Poon, P.S.; Banerjee, A.K.; Vera, W.J.; Bedoya, L. Reagents for bromination; Application in the synthesis of diterpenes, sesquiterpenes and bioactive compounds. Curr. Org. Chem. 2017, 21, 889–907. [Google Scholar] [CrossRef][Green Version]
- Kim, D.Y.; Choi, B.Y. Costunolide - A bioactive sesquiterpene lactone with diverse therapeutic potential. Int. J. Mol. Sci. 2019, 20, 2926. [Google Scholar] [CrossRef]
- Lin, X.; Peng, Z.; Su, C. Potential anti-cancer activities and mechanisms of costunolide and dehydrocostuslactone. Int. J. Mol. Sci. 2015, 16, 10888–10906. [Google Scholar] [CrossRef]
- Jin, X.; Wang, C.; Wang, L. Costunolide inhibits osteosarcoma growth and metastasis via suppressing STAT3 signal pathway. Biomed. Pharmacother. 2020, 121, 109659. [Google Scholar] [CrossRef]
- Kolosenko, I.; Yu, Y.; Busker, S.; Dyczynski, M.; Liu, J.; Haraldsson, M.; Palm Apergi, C.; Helleday, T.; Tamm, K.P.; Page, B.D.G.; et al. Identification of novel small molecules that inhibit STAT3-dependent transcription and function. PLoS ONE 2017, 12, 0178844. [Google Scholar] [CrossRef]
- Yan, Z.; Xu, T.; An, Z.; Hu, Y.; Chen, W.; Ma, J.; Shao, C.; Zhu, F. Costunolide induces mitochondria-mediated apoptosis in human gastric adenocarcinoma BGC-823 cells. BMC Complement. Altern. Med. 2019, 19, 151. [Google Scholar] [CrossRef]
- Ahmad, F.; Dixit, D.; Sharma, V.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Nrf2-driven TERT regulates pentose phosphate pathway in glioblastoma. Cell Death Dis. 2016, 7, 2213. [Google Scholar] [CrossRef]
- Ahmad, F.; Patrick, S.; Sheikh, T.; Sharma, V.; Pathak, P.; Malgulwar, P.B.; Kumar, A.; Joshi, S.D.; Sarkar, C.; Sen, E. Telomerase reverse transcriptase (TERT) - enhancer of zeste homolog 2 (EZH2) network regulates lipid metabolism and DNA damage responses in glioblastoma. J. Neurochem. 2017, 143, 671–683. [Google Scholar] [CrossRef]
- Menendez, J.A.; Lupu, R. Fatty acid synthase (FASN) as a therapeutic target in breast cancer. Expert Opin. Ther. Targets 2017, 21, 1001–1016. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Bell, E.H.; Mischel, P.; Chakravarti, A. Targeting SREBP-1-driven lipid metabolism to treat cancer. Curr. Pharm. Des. 2014, 20, 2619–2626. [Google Scholar] [CrossRef] [PubMed]
- Bost, F.; Kaminski, L. The metabolic modulator PGC-1α in cancer. Am. J. Cancer Res. 2019, 9, 198–211. [Google Scholar] [PubMed]
- Park, E.; Song, J.H.; Kim, M.S.; Park, S.-H.; Kim, T.S. Costunolide, a sesquiterpene lactone, inhibits the differentiation of pro-inflammatory CD4+ T cells through the modulation of mitogen-activated protein kinases. Int. Immunopharmacol. 2016, 40, 508–516. [Google Scholar] [CrossRef] [PubMed]
- Turk, A.; Ahn, J.H.; Jo, Y.H.; Song, J.Y.; Khalife, H.K.; Gali-Muhtasib, H.; Kim, Y.; Hwang, B.Y.; Lee, M.K. NF-κB inhibitory sesquiterpene lactones from Lebanese Laurus nobilis. Phytochem. Lett. 2019, 30, 120–123. [Google Scholar] [CrossRef]
- Saraswati, S.; Alhaider, A.A.; Abdelgadir, A.M. Costunolide suppresses an inflammatory angiogenic response in a subcutaneous murine sponge model. APMIS 2018, 126, 257–266. [Google Scholar] [CrossRef]
- Xie, Y.; Li, Q.-J.; Wang, Z.-G.; Hu, H.-L. Effects of active components from vladimiriae radix and their combinations on ethanol-induced acute gastric ulcer in mice. Chin. J. New Drugs 2019, 28, 2754–2760. [Google Scholar]
- Chen, Z.; Zhang, D.; Li, M.; Wang, B. Costunolide ameliorates lipoteichoic acid-induced acute lung injury via attenuating MAPK signaling pathway. Int. Immunopharm. 2018, 61, 283–289. [Google Scholar] [CrossRef]
- Chen, Y.-T.; Du, Y.; Zhao, B.; Gan, L.-X.; Yu, K.-K.; Sun, L.; Wang, J.; Qian, F. Costunolide alleviates HKSA-induced acute lung injury via inhibition of macrophage activation. Acta Pharmacol. Sin. 2019, 40, 1040–1048. [Google Scholar] [CrossRef]
- Liu, B.; Rong, Y.; Sun, D.; Li, W.; Chen, H.; Cao, B.; Wang, T. Costunolide inhibits pulmonary fibrosis via regulating NF-κB and TGF-β1/Smad2/Nrf2-NOX4 signaling pathways. Biochem. Biophys. Res. Commun. 2019, 510, 329–333. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, X.; Zhao, L.; Shi, M.; Wei, Z.; Yang, Z.; Guo, C.; Fu, Y. Costunolide protects lipopolysaccharide/D-galactosamine–induced acute liver injury in mice by inhibiting NF-κB signaling pathway. J. Surg. Res. 2017, 220, 40–45. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Moqbel, S.A.A.; Xu, L.; Ran, J.; Ma, C.; Xu, K.; Bao, J.; Jiang, L.; Chen, W.; Xiong, Y.; et al. Costunolide inhibits matrix metalloproteinases expression and osteoarthritis via the NF-κB and Wnt/β-catenin signaling pathways. Mol. Med. Rep. 2019, 20, 312–322. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.-K.; Park, S.-J.; Nam, S.-Y.; Kang, S.; Hwang, J.; Lee, S.-J.; Im, D.-S. Anti-allergic effects of sesquiterpene lactones from Saussurea costus (Falc.) Lipsch. determined using in vivo and in vitro experiments. J. Ethnopharmacol. 2018, 213, 256–261. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.E.; Choi, H.C.; Nam, G.; Choi, B.Y. Costunolide promotes the proliferation of human hair follicle dermal papilla cells and induces hair growth in C57 BL/6 mice. J. Cosmet. Dermatol. 2019, 18, 414–421. [Google Scholar] [CrossRef]
- Cala, A.; Zorrilla, J.G.; Rial, C.; Molinillo, J.M.G.; Varela, R.M.; Macías, F.A. Easy access to alkoxy, amino, carbamoyl, hydroxy, and thiol derivatives of sesquiterpene lactones and evaluation of their bioactivity on parasitic weeds. J. Agric. Food Chem. 2019, 67, 10764–10773. [Google Scholar] [CrossRef]
- Elsebai, M.F.; Mocan, A.; Atanasov, A.G. Cynaropicrin: A comprehensive research review and therapeutic potential as an anti-hepatitis C virus agent. Front. Pharmacol. 2016, 7, 472. [Google Scholar] [CrossRef]
- Suchy, M.; Herout, V.; Šorm, F. On terpenes. CXVI. Structure of cynaropicrin. Collect. Czech. Chem. Commun. 1960, 25, 2777–2782. [Google Scholar] [CrossRef]
- Eljounaidi, K.; Comino, C.; Moglia, A.; Cankar, K.; Genre, A.; Hehn, A.; Bourgaud, F.; Beekwilder, J.; Lanteri, S. Accumulation of cynaropicrin in globe artichoke and localization of enzymes involved in its biosynthesis. Plant Sci. 2015, 239, 128–136. [Google Scholar] [CrossRef]
- Colantuono, A.; Ferracane, R.; Vitaglione, P. Potential bioaccessibility and functionality of polyphenols and cynaropicrin from breads enriched with artichoke stem. Food Chem. 2018, 245, 838–844. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, J.; Han, X.; Xu, J.; Wu, Y.; Fang, J. Promotion of HeLa cells apoptosis by cynaropicrin involving inhibition of thioredoxin reductase and induction of oxidative stress. Free Rad. Biol. Med. 2019, 135, 216–226. [Google Scholar] [CrossRef]
- Formisano, C.; Sirignano, C.; Rigano, D.; Chianese, G.; Zengin, G.; Seo, E.J.; Efferth, T.; Taglialatela-Scafati, O. Antiproliferative activity against leukemia cells of sesquiterpene lactones from the Turkish endemic plant Centaurea drabifolia subsp. detonsa. Fitoterapia 2017, 120, 98–102. [Google Scholar] [CrossRef] [PubMed]
- Nawrot, J.; Budzianowski, J.; Nowak, G. Phytochemical profiles of the leaves of Stizolophus balsamita and Psephellus sibiricus and their chemotaxonomic implications. Phytochemistry 2019, 159, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Cis, J.; Nowak, G.; Kisiel, W. Antifeedant properties and chemotaxonomic implications of sesquiterpene lactones and syringin from Rhaponticum pulchrum. Biochem. Syst. Ecol. 2006, 34, 862–867. [Google Scholar] [CrossRef]
- Schinor, E.C.; Salvador, M.J.; Ito, I.Y.; de Albuquerque, S.; Dias, D.A. Trypanocidal and antimicrobial activities of Moquinia kingii. Phytomedicine 2004, 11, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.Z.; Choi, S.U.; Lee, K.R. Cytotoxic sesquiterpene lactones from Saussurea calcicola. Arch. Pharmacol. Res. 2005, 28, 1142–1146. [Google Scholar] [CrossRef]
- Cho, J.Y.; Kim, A.R.; Jung, J.H.; Chun, T.; Rhee, M.H.; Yoo, E.S. Cytotoxic and pro-apoptotic activities of cynaropicrin, a sesquiterpene lactone, on the viability of leukocyte cancer cell lines. Eur. J. Pharmacol. 2004, 492, 85–94. [Google Scholar] [CrossRef]
- Pandey, M.M.; Rastogi, S.; Rawat, A.K.S. Saussurea costus: Botanical, chemical and pharmacological review of an ayurvedic medicinal plant. J. Ethnopharmacol. 2007, 110, 379–390. [Google Scholar] [CrossRef]
- Bhattacharyya, P.R.; Barua, N.C.; Ghosh, A.C. Cynaropicrin from Tricholepis glaberrima: A potential insect feeding deterrent compound. Ind. Crops Prod. 1995, 4, 291–294. [Google Scholar] [CrossRef]
- Brás, T.; Neves, L.A.; Crespo, J.G.; Duarte, M.F. Effect of extraction methodologies and solvent selection upon cynaropicrin extraction from Cynara cardunculus leaves. Sep. Purif. Technol. 2019, 236, 116283. [Google Scholar] [CrossRef]
- Zimmermann, S.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Cynaropicrin: The first plant natural product with in vivo activity against Trypanosoma brucei. Planta Med. 2012, 78, 553–556. [Google Scholar] [CrossRef]
- Zimmermann, S.; Oufir, M.; Leroux, A.; Krauth-Siegel, R.L.; Becker, K.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Cynaropicrin targets the trypanothione redox system in Trypanosoma brucei. Bioorg. Med. Chem. 2013, 21, 7202–7209. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, C.F.; Batista, D.d.G.; De Araújo, J.S.; Batista, M.M.; Lionel, J.; de Souza, E.M.; Hammera, E.R.; Silva, P.B.; De Mieri, M.; Adams, M.; et al. Activities of psilostachyin A and cynaropicrin against Trypanosoma cruzi in vitro and in vivo. Antimicrob. Agents Chemother. 2013, 57, 5307–5314. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, S.; Fouché, G.; De Mieri, M.; Yoshimoto, Y.; Usuki, T.; Nthambeleni, R.; Parkinson, C.J.; van der Westhuyzen, C.; Kaiser, M.; Hamburger, M.; et al. Structure-activity relationship study of sesquiterpene lactones and their semi-synthetic amino derivatives as potential antitrypanosomal products. Molecules 2014, 19, 3523–3538. [Google Scholar] [CrossRef] [PubMed]
- Usuki, T.; Sato, M.; Hara, S.; Yoshimoto, Y.; Kondo, R.; Zimmermann, S.; Kaiser, M.; Brun, R.; Hamburger, M.; Adams, M. Antitrypanosomal structure-activity-relationship study of synthetic cynaropicrin derivatives. Bioorg. Med. Chem. Lett. 2014, 24, 794–798. [Google Scholar] [CrossRef]
- Tanaka, Y.T.; Tanaka, K.; Kojima, H.; Hamada, T.; Masutani, T.; Tsuboi, M.; Akao, Y. Cynaropicrin from Cynara scolymus L. suppresses photoaging of skin by inhibiting the transcription activity of nuclear factor-kappa B. Bioorg. Med. Chem. Lett. 2013, 23, 518–523. [Google Scholar] [CrossRef]
- Willuhn, G. Arnica flowers: Pharmacology, toxicolgy, and analysis of the sesquiterpene lactones—Their main active substances. ACS Symp. Ser. 1998, 691, 118–132. [Google Scholar]
- Iannitti, T.; Morales-Medina, J.C.; Bellavite, P.; Rottigni, V.; Palmieri, B. Effectiveness and safety of Arnica montana in post-surgical setting, pain and inflammation. Am. J. Ther. 2016, 23, 184–197. [Google Scholar] [CrossRef]
- Todorova, M.; Trendafilova, A.; Vitkova, A.; Petrova, M.; Zayova, E.; Antonova, D. Developmental and environmental effects on sesquiterpene lactones in cultivated Arnica montana L. Chem. Biodiv. 2016, 13, 976–981. [Google Scholar] [CrossRef]
- Widen, J.C.; Kempema, A.M.; Villalta, P.W.; Harki, D.A. Targeting NF-κB p65 with a helenalin inspired bis-electrophile. Chem. Biol. 2017, 12, 102–113. [Google Scholar] [CrossRef]
- Li, Y.; Zeng, Y.; Huang, Q.; Wen, S.; Wei, Y.; Chen, Y.; Zhang, X.; Bai, F.; Lu, Z.; Wei, J.; et al. Helenalin from Centipeda minima ameliorates acute hepatic injury by protecting mitochondria function, activating Nrf2 pathway and inhibiting NF-κB activation. Biomed. Pharmacother. 2019, 119, 109435. [Google Scholar] [CrossRef]
- Kriplani, P.; Guarve, K.; Baghael, U.S. Arnica montana L.—A plant of healing: Review. J. Pharm. Pharmacol. 2017, 69, 925–945. [Google Scholar] [CrossRef] [PubMed]
- Chadwick, M.; Trewin, H.; Gawthrop, F.; Wagstaff, C. Sesquiterpenoids lactones: Benefits to plants and people. Int. J. Mol. Sci. 2013, 14, 12780–12805. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.R.; Yeh, Y.M.; Wang, T.C. Potent inhibition of human telomerase by helenalin. Cancer Lett. 2005, 227, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Berges, C.; Fuchs, D.; Opelz, G.; Daniel, V.; Naujokat, C. Helenalin suppresses essential immune functions of activated CD4+ T cells by multiple mechanisms Mol. Immunol. 2009, 46, 2892–2901. [Google Scholar] [CrossRef]
- Lyss, G.; Knorre, A.; Schmidt, T.J.; Pahl, H.L.; Merfort, I. The anti-inflammatory sesquiterpene lactone helenalin inhibits the transcription factor NF-κB by directly targeting p65. J. Biol. Chem. 1998, 273, 33508–33516. [Google Scholar] [CrossRef]
- Zwicker, P.; Schultze, N.; Niehs, S.; Albrecht, D.; Methling, K.; Wurster, M.; Wachlin, G.; Lalk, M.; Lindequist, U.; Haertel, B. Differential effects of helenalin, an anti-inflammatory sesquiterpene lactone, on the proteome, metabolome and the oxidative stress response in several immune cell types. Toxicol. In Vitro 2017, 40, 45–54. [Google Scholar] [CrossRef]
- Büchele, B.; Zugmaier, W.; Lunov, O.; Syrovets, T.; Merfort, I.; Simmet, T. Surface plasmon resonance analysis of nuclear factor-κB protein interactions with the sesquiterpene lactone helenalin. Anal. Biochem. 2010, 401, 30–37. [Google Scholar] [CrossRef]
- Tornhamre, S.; Schmidt, T.J.; Nasman-Glaser, B. Inhibitory effects of helenalin and related compounds on 5-lipoxygenase and leukotriene C(4) synthase in human blood cells. Biochem Pharmacol. 2001, 62, 903–911. [Google Scholar] [CrossRef]
- Schröder, H.; Lösche, W.; Strobach, H.; Leven, W.; Willuhn, G.; Till, U.; Schrör, K. Helenalin and 11α,13-dihydrohelenalin, two constituents from Arnica montana L., inhibit human platelet function via thiol-dependent pathway. Thromb. Res. 1990, 57, 839–845. [Google Scholar] [CrossRef]
- Macêdo, S.B.; Ferreira, L.R.; Perazzo, F.F.; Tavares Carvalho, J.C. Anti-inflammatory activity of Arnica montana 6cH: Preclinical study in animals. Homeopathy 2004, 93, 84–87. [Google Scholar] [CrossRef]
- Castro, F.C.; Magre, A.; Cherpinski, R.; Zelante, P.M.; Neves, L.M.; Esquisatto, M.A.; Mendonça, F.A.; Santos, G.M. Effects of microcurrent application alone or in combination with topical Hypericum perforatum L. and Arnica montana L. on surgically induced wound healing in Wistar rats. Homeopathy 2012, 101, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Widrig, R.; Suter, A.; Saller, R.; Melzer, J. Choosing between NSAID and Arnica for topical treatment of hand osteoarthritis in a randomised, double-blind study. Rheumatol. Int. 2007, 27, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, D.; Brouillette, E.; Jaspar, F.; Malouin, F.; Mainil, J.; Bureau, F.; Lekeux, P. Helenalin reduces Staphylococcus aureus infection in vitro and in vivo. Vet. Microbiol. 2007, 119, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Valan, M.F.; Britto, J.B.; Venkataraman, R. Phytoconstituents with hepatoprotective activity. Int. J. Chem. Sci. 2010, 8, 1421–1432. [Google Scholar]
- Usui, K.; Ikeda, T.; Horibe, Y.; Nakao, M.; Hoshino, T.; Mizushima, T. Identification of HSP70- inducing activity in Arnica montana extract and purification and characterization of HSP70-inducers. J. Dermatol. Sci. 2015, 78, 67–75. [Google Scholar] [CrossRef]
- Lin, X.; Huang, Q.; Bai, F.; Wei, J.; Huang, R.; Wen, S.; Wei, Y.; Zhang, X. Preparation Method of Traditional Chinese Medicine Centipeda minima Sesquiterpene Monomer Helenalin, and Its Application in Preparing Drug for Treating Hepatic Fibrosis and. Inflammation. Patent No. CN 110283151, 27 September 2019. [Google Scholar]
- Lin, X.; Huang, Q.; Bai, F.; Wei, J.; Huang, R.; Wen, S.; Wei, Y.; Zhang, X. Application of Helenalin for Inhibiting Hepatic Stellate Cell Activation from Centipeda. minima. Patent No. CN 110179790, 30 August 2019. [Google Scholar]
- Messaoudi, M.; Chahmi, N.; El-Mzibri, M.; Gmouh, S.; Amzazi, S.; Benbacer, L.; El-Hassouni, M. Cytotoxic effect and chemical composition of Inula viscosa from three different regions of Morocco. Eur. J. Med. Plants 2016, 16, 1–9. [Google Scholar] [CrossRef]
- Hernández, V.; Recio, M.D.C.; Máñz, S.; Prieto, J.M.; Giner, R.M.; Ríos, J.L. A mechanistic approach to the in vivo anti-inflammatory activity of sesquiterpenoid compounds isolated from Inula viscosa. Planta Med. 2001, 67, 726–731. [Google Scholar] [CrossRef]
- Dolejš, L.; Souček, M.; Horák, M.; Herout, V.; Šorm, F. On terpenes. XCIV. The structure of lactucin. Collect. Czech. Chem. Commun. 1958, 23, 2195–2200. [Google Scholar] [CrossRef]
- Ruban, G.; Zabel, V.; Gensch, K.H.; Smalla, H. The crystal structure and absolute configuration of lactucin. Acta Cryst. 1978, 34, 1163–1167. [Google Scholar] [CrossRef]
- Besharat, S.; Besharat, M.; Jabbari, A. Wild lettuce (Lactuca virosa) toxicity. BMJ Case Rep. 2009, 2009, 10–13. [Google Scholar] [CrossRef]
- De Kraker, J.W.; Franssen, M.C.R.; Dalm, M.C.F.; De Groot, A.; Bouwmeester, H.J. Biosynthesis of germacrene a carboxylic acid in chicory roots. Demonstration of a cytochrome p450 (+)-germacrene a hydroxylase and NADP+-dependent sesquiterpenoid dehydrogenase(s) involved in sesquiterpene lactone biosynthesis. Plant Physiol. 2001, 125, 1930–1940. [Google Scholar] [CrossRef] [PubMed]
- Sessa, R.A.; Bennett, M.H.; Lewis, M.J.; Mansfield, J.W.; Beale, M.H. Metabolite profiling of sesquiterpene lactones from Lactuca Species. J. Biol. Chem. 2000, 275, 26877–26884. [Google Scholar] [CrossRef] [PubMed]
- Testone, G.; Mele, G.; di Giacomo, E.; Tenore, G.C.; Gonnella, M.; Nicolodi, C.; Frugis, G.; Iannelli, M.A.; Arnesi, G.; Schiappa, A.; et al. Transcriptome driven characterization of curly- and smooth-leafed endives reveals molecular differences in the sesquiterpenoid pathway. Hortic. Res. 2019, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.W.; Yang, D.S.; Kays, S.J.; Lee, G.P.; Park, K.W. Sesquiterpene lactones and bitterness in korean leaf lettuce cultivars. HortScience 2009, 44, 246–249. [Google Scholar] [CrossRef]
- Kim, H.D.; Hong, K.B.; Noh, D.O.; Suh, H.J. Sleep-inducing effect of lettuce (Lactuca sativa) varieties on pentobarbital-induced sleep. Food Sci. Biotechnol. 2017, 26, 807–814. [Google Scholar] [CrossRef]
- Bahmani, M.; Shahinfard, N.; Rafieian-Kopaei, M.; Saki, K.; Shahsavari, S.; Taherikalani, M.; Ghafourian, S.; Baharvand-Ahmadi, B. Chicory: A review on ethnobotanical effects of Cichorium intybus L. J. Chem. Pharm. Sci. 2015, 8, 672–682. [Google Scholar]
- Ludwig, H. Ueber die Bestandtheile des Lactucariums. Arch. Pharm. Pharm. Med. Chem. 1847, 100, 1–19. [Google Scholar] [CrossRef]
- Christison, R. A Dispensatory, or Commentary on the Pharmacopoeias of Great Britain; Adam and Charles Black Ed.: Edinburgh, UK, 1842. [Google Scholar]
- Thomson, A.T. A Conspectus of the Pharmacopoeias of the London, Edinburgh and Dublin Collegues of Physicians and of the United States Pharmacopoeia Being a Practical Compendium of Materia Medica and Pharmacy; Lee, C.A., Ed.; Henry, G. Langley, 8 Astor House: New York, NY, USA, 1844. [Google Scholar]
- Wesołowska, A.; Nikiforuk, A.; Michalska, K.; Kisiel, W.; Chojnacka-Wójcik, E. Analgesic and sedative activities of lactucin and some lactucin-like guaianolides in mice. J. Ethnopharmacol. 2006, 107, 254–258. [Google Scholar] [CrossRef]
- Yakoot, M.; Helmy; Fawal, K. Pilot study of the efficacy and safety of lettuce seed oil in patients with sleep disorders. Int. J. Gen. Med. 2011, 4, 451–456. [Google Scholar] [CrossRef]
- Schmidt, B.; Ilic, N.; Poulev, A.; Raskin, I. Toxicological evaluation of chicory extract (humans). Food Chem. Toxicol. 2007, 45, 1–18. [Google Scholar] [CrossRef]
- Ghantous, A.; Sinjab, A.; Herceg, Z.; Darwiche, N. Parthenolide: From plant shoots to cancer roots. Drug Discov. Today 2013, 18, 894–905. [Google Scholar] [CrossRef] [PubMed]
- Kreuger, M.R.O.; Grootjans, S.; Biavatti, M.; Vandanabeele, P.; Dherde, K. Sesquiterpene lactones as drugs with multiple targets in cancer treatment: Focus on parthenolide. Anticancer Drugs 2011, 23, 883–896. [Google Scholar] [CrossRef]
- Jafari, N.; Nazeri, S.; Enferadi, S.T. Parthenolide reduces metastasis by inhibition of vimentin expression and induces apoptosis by suppression elongation factor α−1 expression. Phytomedicine 2018, 41, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Freund, R.R.A.; Gobrecht, P.; Fischer, D.; Arndt, H.-D. Advances in chemistry and bioactivity of parthenolide. Nat. Prod. Rep 2020. [Google Scholar] [CrossRef] [PubMed]
- Seca, A.M.L.; Silva, A.M.S.; Pinto, D.C.G.A. Parthenolide and parthenolide-like sesquiterpene lactones as multiple targets drugs: Current knowledges and new developments. In Studies in Natural Products Chemistry; Atta-Ur-Rahman, Ed.; Elsevier Science Publishers: Amsterdam, The Netherlands, 2017; Volume 52, pp. 337–372. [Google Scholar] [CrossRef]
- Ren, Y.; Yu, J.; Kinghorn, A.D. Development of anticancer agents from plant-derived sesquiterpene lactones. Curr. Med. Chem. 2016, 23, 2397–2420. [Google Scholar] [CrossRef]
- Babaei, G.; Aliarab, A.; Abroon, S.; Rasmi, Y.; Aziz, S.G. Application of sesquiterpene lactone: A new promising way for cancer therapy based on anticancer activity. Biomed. Pharmacother. 2018, 106, 239–246. [Google Scholar] [CrossRef]
- Dey, S.; Sarkar, M.; Giri, B. Anti-inflammatory and anti-tumor activities of parthenolide: An update. Chem. Biol. Ther. 2016, 1, 107. [Google Scholar] [CrossRef]
- Saadane, A.; Eastman, J.; Berger, M.; Bonfield, T.M. Parthenolide inhibits ERK and AP-1 which are dysregulated and contribute to excessive IL-8 expression and secretion in cystic fibrosis cells. J. Inflamm. 2011, 8, 26. [Google Scholar] [CrossRef]
- Carlisi, D.; D’Anneo, A.; Angileri, L.; Lauricella, M.; Emanuele, S.; Santulli, A.; Vento, R.; Tesoriere, G. Parthenolide sensitizes hepatocellular carcinoma cells to TRAIL by inducing the expression of death receptors through inhibition of STAT3 activation. J. Cell Physiol. 2011, 226, 1632–1641. [Google Scholar] [CrossRef]
- Nakshatri, P.; Rice, S.E.; Bhat-Nakshatri, P. Antitumor agent parthenolide reverses resistance of breast cancer cells to tumor necrosis factor-related apoptosis-inducing ligand through sustained activation of c-Jun N-terminal kinase. Oncogene 2004, 23, 7330–7344. [Google Scholar] [CrossRef]
- Rüngeler, P.; Castro, V.; Mora, G.; Gören, N.; Vichnewski, W.; Pah, H.L.; Merfort, I.; Schmidt, T.J. Inhibition of transcription factor NFkappaB by sesquiterpene lactones: A proposed molecular mechanism of action. Bioorg. Med. Chem. 1999, 7, 2343–2352. [Google Scholar] [CrossRef]
- Steele, A.J.; Jones, D.T.; Ganeshaguru, K.; Duke, V.; Yogashangary, B.C.; North, J.M.; Lowdell, M.W.; Kottaridis, P.D.; Mehta, A.; Prentice, A.G.; et al. The sesquiterpene lactone parthenolide induces selective apoptosis of B-chronic lymphocytic leukemia cells in vitro. Leukemia 2006, 20, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- D’Anneo, A.; Carlisi, D.; Lauricella, M.; Puleio, R.; Martinez, R.; Di Bella, S.; Di Marco, P.; Emanuele, S.; Di Fiore, R.; Guercio, A.; et al. Parthenolide generates reactive oxygen species and autophagy inMDA-MB231 cells. A soluble parthenolide analogue inhibits tumour growth and metastasis in a xenograft model of breast cancer. Cell Death Dis. 2013, 4, 89. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Karnischky, L.; Li, X.; Peterson, D.R.; Howard, D.S.; Jordan, C.T. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood 2005, 105, 4163–4169. [Google Scholar] [CrossRef] [PubMed]
- Zunino, S.J.; Ducore, J.M.; Storms, D.H. Parthenolide induces significant apoptosis and production of reactive oxygen species in high-risk pre-B leukemia cells. Cancer Lett. 2007, 254, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Gopal, Y.N.; Arora, T.S.; Van Dyke, M.W. Parthenolide specifically depletes histone deacetylase 1 protein and induces cell death through ataxia telangiectasia mutated. Chem. Biol. 2007, 14, 813–823. [Google Scholar] [CrossRef]
- Liu, Z.; Liu, S.; Xie, Z.; Pavlovicz, R.E.; Wu, J.; Chen, P.; Aimiuwu, J.; Pang, J.; Bhasin, D.; Neviani, P.; et al. Modulation of DNA methylation by a sesquiterpene lactone parthenolide. J. Pharmacol. Exp. Ther. 2009, 329, 505–514. [Google Scholar] [CrossRef]
- Fonrose, X.; Ausseil, F.; Soleilhac, E.; Masson, V.; David, B.; Pouny, I.; Cintrat, J.C.; Rousseau, B.; Barette, C.; Massiot, G.; et al. Parthenolide inhibits tubulin carboxypeptidase activity. Cancer Res. 2007, 67, 3371–3378. [Google Scholar] [CrossRef]
- Mathema, V.B.; Koh, Y.S.; Thakuri, B.C.; Sillanpa, M. Parthenolide, a sesquiterpene lactone, expresses multiple anti-cancer and anti-inflammatory activities. Inflammation 2012, 35, 560–565. [Google Scholar] [CrossRef]
- Pei, S.; Jordan, C.T. How close are we to targeting the leukemia stem cell? Best Pract. Res. Clin. Haematol. 2012, 25, 415–418. [Google Scholar] [CrossRef]
- Gach, K.; Długosz, A.; Janecka, A. The role of oxidative stress in anticancer activity of sesquiterpene lactones. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2015, 388, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Liu, L.; Lee, S.O.; Kim, Y.T.; You, K.R.; Kim, D.G. Susceptibility of cholangiocarcinoma cells to parthenolide-induced apoptosis. Cancer Res. 2005, 65, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.L.; Trang, K.T.; Kim, A.H.; Kim, I.H.; Lee, S.O.; Lee, S.T.; Kim, D.G.; Kim, S.W. Parthenolide suppresses tumor growth in a xenograft model of colorectal cancer cells by inducing mitochondrial dysfunction and apoptosis. Int. J. Oncol. 2012, 41, 1547–1553. [Google Scholar] [CrossRef] [PubMed]
- Yun, B.R.; Lee, M.J.; Kim, J.H.; Kim, I.H.; Yu, G.R.; Kim, D.G. Enhancement of parthenolide-induced apoptosis by a PKC-alpha inhibition through heme oxygenase-1 blockage in cholangiocarcinoma cells. Exp. Mol. Med. 2010, 42, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.L.; Liu, Y.C.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O.; Lee, S.T.; Kim, D.; Kim, S.W. Parthenolide induces apoptosis in colitis-associated colon cancer, inhibiting NF-κB signaling. Oncol. Lett. 2015, 9, 2315–2342. [Google Scholar] [CrossRef] [PubMed]
- Baskaran, N.; Selvam, G.S.; Yuvaraj, S.; Abhishek, A. Parthenolide attenuates 7,12-dimethylbenz[a]anthracene induced hamster buccal pouch carcinogenesis. Mol. Cell. Biochem. 2018, 440, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, H.; Shimizu, K. Involvement of Akt/NF-κB pathway in antitumor effects of parthenolide on glioblastoma cells in vitro and in vivo. BMC Cancer. 2012, 12, 453. [Google Scholar] [CrossRef]
- Zhang, M.; Liu, R.T.; Zhang, P.; Zhang, N.; Yang, C.L.; Yue, L.T.; Li, X.L.; Liu, Y.; Li, H.; Du, J.; et al. Parthenolide inhibits the initiation of experimental autoimmune neuritis. J. Neuroimmunol. 2017, 15, 154–161. [Google Scholar] [CrossRef]
- Fiebich, B.L.; Lieb, K.; Engels, S.; Heinrich, M. Inhibition of LPS-induced p42/44 MAP kinase activation and iNOS/NO synthesis by parthenolide in rat primary microglial cells. J. Neuroimmunol. 2002, 132, 18–24. [Google Scholar] [CrossRef]
- Gobrecht, P.; Andreadaki, A.; Diekmann, H.; Heskamp, A.; Leibinger, M.; Fischer, D. Promotion of functional nerve regeneration by inhibition of microtubule detyrosination. J. Neurosci. 2016, 36, 3890–3902. [Google Scholar] [CrossRef]
- Diekmann, H.; Fischer, D. Parthenolide: A novel pharmacological approach to promote nerve regeneration. Neural. Regen. Res. 2016, 11, 1566–1567. [Google Scholar] [CrossRef] [PubMed]
- Li, X.H.; Xiao, T.; Yang, J.H.; Qin, Y.; Gao, J.J.; Liu, H.J.; Zhou, H.G. Parthenolide attenuated bleomycin-induced pulmonary fibrosis via the NF-κB/Snail signaling pathway. Respir. Res. 2018, 19, 111. [Google Scholar] [CrossRef] [PubMed]
- Van der Heiden, K.; Cuhlmann, S.; Luong, A.; Zakkar, M.; Evans, P.C. Role of nuclear factor kappaB in cardiovascular health and disease. Clin. Sci. (Lond.). 2010, 23, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Latanich, C.A.; Toledo-Pereyra, L.H. Searching for NF-kappaB-based treatments of ischemia reperfusion injury. J. Investig. Surg. 2009, 22, 301–315. [Google Scholar] [CrossRef] [PubMed]
- Zingarelli, B.; Hake, P.W.; Denenberg, A.; Wong, H.R. Sesquiterpene lactone parthenolide, an inhibitor of IkappaB kinase complex and nuclear factor-kappaB, exerts beneficial effects in myocardial reperfusion injury. Shock 2002, 17, 127–134. [Google Scholar] [CrossRef]
- Tsai, T.Y.; Lou, S.L.; Cheng, K.S.; Wong, K.L.; Wang, M.L.; Su, T.H.; Chan, P.; Leu, Y.M. Repressed Ca2+ clearance in parthenolide-treated murine brain bEND.3 endothelial cells. Eur. J. Pharmacol. 2015, 769, 280–286. [Google Scholar] [CrossRef]
- Nasim, S.; Crooks, P.A. Antileukemic activity of aminoparthenolide analogs. Bioorg. Med. Chem. Lett. 2008, 18, 3870–3873. [Google Scholar] [CrossRef]
- Neelakantan, S.; Nasim, S.; Guzman, M.L.; Jordan, C.T.; Crooks, P.A. Aminoparthenolides as novel anti-leukemic agents: Discovery of the NF-kappaB inhibitor, DMAPT (LC-1). Bioorg. Med. Chem. Lett. 2009, 19, 4346–4349. [Google Scholar] [CrossRef]
- Guzman, M.L.; Rossi, R.M.; Neelakantan, S.; Li, X.; Corbett, C.A.; Hassane, D.C.; Becker, M.W.; Bennett, J.M.; Sullivan, E.; Lachowicz, J.L.; et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood 2007, 110, 4427–4431. [Google Scholar] [CrossRef]
- Carlisi, D.G.; Di Fiore, B.R.; Scerri, C.; Drago-Ferrante, R.; Tesoriere, V.G. Parthenolide and DMAPT exert cytotoxic effects on breast cancer stem-like cells by inducing oxidative stress, mitochondrial dysfunction and necrosis. Cell Death Dis. 2016, 7, 2194. [Google Scholar] [CrossRef]
- Song, J.M.; Qian, X.; Upadhyayya, P.; Hong, K.H.M.; Kassie, F. Dimethylaminoparthenolide, a water soluble parthenolide, suppresses lung tumorigenesis through down-regulating the STAT3 signaling pathway. Curr. Cancer Drug Targets 2014, 14, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Nakshatri, H.; Appaiah, H.N.; Anjanappa, M.; Gilley, D.; Tanaka, H.; Badve, S.; Crooks, P.A.; Mathews, W.; Sweeney, C.; Bhat-Nakshatri, P. NF-κB-dependent and -independent epigenetic modulation using the novel anti-cancer agent DMAPT. Cell Death Dis. 2015, 22, 1608. [Google Scholar] [CrossRef] [PubMed]
- Adis Insight. Available online: http://adisinsight.springer.com/drugs/800029612 (accessed on 15 January 2020).
- Morel, K.L.; Ormsby, R.J.; Klebe, S.; Sweeney, C.J.; Sykes, P.J. Parthenolide selectively sensitizes prostate tumor tissue to radiotherapy while protecting healthy tissues in vivo. Radiat. Res. 2017, 187, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Morel, K.L.; Ormsby, R.J.; Klebe, S.; Sweeney, C.J.; Sykes, P.J. DMAPT is an effective radioprotector from long-term radiation-induced damage to normal mouse tissues in vivo. Radiat. Res. 2019, 192, 231–239. [Google Scholar] [CrossRef]
- Mendonca, M.; Turchan, W.; Alpucha, M.; Watson, C.N.; Estabrook, N.; Chin-Sinex, H.; Shapiro, J.B.; Imasuen-Williams, I.E.; Rangela, G.; Gilley, D.; et al. DMAPT inhibits NF-κB activity and increases sensitivity of prostate cancer cells to X-rays in vitro and in tumor xenografts in vivo. Free Radic. Biol. Med. 2017, 112, 318–326. [Google Scholar] [CrossRef]
- Li, X.; Payne, D.T.; Ampolu, B.; Bland, N.; Brown, J.T.; Dutton, M.J.; Fitton, C.A.; Gulliver, A.; Hale, L.; Hamza, D.; et al. Derivatisation of parthenolide to address chemoresistant chronic lymphocytic leukaemia. Med. Chem. Commun. 2019, 10, 1379. [Google Scholar] [CrossRef]
- Baranello, M.P.; Bauer, L.; Danielle, S.W.; Benoit, D. Poly (styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromolecules 2014, 15, 2629–2641. [Google Scholar] [CrossRef]
- Karmakar, A.; Xu, Y.; Mustafa, T.; Kannarpady, G.; Bratton, S.M.; Radominska-Pandya, A.; Crooks, P.A.; Biris, A.S. Nanodelivery of parthenolide using functionalized nanographene enhances its anticancer activity. RSC Adv. 2015, 5, 2411. [Google Scholar] [CrossRef]
- Jin, X.; Zhou, J.; Zhang, Z.; Lv, H. The combined administration of parthenolide and ginsenoside CK in long circulation liposomes with targeted tLyp-1 ligand induce mitochondria-mediated lung cancer apoptosis. artificial cells. Artif. Cells Nanomed. Biotechnol. 2018, 46, S931–S942. [Google Scholar] [CrossRef]
- Darwish, N.H.E.; Sudha, T.; Godugu, K.; Bharali, D.J.; Elbaz, O.; El-Ghaffar, H.A.A.; Azmy, E.; Anber, N.; Mousa, S.A. Novel targeted nano-parthenolide molecule against NF-kB in acute myeloid leukemia. Molecules 2019, 24, 2103. [Google Scholar] [CrossRef]
- Kim, S.L.; Liu, Y.C.; Park, Y.R.; Seo, S.Y.; Kim, S.H.; Kim, I.H.; Lee, S.O.; Lee, S.; Kim, D.G.; Kim, S.W. Parthenolide enhances sensitivity of colorectal cancer cells to TRAIL by inducing death receptor 5 and promotes TRAIL-induced apoptosis. Int. J. Oncol. 2015, 46, 1121–1130. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, B.K.; Yip-Schneider, M.T.; Waters, J.A.; Beane, J.D.; Crooks, P.A.; Schmidt, C.M. Dimethylamino parthenolide enhances the inhibitory effects of gemcitabine in human pancreatic cancer cells. J. Gastrointest. Surg. 2012, 16, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Njoku, V.; Ralstin, M.; Holcomb, B.; Crooks, P.A.; Neelakantan, S.; Sweeney, C.J.; Schmidt, C.M. Effect of celecoxib and the novel anti-cancer agent, dimethylamino-parthenolide, in a developmental model of pancreatic cancer. Pancreas 2008, 37, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Yip-Schneider, M.T.; Wu, H.; Hruban, R.H.; Lowy, A.M.; Crooks, P.A.; Schmidt, C.M. Efficacy of dimethylaminoparthenolide and sulindac in combination with gemcitabine in a genetically engineered mouse model of pancreatic cancer. Pancreas 2013, 42, 160–167. [Google Scholar] [CrossRef]
- Yip-Schneider, M.T.; Wu, H.; Stantz, K.; Agaram, N.; Crooks, P.A.; Schmidt, C.M. Dimethylaminoparthenolide and gemcitabine: A survival study using a genetically engineered mouse model of pancreatic cancer. BMC Cancer 2013, 13, 194. [Google Scholar] [CrossRef]
- Carlisi, D.; Lauricella, M.; D’Anneo, A.; Buttitta, G.; Emanuele, S.; di Fiore, R.; Martinez, R.; Rolfo, C.; Vento, R.; Tesoriere, G. The synergistic effect of SAHA and parthenolide in MDA-MB231 breast cancer cells. J. Cell Physiol. 2015, 230, 1276–1289. [Google Scholar] [CrossRef]
- Lamture, G.; Crooks, P.A.; Borrelli, M.J. Actinomycin-D and dimethylamino-parthenolide synergism in treating human pancreatic cancer cells. Drug Dev. Res. 2018, 79, 287–294. [Google Scholar] [CrossRef]
- Rasmussen, U.; Broogger, C.S.; Sandberg, F. Thapsigargine and thapsigargicine, two new histamine liberators from Thapsia garganica L. Acta Pharm. Suec. 1978, 15, 133–140. [Google Scholar]
- Andersen, T.B.; López, C.Q.; Manczak, T.; Martinez, K.; Simonsen, H.T. Thapsigargin - from Thapsia L. to mipsagargin. Molecules 2015, 20, 6113–6127. [Google Scholar] [CrossRef]
- Doan, N.T.Q.; Paulsen, E.S.; Sehgal, P.; Møller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef]
- Mohamed Ibrahim, A.M.; Martinez-Swatson, K.A.; Benkaci-Ali, F.; Cozzi, F.; Zoulikha, F.; Simonsen, H.T. Effects of gamma irradiation and comparison of different extraction methods on sesquiterpene lactone yields from the medicinal plant Thapsia garganica L. (Apiaceae). J. Appl. Res. Med. Aromat. Plants 2018, 8, 26–32. [Google Scholar] [CrossRef]
- Crestey, F.; Toma, M.; Christensen, S.B. Concise synthesis of thapsigargin from nortrilobolide. Tetrahedron Lett. 2015, 56, 5896–5898. [Google Scholar] [CrossRef]
- Chen, D.; Evans, P.A. A concise, efficient and scalable total synthesis of thapsigargin and nortrilobolide from (R)-(-)-carvone. J. Am. Chem. Soc. 2017, 139, 6046–6049. [Google Scholar] [CrossRef] [PubMed]
- Denmeade, S.R.; Isaacs, J.T. The SERCA pump as a therapeutic target: Making a “smart bomb” for prostate cancer. Cancer Biol. Ther. 2005, 4, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.; Meng, M.; Tian, Z.; Xie, F.; Yin, Q.; Dai, C.; Wang, J.; Zhang, Q.; Liu, Y.; Liu, C. Pharmacological preconditioning with the cellular stress inducer thapsigargin protects against experimental sepsis. Pharmacol. Res. 2019, 141, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Michelangeli, F.; East, J.M. A diversity of SERCA Ca2+ pump inhibitors. Biochem. Soc. Transact. 2011, 39, 789–797. [Google Scholar] [CrossRef]
- Wootton, L.L.; Michelangeli, F. The effects of the phenylalanine to valine mutation on the sensitivity of sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) Ca2+ pump isoforms 1, 2, and 3 to thapsigargin and other inhibitors. J. Biol. Chem. 2006, 281, 6970–6976. [Google Scholar] [CrossRef]
- Skytte, D.M.; Moller, J.V.; Liu, H.; Nielsen, H.O.; Svenningsen, L.E.; Jensen, C.M.; Olsen, C.E.; Christensen, S.B. Elucidation of the topography of the thapsigargin binding site in the sarco-endoplasmic calcium ATPase. Bioorg. Med. Chem. 2010, 18, 5634–5646. [Google Scholar] [CrossRef]
- Shiraishi, M.; Hirasawa, N.; Kobayashi, Y.; Oikawa, S.; Murakami, A.; Ohuchi, K. Participation of mitogen-activated protein kinase in thapsigargin- and TPA-induced histamine production in murine macrophage RAW 264.7 cells. Brit. J. Pharmacol. 2000, 129, 515–524. [Google Scholar] [CrossRef]
- Muramatsu, Y.; Maemoto, T.; Iwashita, A.; Matsuoka, N. Novel neuroprotective compound SCH-20148 rescues thymocytes and SH-SY5Y cells from thapsigargin-induced mitochondrial membrane potential reduction and cell death. Eur. J. Pharmacol. 2007, 563, 40–48. [Google Scholar] [CrossRef]
- Wang, C.; Li, T.; Tang, S.; Zhao, D.; Zhang, C.; Zhang, S.; Deng, S.; Zhou, Y.; Xiao, X. Thapsigargin induces apoptosis when autophagy is inhibited in HepG2 cells and both processes are regulated by ROS-dependent pathway. Environ. Toxicol. Pharmacol. 2016, 41, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Silva, Z.; Verissimo, T.; Videira, P.A.; Novo, C. Protein disulfide isomerases: Impact of thapsigargin treatment on their expression in melanoma cell lines. Int. J. Biol. Macromol. 2015, 79, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, D.R.; Cleveland, J.C.J.; Mitchell, M.B.; Rowland, R.T.; Banerjee, A.; Harken, A.H. Constractive priming of myocardium against ishemia-reperfusion injury. Shock 1996, 6, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Lee, J.E.; Elumalai, S.; Moon, J.S.; Won, K.C. Myricetin prevents thapsigargin-induced CDK5-P66Shc signalosome mediated pancreatic β-cell dysfunction. Free Rad. Biol. Med. 2019, 141, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Janyou, A.; Changtam, C.; Suksamrarn, A.; Tocharus, C.; Tocharus, J. Suppression effects of O-demethyldemethoxycurcumin on thapsigargin triggered on endoplasmic reticulum stress in SK-N-SH cells. Neurotoxicology 2015, 50, 92–100. [Google Scholar] [CrossRef]
- Einbond, L.S.; Wu, H.A.; Sandu, C.; Ford, M.; Mighty, J.; Antonetti, V.; Redenti, S.; Ma, H. Digitoxin enhances the growth inhibitory effects of thapsigargin and simvastatin on ER negative human breast cancer cells. Fitoterapia 2016, 109, 146–154. [Google Scholar] [CrossRef]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Nat. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef]
- Zhong, W.; Chebolu, S.; Darmani, N.A. Thapsigargin-induced activation of Ca2+-CaMKII-ERK in brainstem contributes to substance P release and induction of emesis in the least shrew. Neuropharmacology 2016, 103, 195–210. [Google Scholar] [CrossRef]
- Kmonickova, E.; Harmatha, J.; Vokac, K.; Kostecka, P.; Farghali, H.; Zidek, Z. Sesquiterpene lactone trilobolide activates production of interferon-gamma and nitric oxide. Fitoterapia 2010, 81, 1213–1219. [Google Scholar] [CrossRef]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-activated prodrug: Results of a first-in-man phase I clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A phase II, multicenter, single-arm study of mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [PubMed]
- Park, H.H.; Kim, S.G.; Kim, M.J.; Lee, J.; Choi, B.K.; Jin, M.H.; Lee, E. Suppressive effect of tomentosin on the production of inflammatory mediators in RAW264.7 cells. Biol. Pharm. Bull. 2014, 37, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Merghoub, N.; El Btaouri, H.; Benbacer, L.; Gmouh, S.; Trentesaux, C.; Brassart, B.; Attaleb, M.; Madoulet, C.; Wenner, T.; Amzazi, S.; et al. Tomentosin induces telomere shortening and caspase-dependant apoptosis in cervical cancer cells. J. Cell. Biochem. 2017, 118, 1689–1698. [Google Scholar] [CrossRef]
- Göpfert, J.C.; Heil, N.; Conrad, J.; Spring, O. Cytological development and sesquiterpene lactone secretion in capitate glandular trichomes of sunflower. Plant Biol. 2005, 7, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.M.; Lee, J.; Nam, M.J.; Choi, Y.S.; Park, S.H. Tomentosin displays anti-carcinogenic effect in human osteosarcoma MG-63 cells via the induction of intracellular reactive oxygen species. Int. J. Mol. Sci. 2019, 20, 1508. [Google Scholar] [CrossRef] [PubMed]
- De Laurentis, N.; Losacco, V.; Milillo, M.A.; Lai, O. Chemical investigations of volatile constituents of Inula viscosa (L.) Aiton (Asteraceae) from different areas of Apulia, Southern Italy. Delpinoa 2002, 44, 115–119. [Google Scholar]
- Bar-Shalom, R.; Bergman, M.; Grossman, S.; Azzam, N.; Sharvit, L.; Fares, F. Inula Viscosa extract inhibits growth of colorectal cancer cells in vitro and in vivo through induction of apoptosis. Front. Oncol. 2019, 9, 1–14. [Google Scholar] [CrossRef]
- Cohen, Y.; Wang, W.; Ben-Daniel, B.H.; Ben-Daniel, Y. Extracts of Inula viscosa control downy mildew of grapes caused by Plasmopara viticola. Phytopathology 2006, 96, 417–424. [Google Scholar] [CrossRef]
- Ahern, J.R.; Whitney, K.D. Stereochemistry affects sesquiterpene lactone bioactivity against an herbivorous grasshopper. Chemoecology 2014, 24, 35–39. [Google Scholar] [CrossRef]











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moujir, L.; Callies, O.; Sousa, P.M.C.; Sharopov, F.; Seca, A.M.L. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Appl. Sci. 2020, 10, 3001. https://doi.org/10.3390/app10093001
Moujir L, Callies O, Sousa PMC, Sharopov F, Seca AML. Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Applied Sciences. 2020; 10(9):3001. https://doi.org/10.3390/app10093001
Chicago/Turabian StyleMoujir, Laila, Oliver Callies, Pedro M. C. Sousa, Farukh Sharopov, and Ana M. L. Seca. 2020. "Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases" Applied Sciences 10, no. 9: 3001. https://doi.org/10.3390/app10093001
APA StyleMoujir, L., Callies, O., Sousa, P. M. C., Sharopov, F., & Seca, A. M. L. (2020). Applications of Sesquiterpene Lactones: A Review of Some Potential Success Cases. Applied Sciences, 10(9), 3001. https://doi.org/10.3390/app10093001