Oxidative Phosphorylation—an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis


Abstract
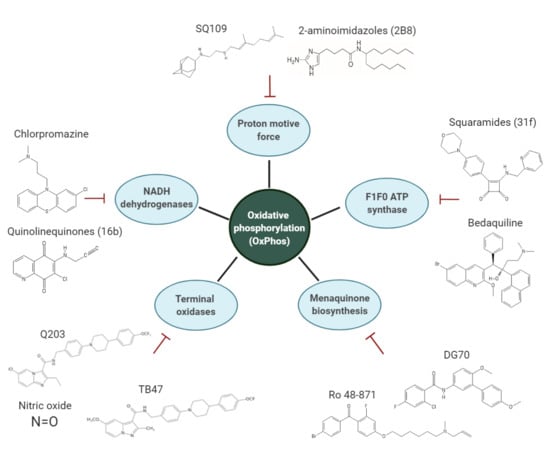
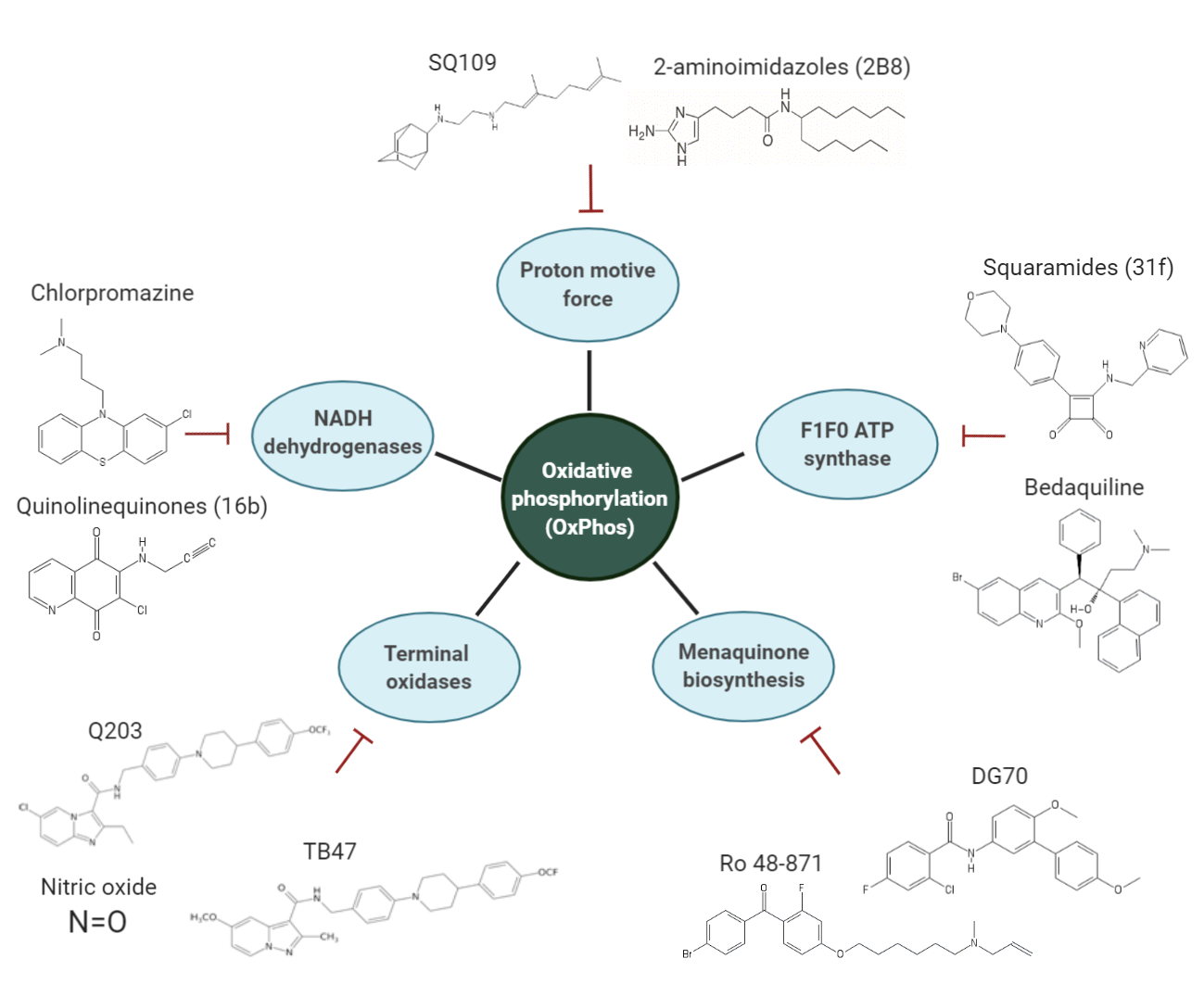
1. Introduction
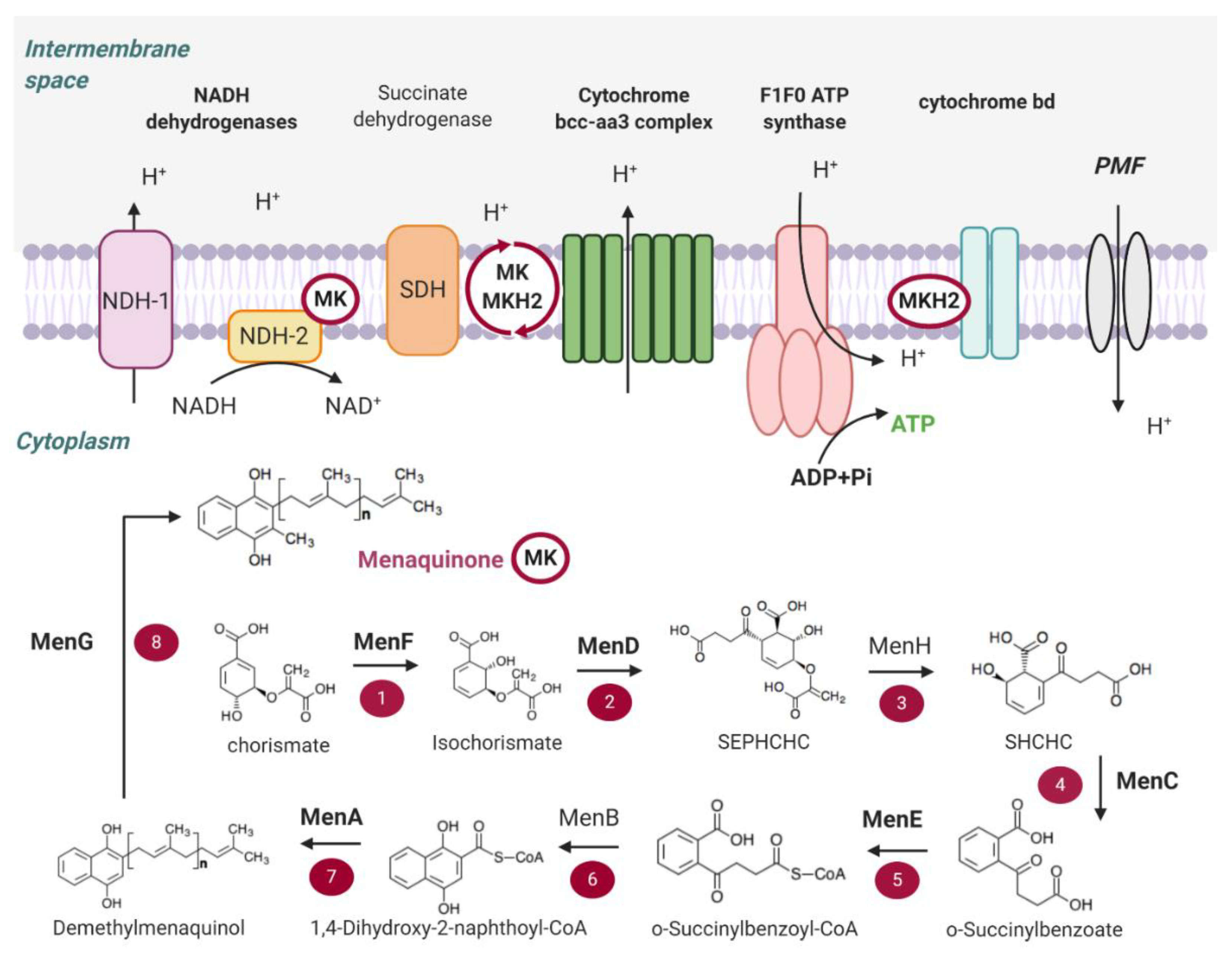
2. Main Components of the OxPhos Pathway in M. tb
2.1. NADH Dehydrogenases
2.2. Succinate Dehydrogenase
2.3. Menaquinone (MK) Biosynthesis
2.4. Terminal Oxidases
2.5. ATP Synthase
2.6. Proton Motive Force (PMF)
3. Overview of the Inhibitors Targeting the OxPhos Pathway
3.1. NADH Dehydrogenases
3.2. Menaquinone (MK) Biosynthesis
3.2.1. MenD
3.2.2. MenE
3.2.3. MenA
3.2.4. MenG
3.3. Inhibitors of the Cyt-bcc-aa3 Complex
3.4. Inhibitors of Cyt-bd Oxidase
3.5. Inhibitors of the F1F0 ATP Synthase
3.6. PMF
3.7. Respiratory Poisoning
4. Combinations Including ETC Inhibitors
5. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO | Global Tuberculosis Report 2019. Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 17 February 2020).
- McKinney, J.D. In vivo veritas: The search for TB drug targets goes live. Nat. Med. 2000, 6, 1330–1333. [Google Scholar] [CrossRef] [PubMed]
- WHO | WHO Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment. Available online: http://www.who.int/tb/publications/2019/consolidated-guidelines-drug-resistant-TB-treatment/en/ (accessed on 17 February 2020).
- Sotgiu, G.; Centis, R.; D’ambrosio, L.; Migliori, G.B. Tuberculosis treatment and drug regimens. Cold Spring Harb. Perspect. Med. 2015, 5, a017822. [Google Scholar] [CrossRef] [PubMed]
- Borisov, S.; Danila, E.; Maryandyshev, A.; Dalcolmo, M.; Miliauskas, S.; Kuksa, L.; Manga, S.; Skrahina, A.; Diktanas, S.; Codecasa, L.R.; et al. Surveillance of adverse events in the treatment of drug-resistant tuberculosis: First global report. Eur. Respir. J. 2019, 54, 1901522. [Google Scholar] [CrossRef] [PubMed]
- Akkerman, O.; Aleksa, A.; Alffenaar, J.-W.; Al-Marzouqi, N.H.; Arias-Guillén, M.; Belilovski, E.; Bernal, E.; Boeree, M.J.; Borisov, S.E.; Bruchfeld, J.; et al. Surveillance of adverse events in the treatment of drug-resistant tuberculosis: A global feasibility study. Int. J. Infect. Dis. 2019, 83, 72–76. [Google Scholar] [CrossRef]
- Janssen Therapeutics, Division of Janssen Products, LP. (2019) Sirturo ®: HIGHLIGHTS OF PRESCRIBING INFORMATION. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/204384s010lbl.pdf (accessed on 26 March 2020).
- European Medicines Agency-Find Medicine-Deltyba. Available online: http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002552/human_med_001699.jsp&mid=WC0b01ac058001d124 (accessed on 31 May 2018).
- U.S. Food and Drug Administration FDA Approves New Drug for Treatment-Resistant Forms of Tuberculosis that Affects the Lungs. Available online: http://www.fda.gov/news-events/press-announcements/fda-approves-new-drug-treatment-resistant-forms-tuberculosis-affects-lungs (accessed on 31 January 2020).
- Singh, R.; Manjunatha, U.; Boshoff, H.I.M.; Ha, Y.H.; Niyomrattanakit, P.; Ledwidge, R.; Dowd, C.S.; Lee, I.Y.; Kim, P.; Zhang, L.; et al. PA-824 kills nonreplicating Mycobacterium tuberculosis by intracellular NO release. Science 2008, 322, 1392–1395. [Google Scholar] [CrossRef]
- Pipeline | Working Group for New TB Drugs. Available online: https://www.newtbdrugs.org/pipeline/clinical (accessed on 17 February 2020).
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting energy metabolism in mycobacterium tuberculosis, a new paradigm in antimycobacterial drug discovery. mBio 2017, 8, e00272-17. [Google Scholar] [CrossRef]
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative phosphorylation as a target space for tuberculosis: Success, caution, and future directions. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef]
- Matsoso, L.G.; Kana, B.D.; Crellin, P.K.; Lea-Smith, D.J.; Pelosi, A.; Powell, D.; Dawes, S.S.; Rubin, H.; Coppel, R.L.; Mizrahi, V. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 2005, 187, 6300–6308. [Google Scholar] [CrossRef]
- Billig, S.; Schneefeld, M.; Huber, C.; Grassl, G.A.; Eisenreich, W.; Bange, F.-C. Lactate oxidation facilitates growth of Mycobacterium tuberculosis in human macrophages. Sci. Rep. 2017, 7, 6484. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.T.; Brosch, R. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Trends Biochem. Sci. 1997, 22, 28–31. [Google Scholar]
- Boshoff, H.I.M.; Barry 3rd, C.E. Tuberculosis—Metabolism and respiration in the absence of growth. Nat. Rev. Microbiol. 2005, 3, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.P.S.; Alonso, S.; Rand, L.; Dick, T.; Pethe, K. The protonmotive force is required for maintaining ATP homeostasis and viability of hypoxic, nonreplicating mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2008, 105, 11945–11950. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Vranckx, L.; Dendouga, N.; Balemans, W.; den Wyngaert, I.V.; Vergauwen, K.; Göhlmann, H.W.H.; Willebrords, R.; Poncelet, A.; Guillemont, J.; et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008, 283, 25273–25280. [Google Scholar] [CrossRef] [PubMed]
- Gengenbacher, M.; Rao, S.P.S.; Pethe, K.; Dick, T. Nutrient-starved, non-replicating Mycobacterium tuberculosis requires respiration, ATP synthase and isocitrate lyase for maintenance of ATP homeostasis and viability. Microbiol. Read. Engl. 2010, 156, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Adams, K.N.; Takaki, K.; Connolly, L.E.; Wiedenhoft, H.; Winglee, K.; Humbert, O.; Edelstein, P.H.; Cosma, C.L.; Ramakrishnan, L. Drug tolerance in replicating mycobacteria mediated by a macrophage-induced efflux mechanism. Cell 2011, 145, 39–53. [Google Scholar] [CrossRef]
- Machado, D.; Couto, I.; Perdigão, J.; Rodrigues, L.; Portugal, I.; Baptista, P.; Veigas, B.; Amaral, L.; Viveiros, M. Contribution of efflux to the emergence of isoniazid and multidrug resistance in Mycobacterium tuberculosis. PLoS ONE 2012, 7, e34538. [Google Scholar] [CrossRef]
- Adams, K.N.; Szumowski, J.D.; Ramakrishnan, L. Verapamil, and its metabolite norverapamil, inhibit macrophage-induced, bacterial efflux pump-mediated tolerance to multiple anti-tubercular drugs. J. Infect. Dis. 2014, 210, 456–466. [Google Scholar] [CrossRef]
- Coelho, T.; Machado, D.; Couto, I.; Maschmann, R.; Ramos, D.; von Groll, A.; Rossetti, M.L.; Silva, P.A.; Viveiros, M. Enhancement of antibiotic activity by efflux inhibitors against multidrug resistant Mycobacterium tuberculosis clinical isolates from Brazil. Front. Microbiol. 2015, 6, 330. [Google Scholar] [CrossRef]
- Li, G.; Zhang, J.; Guo, Q.; Jiang, Y.; Wei, J.; Zhao, L.; Zhao, X.; Lu, J.; Wan, K. Efflux pump gene expression in multidrug-resistant Mycobacterium tuberculosis clinical isolates. PLoS ONE 2015, 10, e0119013. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Villellas, C.; Coeck, N.; Thys, K.; Gevers, T.; Vranckx, L.; Lounis, N.; de Jong, B.C.; Koul, A. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PLoS ONE 2014, 9, e102135. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, E.A.; Yano, T.; Li, L.-S.; Avarbock, D.; Avarbock, A.; Helm, D.; McColm, A.A.; Duncan, K.; Lonsdale, J.T.; Rubin, H. Inhibitors of type II NADH:menaquinone oxidoreductase represent a class of antitubercular drugs. Proc. Natl. Acad. Sci. USA 2005, 102, 4548–4553. [Google Scholar] [CrossRef] [PubMed]
- Harbut, M.B.; Yang, B.; Liu, R.; Yano, T.; Vilchèze, C.; Cheng, B.; Lockner, J.; Guo, H.; Yu, C.; Franzblau, S.G.; et al. Small molecules targeting mycobacterium tuberculosis Type II NADH dehydrogenase exhibit antimycobacterial activity. Angew. Chem. Int. Ed. Engl. 2018, 57, 3478–3482. [Google Scholar] [CrossRef] [PubMed]
- Murugesan, D.; Ray, P.C.; Bayliss, T.; Prosser, G.A.; Harrison, J.R.; Green, K.; Soares de Melo, C.; Feng, T.-S.; Street, L.J.; Chibale, K.; et al. 2-Mercapto-Quinazolinones as inhibitors of Type II NADH Dehydrogenase and Mycobacterium tuberculosis: Structure-activity relationships, mechanism of action and absorption, distribution, metabolism, and excretion characterization. ACS Infect. Dis. 2018, 4, 954–969. [Google Scholar] [CrossRef] [PubMed]
- Sassetti, C.M.; Boyd, D.H.; Rubin, E.J. Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol. 2003, 48, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.E.; Gawronski, J.D.; Dejesus, M.A.; Ioerger, T.R.; Akerley, B.J.; Sassetti, C.M. High-resolution phenotypic profiling defines genes essential for mycobacterial growth and cholesterol catabolism. PLoS Pathog. 2011, 7, e1002251. [Google Scholar] [CrossRef]
- DeJesus, M.A.; Gerrick, E.R.; Xu, W.; Park, S.W.; Long, J.E.; Boutte, C.C.; Rubin, E.J.; Schnappinger, D.; Ehrt, S.; Fortune, S.M.; et al. Comprehensive essentiality analysis of the mycobacterium tuberculosis genome via saturating transposon mutagenesis. mBio 2017, 8, e02133-16. [Google Scholar] [CrossRef]
- Vilchèze, C.; Weinrick, B.; Leung, L.W.; Jacobs, W.R. Plasticity of Mycobacterium tuberculosis NADH dehydrogenases and their role in virulence. Proc. Natl. Acad. Sci. USA 2018, 115, 1599–1604. [Google Scholar] [CrossRef]
- Beites, T.; O’Brien, K.; Tiwari, D.; Engelhart, C.A.; Walters, S.; Andrews, J.; Yang, H.-J.; Sutphen, M.L.; Weiner, D.M.; Dayao, E.K.; et al. Plasticity of the mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat. Commun. 2019, 10, 4970. [Google Scholar] [CrossRef]
- Maklashina, E.; Cecchini, G.; Dikanov, S.A. Defining a direction: Electron transfer and catalysis in Escherichia coli complex II enzymes. Biochim. Biophys. Acta 2013, 1827, 668–678. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hards, K.; Adolph, C.; Harold, L.K.; McNeil, M.B.; Cheung, C.-Y.; Jinich, A.; Rhee, K.Y.; Cook, G.M. Two for the price of one: Attacking the energetic-metabolic hub of mycobacteria to produce new chemotherapeutic agents. Prog. Biophys. Mol. Biol. 2019, S0079610719302111. [Google Scholar] [CrossRef] [PubMed]
- Rutter, J.; Winge, D.R.; Schiffman, J.D. Succinate dehydrogenase—Assembly, regulation and role in human disease. Mitochondrion 2010, 10, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Cook, G.M.; Hards, K.; Vilchèze, C.; Hartman, T.; Berney, M. Energetics of respiration and oxidative phosphorylation in mycobacteria. Microbiol. Spectr. 2014, 2, 389–409. [Google Scholar] [CrossRef] [PubMed]
- Baek, S.-H.; Li, A.H.; Sassetti, C.M. Metabolic regulation of mycobacterial growth and antibiotic sensitivity. PLoS Biol. 2011, 9, e1001065. [Google Scholar] [CrossRef] [PubMed]
- Hartman, T.; Weinrick, B.; Vilchèze, C.; Berney, M.; Tufariello, J.; Cook, G.M.; Jacobs, W.R.J., Jr. Succinate Dehydrogenase is the Regulator of Respiration in Mycobacterium tuberculosis. PLoS Pathog. 2014, 10, e1004510. [Google Scholar] [CrossRef]
- Lemos, R.S.; Fernandes, A.S.; Pereira, M.M.; Gomes, C.M.; Teixeira, M. Quinol:fumarate oxidoreductases and succinate:quinone oxidoreductases: Phylogenetic relationships, metal centres and membrane attachment. Biochim. Biophys. Acta 2002, 1553, 158–170. [Google Scholar] [CrossRef]
- Pecsi, I.; Hards, K.; Ekanayaka, N.; Berney, M.; Hartman, T.; Jacobs, W.R.; Cook, G.M. Essentiality of succinate dehydrogenase in mycobacterium smegmatis and its role in the generation of the membrane potential under hypoxia. mBio 2014, 5, e01093-14. [Google Scholar] [CrossRef]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [CrossRef]
- Meganathan, R. Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): A perspective on enzymatic mechanisms. In Vitamins & Hormones; Cofactor Biosynthesis; Academic Press: Cambridge, MA, USA, 2001; Volume 61, pp. 173–218. [Google Scholar]
- Dhiman, R.K.; Mahapatra, S.; Slayden, R.A.; Boyne, M.E.; Lenaerts, A.; Hinshaw, J.C.; Angala, S.K.; Chatterjee, D.; Biswas, K.; Narayanasamy, P.; et al. Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol. 2009, 72, 85–97. [Google Scholar] [CrossRef]
- Kurosu, M.; Crick, D. MenA is a promising drug target for developing novel lead molecules to combat mycobacterium tuberculosis. Med. Chem. 2009, 5, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Liu, N.; Zhang, H.; Knudson, S.E.; Li, H.-J.; Lai, C.-T.; Simmerling, C.; Slayden, R.A.; Tonge, P.J. CoA Adducts of 4-Oxo-4-phenylbut-2-enoates: Inhibitors of MenB from the M. tuberculosis Menaquinone Biosynthesis Pathway. ACS Med. Chem. Lett. 2011, 2, 818–823. [Google Scholar] [CrossRef] [PubMed]
- Debnath, J.; Siricilla, S.; Wan, B.; Crick, D.C.; Lenaerts, A.J.; Franzblau, S.G.; Kurosu, M. Discovery of selective menaquinone biosynthesis inhibitors against Mycobacterium tuberculosis. J. Med. Chem. 2012, 55, 3739–3755. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhang, H.; Tonge, P.J.; Tan, D.S. Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett. 2008, 18, 5963–5966. [Google Scholar] [CrossRef]
- Sukheja, P.; Kumar, P.; Mittal, N.; Li, S.-G.; Singleton, E.; Russo, R.; Perryman, A.L.; Shrestha, R.; Awasthi, D.; Husain, S.; et al. A novel small-molecule inhibitor of the mycobacterium tuberculosis demethylmenaquinone methyltransferase meng is bactericidal to both growing and nutritionally deprived persister cells. mBio 2017, 8, e02022-16. [Google Scholar] [CrossRef]
- Truglio, J.J.; Theis, K.; Feng, Y.; Gajda, R.; Machutta, C.; Tonge, P.J.; Kisker, C. Crystal structure of Mycobacterium tuberculosis MenB, a key enzyme in vitamin K2 biosynthesis. J. Biol. Chem. 2003, 278, 42352–42360. [Google Scholar] [CrossRef]
- Morand, O.H.; Aebi, J.D.; Dehmlow, H.; Ji, Y.H.; Gains, N.; Lengsfeld, H.; Himber, J. Ro 48-8.071, a new 2,3-oxidosqualene:lanosterol cyclase inhibitor lowering plasma cholesterol in hamsters, squirrel monkeys, and minipigs: Comparison to simvastatin. J. Lipid Res. 1997, 38, 373–390. [Google Scholar]
- Goldman, R.C. Why are membrane targets discovered by phenotypic screens and genome sequencing in Mycobacterium tuberculosis? Tuberc. Edinb. Scotl. 2013, 93, 569–588. [Google Scholar] [CrossRef]
- Megehee, J.A.; Hosler, J.P.; Lundrigan, M.D. Evidence for a cytochrome bcc-aa3 interaction in the respiratory chain of Mycobacterium smegmatis. Microbiol. Read. Engl. 2006, 152, 823–829. [Google Scholar] [CrossRef][Green Version]
- Kim, M.-S.; Jang, J.; Ab Rahman, N.B.; Pethe, K.; Berry, E.A.; Huang, L.-S. Isolation and characterization of a hybrid respiratory supercomplex consisting of mycobacterium tuberculosis cytochrome bcc and mycobacterium smegmatis cytochrome aa3. J. Biol. Chem. 2015, 290, 14350–14360. [Google Scholar] [CrossRef]
- Gong, H.; Li, J.; Xu, A.; Tang, Y.; Ji, W.; Gao, R.; Wang, S.; Yu, L.; Tian, C.; Li, J.; et al. An electron transfer path connects subunits of a mycobacterial respiratory supercomplex. Science 2018, 362, eaat8923. [Google Scholar] [CrossRef] [PubMed]
- Wiseman, B.; Nitharwal, R.G.; Fedotovskaya, O.; Schäfer, J.; Guo, H.; Kuang, Q.; Benlekbir, S.; Sjöstrand, D.; Ädelroth, P.; Rubinstein, J.L.; et al. Structure of a functional obligate complex III 2 IV 2 respiratory supercomplex from Mycobacterium smegmatis. Nat. Struct. Mol. Biol. 2018, 25, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [PubMed]
- Allen, R.J.; Brenner, E.P.; VanOrsdel, C.E.; Hobson, J.J.; Hearn, D.J.; Hemm, M.R. Conservation analysis of the CydX protein yields insights into small protein identification and evolution. BMC Genom. 2014, 15, 946. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.K.; Hatch, L.; Cleeter, M.W.J.; Gibson, F.; Cox, G.B.; Wu, G. Cytochrome bd biosynthesis in Escherichia coli: The sequences of the cydC and cydD genes suggest that they encode the components of an ABC membrane transporter. Mol. Microbiol. 1993, 10, 421–430. [Google Scholar] [CrossRef]
- Shepherd, M. The CydDC ABC transporter of Escherichia coli: New roles for a reductant efflux pump. Biochem. Soc. Trans. 2015, 43, 908–912. [Google Scholar] [CrossRef][Green Version]
- Mascolo, L.; Bald, D. Cytochrome bd in Mycobacterium tuberculosis: A respiratory chain protein involved in the defense against antibacterials. Prog. Biophys. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Berney, M.; Hartman, T.E.; Jacobs, W.R. A Mycobacterium tuberculosis Cytochrome bd Oxidase Mutant Is Hypersensitive to Bedaquiline. mBio 2014, 5, e01275-14. [Google Scholar] [CrossRef]
- Kalia, N.P.; Hasenoehrl, E.J.; Rahman, N.B.A.; Koh, V.H.; Ang, M.L.T.; Sajorda, D.R.; Hards, K.; Grüber, G.; Alonso, S.; Cook, G.M.; et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl. Acad. Sci. USA 2017, 114, 7426–7431. [Google Scholar] [CrossRef]
- Safarian, S.; Rajendran, C.; Müller, H.; Preu, J.; Langer, J.D.; Ovchinnikov, S.; Hirose, T.; Kusumoto, T.; Sakamoto, J.; Michel, H. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 2016, 352, 583–586. [Google Scholar] [CrossRef]
- Safarian, S.; Hahn, A.; Mills, D.J.; Radloff, M.; Eisinger, M.L.; Nikolaev, A.; Meier-Credo, J.; Melin, F.; Miyoshi, H.; Gennis, R.B.; et al. Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 2019, 366, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Sviriaeva, E.; Pethe, K. Targeting the cytochrome oxidases for drug development in mycobacteria. Prog. Biophys. Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- D’mello, R.; Hill, S.; Poole, R.K. The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: Implications for regulation of activity in vivo by oxygen inhibition. Microbiology 1996, 142, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Gennis, R.B. The cytochrome d complex is a coupling site in the aerobic respiratory chain of Escherichia coli. J. Biol. Chem. 1985, 260, 14003–14008. [Google Scholar] [PubMed]
- Jasaitis, A.; Borisov, V.B.; Belevich, N.P.; Morgan, J.E.; Konstantinov, A.A.; Verkhovsky, M.I. Electrogenic reactions of cytochrome bd. Biochemistry 2000, 39, 13800–13809. [Google Scholar] [CrossRef]
- Gopinath, V.; Raghunandanan, S.; Gomez, R.L.; Jose, L.; Surendran, A.; Ramachandran, R.; Pushparajan, A.R.; Mundayoor, S.; Jaleel, A.; Kumar, R.A. Profiling the proteome of mycobacterium tuberculosis during dormancy and reactivation. Mol. Cell. Proteom. 2015, 14, 2160–2176. [Google Scholar] [CrossRef]
- Cortes, T.; Schubert, O.T.; Banaei-Esfahani, A.; Collins, B.C.; Aebersold, R.; Young, D.B. Delayed effects of transcriptional responses in Mycobacterium tuberculosis exposed to nitric oxide suggest other mechanisms involved in survival. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef]
- Shi, L.; Sohaskey, C.D.; Kana, B.D.; Dawes, S.; North, R.J.; Mizrahi, V.; Gennaro, M.L. Changes in energy metabolism of Mycobacterium tuberculosis in mouse lung and under in vitro conditions affecting aerobic respiration. Proc. Natl. Acad. Sci. USA 2005, 102, 15629–15634. [Google Scholar] [CrossRef]
- Arora, K.; Ochoa-Montano, B.; Tsang, P.S.; Blundell, T.L.; Dawes, S.S.; Mizrahi, V.; Bayliss, T.; Mackenzie, C.J.; Cleghorn, L.A.T.; Ray, P.C.; et al. Respiratory flexibility in response to inhibition of cytochrome c oxidase in mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6962–6965. [Google Scholar] [CrossRef]
- Foo, C.S.; Lupien, A.; Kienle, M.; Vocat, A.; Benjak, A.; Sommer, R.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; Piton, J.; et al. Arylvinylpiperazine Amides, a New Class of Potent Inhibitors Targeting QcrB of Mycobacterium tuberculosis. mBio 2018, 9. [Google Scholar] [CrossRef]
- Lupien, A.; Foo, C.S.-Y.; Savina, S.; Vocat, A.; Piton, J.; Monakhova, N.; Benjak, A.; Lamprecht, D.A.; Steyn, A.J.C.; Pethe, K.; et al. New 2-Ethylthio-4-methylaminoquinazoline derivatives inhibiting two subunits of cytochrome bc1 in Mycobacterium tuberculosis. PLoS Pathog. 2020, 16, e1008270. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Lill, H.; Bald, D. ATP synthase in mycobacteria: Special features and implications for a function as drug target. Biochim. Biophys. Acta BBA-Bioenerg. 2014, 1837, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.E. The ATP synthase: The understood, the uncertain and the unknown. Biochem. Soc. Trans. 2013, 41, 1–16. [Google Scholar] [CrossRef] [PubMed]
- von Ballmoos, C.; Cook, G.M.; Dimroth, P. Unique rotary atp synthase and its biological diversity. Annu. Rev. Biophys. 2008, 37, 43–64. [Google Scholar] [CrossRef]
- Higashi, T.; Kalra, V.K.; Lee, S.H.; Bogin, E.; Brodie, A.F. Energy-transducing membrane-bound coupling factor-ATPase from Mycobacterium phlei. I. Purification, homogeneity, and properties. J. Biol. Chem. 1975, 250, 6541–6548. [Google Scholar]
- Haagsma, A.C.; Driessen, N.N.; Hahn, M.-M.; Lill, H.; Bald, D. ATP synthase in slow- and fast-growing mycobacteria is active in ATP synthesis and blocked in ATP hydrolysis direction. FEMS Microbiol. Lett. 2010, 313, 68–74. [Google Scholar] [CrossRef]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.H.; Neefs, J.-M.; Winkler, H.; Gestel, J.V.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A Diarylquinoline drug active on the ATP synthase of mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef]
- Bald, D.; Koul, A. Respiratory ATP synthesis: The new generation of mycobacterial drug targets? FEMS Microbiol. Lett. 2010, 308, 1–7. [Google Scholar] [CrossRef]
- Zhang, A.T.; Montgomery, M.G.; Leslie, A.G.W.; Cook, G.M.; Walker, J.E. The structure of the catalytic domain of the ATP synthase from Mycobacterium smegmatis is a target for developing antitubercular drugs. Proc. Natl. Acad. Sci. USA 2019, 116, 4206–4211. [Google Scholar] [CrossRef]
- Joon, S.; Ragunathan, P.; Sundararaman, L.; Nartey, W.; Kundu, S.; Manimekalai, M.S.S.; Bogdanović, N.; Dick, T.; Grüber, G. The NMR solution structure of Mycobacterium tuberculosis F- ATP synthase subunit ε provides new insight into energy coupling inside the rotary engine. FEBS J. 2018, 285, 1111–1128. [Google Scholar] [CrossRef]
- Saw, W.-G.; Wu, M.-L.; Ragunathan, P.; Biuković, G.; Lau, A.-M.; Shin, J.; Harikishore, A.; Cheung, C.-Y.; Hards, K.; Sarathy, J.P.; et al. Disrupting coupling within mycobacterial F-ATP synthases subunit ε causes dysregulated energy production and cell wall biosynthesis. Sci. Rep. 2019, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Hotra, A.; Suter, M.; Biuković, G.; Ragunathan, P.; Kundu, S.; Dick, T.; Grüber, G. Deletion of a unique loop in the mycobacterial F-ATP synthase γ subunit sheds light on its inhibitory role in ATP hydrolysis-driven H+ pumping. FEBS J. 2016, 283, 1947–1961. [Google Scholar] [CrossRef] [PubMed]
- Ragunathan, P.; Sielaff, H.; Sundararaman, L.; Biuković, G.; Manimekalai, M.S.S.; Singh, D.; Kundu, S.; Wohland, T.; Frasch, W.; Dick, T.; et al. The uniqueness of subunit α of mycobacterial F-ATP synthases: An evolutionary variant for niche adaptation. J. Biol. Chem. 2017, 292, 11262–11279. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, N.; Unemoto, T.; Kobayashi, H. Proton motive force is not obligatory for growth of Escherichia coli. J. Bacteriol. 1984, 160, 1074–1077. [Google Scholar] [CrossRef] [PubMed]
- Melo, A.M.P.; Bandeiras, T.M.; Teixeira, M. New insights into type II NAD (P) H:quinone oxidoreductases. Microbiol. Mol. Biol. Rev. 2004, 68, 603–616. [Google Scholar] [CrossRef]
- Yano, T.; Kassovska-Bratinova, S.; Teh, J.S.; Winkler, J.; Sullivan, K.; Isaacs, A.; Schechter, N.M.; Rubin, H. Reduction of clofazimine by mycobacterial type 2 NADH: Quinone oxidoreductase: A pathway for the generation of bactericidal levels of reactive oxygen species. J. Biol. Chem. 2011, 286, 10276–10287. [Google Scholar] [CrossRef]
- Xu, J.; Wang, B.; Fu, L.; Zhu, H.; Guo, S.; Huang, H.; Yin, D.; Zhang, Y.; Lu, Y. In vitro and in vivo activities of the Riminophenazine TBI-166 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019, 63, e02155-18. [Google Scholar] [CrossRef]
- Bourdon, J.L. Contribution to the study of the antibiotic properties of chlorpromazine or 4560 RP. Ann. Inst. Pasteur 1961, 101, 876–886. [Google Scholar]
- Amaral, L.; Kristiansen, J.E.; Abebe, L.S.; Millett, W. Inhibition of the respiration of multi-drug resistant clinical isolates of Mycobacterium tuberculosis by thioridazine: Potential use for initial therapy of freshly diagnosed tuberculosis. J. Antimicrob. Chemother. 1996, 38, 1049–1053. [Google Scholar] [CrossRef]
- Bettencourt, M.V.; Bosne-David, S.; Amaral, L. Comparative in vitro activity of phenothiazines against multidrug-resistant Mycobacterium tuberculosis. Int. J. Antimicrob. Agents 2000, 16, 69–71. [Google Scholar] [CrossRef]
- Ratnakar, P.; Murthy, P.S. Antitubercular activity of trifluoperazine, a calmodulin antagonist. FEMS Microbiol. Lett. 1992, 76, 73–76. [Google Scholar] [CrossRef] [PubMed]
- Gadre, D.V.; Talwar, V. In vitro susceptibility testing of Mycobacterium tuberculosis strains to trifluoperazine. J. Chemother. Florence Italy 1999, 11, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Kristiansen, J.E.; Viveiros, M.; Atouguia, J. Activity of phenothiazines against antibiotic-resistant Mycobacterium tuberculosis: A review supporting further studies that may elucidate the potential use of thioridazine as anti-tuberculosis therapy. J. Antimicrob. Chemother. 2001, 47, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Amaral, L.; Viveiros, M. Thioridazine: A non-antibiotic drug highly effective, in combination with first line anti-tuberculosis drugs, against any form of antibiotic resistance of mycobacterium tuberculosis due to its multi-mechanisms of action. Antibiotics 2017, 6, 3. [Google Scholar] [CrossRef]
- Crowle, A.J.; Douvas, G.S.; May, M.H. Chlorpromazine: A drug potentially useful for treating mycobacterial infections. Chemotherapy 1992, 38, 410–419. [Google Scholar] [CrossRef]
- Salie, S.; Hsu, N.-J.; Semenya, D.; Jardine, A.; Jacobs, M. Novel non-neuroleptic phenothiazines inhibit Mycobacterium tuberculosis replication. J. Antimicrob. Chemother. 2014, 69, 1551–1558. [Google Scholar] [CrossRef]
- He, C.-X.; Meng, H.; Zhang, X.; Cui, H.-Q.; Yin, D.-L. Synthesis and bio-evaluation of phenothiazine derivatives as new anti-tuberculosis agents. Chin. Chem. Lett. 2015, 26, 951–954. [Google Scholar] [CrossRef]
- Jardine, M.A.; Jacobs, M. Phenothiazine Derivatives and Their Use against Tuberculosis. WO2014080378A1, 30 May 2014. [Google Scholar]
- Trivedi, A.R.; Siddiqui, A.B.; Shah, V.H. Design, synthesis, characterization and antitubercular activity of some 2-heterocycle-substituted phenothiazines. Arkivoc 2008, 2008, 210–217. [Google Scholar]
- Shirude, P.S.; Paul, B.; Roy Choudhury, N.; Kedari, C.; Bandodkar, B.; Ugarkar, B.G. Quinolinyl Pyrimidines: Potent Inhibitors of NDH-2 as a Novel Class of Anti-TB Agents. ACS Med. Chem. Lett. 2012, 3, 736–740. [Google Scholar] [CrossRef]
- Heikal, A.; Hards, K.; Cheung, C.-Y.; Menorca, A.; Timmer, M.S.M.; Stocker, B.L.; Cook, G.M. Activation of type II NADH dehydrogenase by quinolinequinones mediates antitubercular cell death. J. Antimicrob. Chemother. 2016, 71, 2840–2847. [Google Scholar] [CrossRef]
- Bringmann, G.; Reichert, Y.; Kane, V.V. The total synthesis of streptonigrin and related antitumor antibiotic natural products. Tetrahedron 2004, 60, 3539–3574. [Google Scholar] [CrossRef]
- Colucci, M.A.; Moody, C.J.; Couch, G.D. Natural and synthetic quinones and their reduction by the quinone reductase enzyme NQO1: From synthetic organic chemistry to compounds with anticancer potential. Org. Biomol. Chem. 2008, 6, 637–656. [Google Scholar] [CrossRef] [PubMed]
- Pearce, A.N.; Chia, E.W.; Berridge, M.V.; Clark, G.R.; Harper, J.L.; Larsen, L.; Maas, E.W.; Page, M.J.; Perry, N.B.; Webb, V.L.; et al. Anti-inflammatory thiazine alkaloids isolated from the New Zealand ascidian Aplidium sp.: Inhibitors of the neutrophil respiratory burst in a model of gouty arthritis. J. Nat. Prod. 2007, 70, 936–940. [Google Scholar] [CrossRef] [PubMed]
- Mulchin, B.J.; Newton, C.G.; Baty, J.W.; Grasso, C.H.; Martin, W.J.; Walton, M.C.; Dangerfield, E.M.; Plunkett, C.H.; Berridge, M.V.; Harper, J.L.; et al. The anti-cancer, anti-inflammatory and tuberculostatic activities of a series of 6,7-substituted-5,8-quinolinequinones. Bioorg. Med. Chem. 2010, 18, 3238–3251. [Google Scholar] [CrossRef]
- Santoso, K.T.; Menorca, A.; Cheung, C.-Y.; Cook, G.M.; Stocker, B.L.; Timmer, M.S.M. The synthesis and evaluation of quinolinequinones as anti-mycobacterial agents. Bioorg. Med. Chem. 2019, 27, 3532–3545. [Google Scholar] [CrossRef]
- Barry, V.C.; Belton, J.G.; Conalty, M.L.; Denneny, J.M.; Edward, D.W.; O’sullivan, J.F.; Twomey, D.; Winder, F. A new series of phenazines (rimino-compounds) with high antituberculosis activity. Nature 1957, 179, 1013–1015. [Google Scholar] [CrossRef]
- Zhang, D.; Liu, Y.; Zhang, C.; Zhang, H.; Wang, B.; Xu, J.; Fu, L.; Yin, D.; Cooper, C.B.; Ma, Z.; et al. Synthesis and biological evaluation of novel 2-methoxypyridylamino-substituted riminophenazine derivatives as antituberculosis agents. Molecules 2014, 19, 4380–4394. [Google Scholar] [CrossRef]
- Fang, M.; Toogood, R.D.; Macova, A.; Ho, K.; Franzblau, S.G.; McNeil, M.R.; Sanders, D.A.R.; Palmer, D.R.J. Succinylphosphonate esters are competitive inhibitors of mend that show active-site discrimination between homologous α-ketoglutarate-decarboxylating enzymes. Biochemistry 2010, 49, 2672–2679. [Google Scholar] [CrossRef]
- Kurosu, M.; Narayanasamy, P.; Biswas, K.; Dhiman, R.; Crick, D.C. Discovery of 1,4-dihydroxy-2-naphthoate [corrected] prenyltransferase inhibitors: New drug leads for multidrug-resistant gram-positive pathogens. J. Med. Chem. 2007, 50, 3973–3975. [Google Scholar] [CrossRef]
- Berube, B.J.; Russell, D.; Castro, L.; Choi, S.; Narayanasamy, P.; Parish, T. Novel mena inhibitors are bactericidal against mycobacterium tuberculosis and synergize with electron transport chain inhibitors. Antimicrob. Agents Chemother. 2019, 63, e02661-18. [Google Scholar] [CrossRef]
- Benkovic, S.J.; Baker, S.J.; Alley, M.R.K.; Woo, Y.-H.; Zhang, Y.-K.; Akama, T.; Mao, W.; Baboval, J.; Rajagopalan, P.T.R.; Wall, M.; et al. Identification of borinic esters as inhibitors of bacterial cell growth and bacterial methyltransferases, CcrM and MenH. J. Med. Chem. 2005, 48, 7468–7476. [Google Scholar] [CrossRef] [PubMed]
- Jirgis, E.N.M.; Bashiri, G.; Bulloch, E.M.M.; Johnston, J.M.; Baker, E.N. Structural views along the mycobacterium tuberculosis MenD reaction pathway illuminate key aspects of thiamin diphosphate-dependent enzyme mechanisms. Structure 2016, 24, 1167–1177. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Zhou, R.; Sharma, I.; Li, X.; Kumar, G.; Swaminathan, S.; Tonge, P.J.; Tan, D.S. Stable analogues of OSB-AMP: Potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone biosynthesis. Chembiochem Eur. J. Chem. Biol. 2012, 13, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, W.; Tamura, T. A Quinoline Antibiotic from Rhodococcus erythropolis JCM 6824. J. Antibiot. (Tokyo) 2008, 61, 680–682. [Google Scholar] [CrossRef]
- Dhiman, R.K.; Pujari, V.; Kincaid, J.M.; Ikeh, M.A.; Parish, T.; Crick, D.C. Characterization of MenA (isoprenyl diphosphate:1,4-dihydroxy-2-naphthoate isoprenyltransferase) from Mycobacterium tuberculosis. PLoS ONE 2019, 14, e0214958. [Google Scholar] [CrossRef]
- Choi, S.; Frandsen, J.; Narayanasamy, P. Novel long-chain compounds with both immunomodulatory and MenA inhibitory activities against Staphylococcus aureus and its biofilm. Sci. Rep. 2017, 7, 40077. [Google Scholar] [CrossRef]
- Choi, S.; Larson, M.A.; Hinrichs, S.H.; Bartling, A.M.; Frandsen, J.; Narayanasamy, P. Discovery of bicyclic inhibitors against menaquinone biosynthesis. Future Med. Chem. 2016, 8, 11–16. [Google Scholar] [CrossRef]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 2013, 8, 313–321. [Google Scholar] [CrossRef]
- Moraski, G.C.; Deboosère, N.; Marshall, K.L.; Weaver, H.A.; Vandeputte, A.; Hastings, C.; Woolhiser, L.; Lenaerts, A.J.; Brodin, P.; Miller, M.J. Intracellular and in vivo evaluation of imidazo[2,1-b]thiazole-5-carboxamide anti-tuberculosis compounds. PLoS ONE 2020, 15, e0227224. [Google Scholar] [CrossRef]
- Tantry, S.J.; Markad, S.D.; Shinde, V.; Bhat, J.; Balakrishnan, G.; Gupta, A.K.; Ambady, A.; Raichurkar, A.; Kedari, C.; Sharma, S.; et al. Discovery of Imidazo[1,2-a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial adenosine triphosphate (ATP) synthesis. J. Med. Chem. 2017, 60, 1379–1399. [Google Scholar] [CrossRef]
- Lu, X.; Williams, Z.; Hards, K.; Tang, J.; Cheung, C.-Y.; Aung, H.L.; Wang, B.; Liu, Z.; Hu, X.; Lenaerts, A.; et al. Pyrazolo[1,5- a]pyridine inhibitor of the respiratory cytochrome bcc complex for the treatment of drug-resistant tuberculosis. ACS Infect. Dis. 2019, 5, 239–249. [Google Scholar] [CrossRef] [PubMed]
- Rybniker, J.; Vocat, A.; Sala, C.; Busso, P.; Pojer, F.; Benjak, A.; Cole, S.T. Lansoprazole is an antituberculous prodrug targeting cytochrome bc1. Nat. Commun. 2015, 6, 7659. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekera, N.S.; Berube, B.J.; Shetye, G.; Chettiar, S.; O’Malley, T.; Manning, A.; Flint, L.; Awasthi, D.; Ioerger, T.R.; Sacchettini, J.; et al. Improved phenoxyalkylbenzimidazoles with activity against Mycobacterium tuberculosis appear to target QcrB. ACS Infect. Dis. 2017, 3, 898–916. [Google Scholar] [CrossRef] [PubMed]
- van der Westhuyzen, R.; Winks, S.; Wilson, C.R.; Boyle, G.A.; Gessner, R.K.; Soares de Melo, C.; Taylor, D.; de Kock, C.; Njoroge, M.; Brunschwig, C.; et al. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: A novel antimycobacterial class targeting mycobacterial respiration. J. Med. Chem. 2015, 58, 9371–9381. [Google Scholar] [CrossRef] [PubMed]
- Pissinate, K.; Villela, A.D.; Rodrigues-Junior, V.; Giacobbo, B.C.; Grams, E.S.; Abbadi, B.L.; Trindade, R.V.; Roesler Nery, L.; Bonan, C.D.; Back, D.F.; et al. 2-(Quinolin-4-yloxy)acetamides are active against drug-susceptible and drug-resistant mycobacterium tuberculosis strains. ACS Med. Chem. Lett. 2016, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Giacobbo, B.C.; Pissinate, K.; Rodrigues-Junior, V.; Villela, A.D.; Grams, E.S.; Abbadi, B.L.; Subtil, F.T.; Sperotto, N.; Trindade, R.V.; Back, D.F.; et al. New insights into the SAR and drug combination synergy of 2-(quinolin-4-yloxy)acetamides against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2017, 126, 491–501. [Google Scholar] [CrossRef]
- Cleghorn, L.A.T.; Ray, P.C.; Odingo, J.; Kumar, A.; Wescott, H.; Korkegian, A.; Masquelin, T.; Lopez Moure, A.; Wilson, C.; Davis, S.; et al. Identification of morpholino thiophenes as novel mycobacterium tuberculosis inhibitors, targeting QcrB. J. Med. Chem. 2018, 61, 6592–6608. [Google Scholar] [CrossRef]
- Harrison, G.A.; Bridwell, A.E.M.; Singh, M.; Jayaraman, K.; Weiss, L.A.; Kinsella, R.L.; Aneke, J.S.; Flentie, K.; Schene, M.E.; Gaggioli, M.; et al. Identification of 4-Amino-Thieno[2,3-d]Pyrimidines as QcrB Inhibitors in Mycobacterium tuberculosis. mSphere 2019, 4, e00606-19. [Google Scholar] [CrossRef]
- Meunier, B.; Madgwick, S.A.; Reil, E.; Oettmeier, W.; Rich, P.R. New inhibitors of the quinol oxidation sites of bacterial cytochromes bo and bd. Biochemistry 1995, 34, 1076–1083. [Google Scholar] [CrossRef]
- Moraski, G.C.; Markley, L.D.; Hipskind, P.A.; Boshoff, H.; Cho, S.; Franzblau, S.G.; Miller, M.J. Advent of Imidazo[1,2-a]pyridine-3-carboxamides with potent multi- and extended drug resistant antituberculosis activity. ACS Med. Chem. Lett. 2011, 2, 466–470. [Google Scholar] [CrossRef]
- Abrahams, K.A.; Cox, J.A.G.; Spivey, V.L.; Loman, N.J.; Pallen, M.J.; Constantinidou, C.; Fernández, R.; Alemparte, C.; Remuiñán, M.J.; Barros, D.; et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 2012, 7, e52951. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.A.; Rao, S.P.S.; Ping Tan, M.; Lin, X.; Chyba, J.; Tay, J.; Ng, S.H.; Tan, B.H.; Cherian, J.; Duraiswamy, J.; et al. A high-throughput screen to identify inhibitors of atp homeostasis in non-replicating mycobacterium tuberculosis. ACS Chem. Biol. 2012, 7, 1190–1197. [Google Scholar] [CrossRef]
- Moraski, G.C.; Markley, L.D.; Cramer, J.; Hipskind, P.A.; Boshoff, H.; Bailey, M.; Alling, T.; Ollinger, J.; Parish, T.; Miller, M.J. Advancement of Imidazo[1,2-a]pyridines with improved pharmacokinetics and nanomolar activity against mycobacterium tuberculosis. ACS Med. Chem. Lett. 2013, 4, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Oliver, A.G.; Markley, L.D.; Cho, S.; Franzblau, S.G.; Miller, M.J. Scaffold-switching: An exploration of 5,6-fused bicyclic heteroaromatics systems to afford antituberculosis activity akin to the imidazo[1,2-a]pyridine-3-carboxylates. Bioorg. Med. Chem. Lett. 2014, 24, 3493–3498. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Kim, R.Y.; Seo, M.J.; Lee, S.; Kim, Y.M.; Seo, M.; Seo, J.J.; Ko, Y.; Choi, I.; Jang, J.; et al. Lead optimization of a novel series of imidazo[1,2-a]pyridine amides leading to a clinical candidate (Q203) as a multi- and extensively-drug-resistant anti-tuberculosis agent. J. Med. Chem. 2014, 57, 5293–5305. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Seeger, N.; Miller, P.A.; Oliver, A.G.; Boshoff, H.I.; Cho, S.; Mulugeta, S.; Anderson, J.R.; Franzblau, S.G.; Miller, M.J. Arrival of Imidazo[2,1-b]thiazole-5-carboxamides: Potent anti-tuberculosis agents that target QcrB. ACS Infect. Dis. 2016, 2, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Telacebec (Q203) | Working Group for New TB Drugs. Available online: https://www.newtbdrugs.org/pipeline/compound/telacebec-q203 (accessed on 3 February 2020).
- Bouvier, G.; Simenel, C.; Jang, J.; Kalia, N.P.; Choi, I.; Nilges, M.; Pethe, K.; Izadi-Pruneyre, N. Target engagement and binding mode of an antituberculosis drug to its bacterial target deciphered in whole living cells by NMR. Biochemistry 2019, 58, 526–533. [Google Scholar] [CrossRef]
- Moosa, A.; Lamprecht, D.A.; Arora, K.; Barry, C.E.; Boshoff, H.I.M.; Ioerger, T.R.; Steyn, A.J.C.; Mizrahi, V.; Warner, D.F. Susceptibility of mycobacterium tuberculosis cytochrome bd oxidase mutants to compounds targeting the terminal respiratory oxidase, cytochrome c. Antimicrob. Agents Chemother. 2017, 61, e01338-17. [Google Scholar] [CrossRef]
- Scherr, N.; Bieri, R.; Thomas, S.S.; Chauffour, A.; Kalia, N.P.; Schneide, P.; Ruf, M.-T.; Lamelas, A.; Manimekalai, M.S.S.; Grüber, G.; et al. Targeting the Mycobacterium ulcerans cytochrome bc1:aa3 for the treatment of Buruli ulcer. Nat. Commun. 2018, 9, 5370. [Google Scholar] [CrossRef]
- Tang, J.; Wang, B.; Wu, T.; Wan, J.; Tu, Z.; Njire, M.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Lu, X.; et al. Design, synthesis, and biological evaluation of Pyrazolo[1,5-a]pyridine-3-carboxamides as novel antitubercular agents. ACS Med. Chem. Lett. 2015, 6, 814–818. [Google Scholar] [CrossRef]
- Lu, X.; Tang, J.; Cui, S.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Zhang, X.; Ding, K. Pyrazolo[1,5-a]pyridine-3-carboxamide hybrids: Design, synthesis and evaluation of anti-tubercular activity. Eur. J. Med. Chem. 2017, 125, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Kalia, N.P.; Jin, X.E.F.; Hasenoehrl, E.J.; Berney, M.; Pethe, K. Inhibitors of energy metabolism interfere with antibiotic-induced death in mycobacteria. J. Biol. Chem. 2019, 294, 1936–1943. [Google Scholar] [CrossRef] [PubMed]
- Mdanda, S.; Baijnath, S.; Shobo, A.; Singh, S.D.; Maguire, G.E.M.; Kruger, H.G.; Arvidsson, P.I.; Naicker, T.; Govender, T. Lansoprazole-sulfide, pharmacokinetics of this promising anti-tuberculous agent. Biomed. Chromatogr. 2017, 31, e4035. [Google Scholar] [CrossRef] [PubMed]
- Yates, T.A.; Tomlinson, L.A.; Bhaskaran, K.; Langan, S.; Thomas, S.; Smeeth, L.; Douglas, I.J. Lansoprazole use and tuberculosis incidence in the United Kingdom Clinical Practice Research Datalink: A population based cohort. PLoS Med. 2017, 14, e1002457. [Google Scholar] [CrossRef]
- Chandrasekera, N.S.; Alling, T.; Bailey, M.A.; Files, M.; Early, J.V.; Ollinger, J.; Ovechkina, Y.; Masquelin, T.; Desai, P.V.; Cramer, J.W.; et al. Identification of phenoxyalkylbenzimidazoles with antitubercular activity. J. Med. Chem. 2015, 58, 7273–7285. [Google Scholar] [CrossRef]
- Pitta, E.; Rogacki, M.K.; Balabon, O.; Huss, S.; Cunningham, F.; Lopez-Roman, E.M.; Joossens, J.; Augustyns, K.; Ballell, L.; Bates, R.H.; et al. Searching for New Leads for Tuberculosis: Design, Synthesis, and Biological Evaluation of Novel 2-Quinolin-4-yloxyacetamides. J. Med. Chem. 2016, 59, 6709–6728. [Google Scholar] [CrossRef]
- Phummarin, N.; Boshoff, H.I.; Tsang, P.S.; Dalton, J.; Wiles, S.; Barry 3rd, C.E.; Copp, B.R. SAR and identification of 2-(quinolin-4-yloxy)acetamides as Mycobacterium tuberculosis cytochrome bc 1 inhibitors. Medchemcomm 2016, 7, 2122–2127. [Google Scholar] [CrossRef]
- Subtil, F.T.; Villela, A.D.; Abbadi, B.L.; Rodrigues-Junior, V.S.; Bizarro, C.V.; Timmers, L.F.S.M.; de Souza, O.N.; Pissinate, K.; Machado, P.; López-Gavín, A.; et al. Activity of 2-(quinolin-4-yloxy)acetamides in Mycobacterium tuberculosis clinical isolates and identification of their molecular target by whole-genome sequencing. Int. J. Antimicrob. Agents 2018, 51, 378–384. [Google Scholar] [CrossRef]
- Berry, E.A.; Guergova-Kuras, M.; Huang, L.S.; Crofts, A.R. Structure and function of cytochrome bc complexes. Annu. Rev. Biochem. 2000, 69, 1005–1075. [Google Scholar] [CrossRef]
- Lamprecht, D.A.; Finin, P.M.; Rahman, M.A.; Cumming, B.M.; Russell, S.L.; Jonnala, S.R.; Adamson, J.H.; Steyn, A.J.C. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 2016, 7, 12393. [Google Scholar] [CrossRef]
- Lu, P.; Heineke, M.H.; Koul, A.; Andries, K.; Cook, G.M.; Lill, H.; van Spanning, R.; Bald, D. The cytochrome bd-type quinol oxidase is important for survival of Mycobacterium smegmatis under peroxide and antibiotic-induced stress. Sci. Rep. 2015, 5, 10333. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Asseri, A.H.; Kremer, M.; Maaskant, J.; Ummels, R.; Lill, H.; Bald, D. The anti-mycobacterial activity of the cytochrome bcc inhibitor Q203 can be enhanced by small-molecule inhibition of cytochrome bd. Sci. Rep. 2018, 8, 2625. [Google Scholar] [CrossRef] [PubMed]
- Flentie, K.; Harrison, G.A.; Tükenmez, H.; Livny, J.; Good, J.A.D.; Sarkar, S.; Zhu, D.X.; Kinsella, R.L.; Weiss, L.A.; Solomon, S.D.; et al. Chemical disarming of isoniazid resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2019, 116, 10510–10517. [Google Scholar] [CrossRef] [PubMed]
- Tran, S.L.; Cook, G.M. The F1Fo-ATP synthase of Mycobacterium smegmatis is essential for growth. J. Bacteriol. 2005, 187, 5023–5028. [Google Scholar] [CrossRef]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- Haagsma, A.C.; Podasca, I.; Koul, A.; Andries, K.; Guillemont, J.; Lill, H.; Bald, D. Probing the interaction of the diarylquinoline TMC207 with its target mycobacterial ATP synthase. PLoS ONE 2011, 6, e23575. [Google Scholar] [CrossRef]
- Nesci, S.; Trombetti, F.; Algieri, C.; Pagliarani, A. A therapeutic role for the F1FO-ATP synthase. SLAS Discov. Adv. Life Sci. R D 2019, 24, 893–903. [Google Scholar] [CrossRef]
- Preiss, L.; Langer, J.D.; Yildiz, Ö.; Eckhardt-Strelau, L.; Guillemont, J.E.G.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef]
- Hards, K.; McMillan, D.G.G.; Schurig-Briccio, L.A.; Gennis, R.B.; Lill, H.; Bald, D.; Cook, G.M. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl. Acad. Sci. USA 2018, 115, 7326–7331. [Google Scholar] [CrossRef]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar] [CrossRef]
- Almeida, D.; Ioerger, T.; Tyagi, S.; Li, S.-Y.; Mdluli, K.; Andries, K.; Grosset, J.; Sacchettini, J.; Nuermberger, E. Mutations in pepQ confer low-level resistance to bedaquiline and clofazimine in mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 4590–4599. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Wang, B.; Hu, M.; Huo, F.; Guo, S.; Jing, W.; Nuermberger, E.; Lu, Y. Primary clofazimine and bedaquiline resistance among isolates from patients with multidrug-resistant tuberculosis. Antimicrob. Agents Chemother. 2017, 61, e00239-17. [Google Scholar] [CrossRef] [PubMed]
- Ghajavand, H.; Kargarpour Kamakoli, M.; Khanipour, S.; Pourazar Dizaji, S.; Masoumi, M.; Rahimi Jamnani, F.; Fateh, A.; Siadat, S.D.; Vaziri, F. High prevalence of bedaquiline resistance in treatment-naive tuberculosis patients and verapamil effectiveness. Antimicrob. Agents Chemother. 2019, 63, e02530-18. [Google Scholar] [CrossRef] [PubMed]
- Zimenkov, D.V.; Nosova, E.Y.; Kulagina, E.V.; Antonova, O.V.; Arslanbaeva, L.R.; Isakova, A.I.; Krylova, L.Y.; Peretokina, I.V.; Makarova, M.V.; Safonova, S.G.; et al. Examination of bedaquiline- and linezolid-resistant Mycobacterium tuberculosis isolates from the Moscow region. J. Antimicrob. Chemother. 2017, 72, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Villellas, C.; Coeck, N.; Meehan, C.J.; Lounis, N.; de Jong, B.; Rigouts, L.; Andries, K. Unexpected high prevalence of resistance-associated Rv0678 variants in MDR-TB patients without documented prior use of clofazimine or bedaquiline. J. Antimicrob. Chemother. 2017, 72, 684–690. [Google Scholar] [PubMed]
- Guglielmetti, L.; Tiberi, S.; Burman, M.; Kunst, H.; Wejse, C.; Togonidze, T.; Bothamley, G.; Lange, C. QT prolongation and cardiac toxicity of new tuberculosis drugs in Europe: A tuberculosis network European trialsgroup (TBnet) study. Eur. Respir. J. 2018, 52, 1800537. [Google Scholar] [CrossRef]
- Tong, A.S.T.; Choi, P.J.; Blaser, A.; Sutherland, H.S.; Tsang, S.K.Y.; Guillemont, J.; Motte, M.; Cooper, C.B.; Andries, K.; Van den Broeck, W.; et al. 6-Cyano analogues of bedaquiline as less lipophilic and potentially safer diarylquinolines for tuberculosis. ACS Med. Chem. Lett. 2017, 8, 1019–1024. [Google Scholar] [CrossRef]
- Choi, P.J.; Sutherland, H.S.; Tong, A.S.T.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Lotlikar, M.U.; Upton, A.M.; Guillemont, J.; Motte, M.; et al. Synthesis and evaluation of analogues of the tuberculosis drug bedaquiline containing heterocyclic B-ring units. Bioorg. Med. Chem. Lett. 2017, 27, 5190–5196. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Conole, D.; Blaser, A.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.U.; Denny, W.A.; et al. Structure-activity relationships for analogs of the tuberculosis drug bedaquiline with the naphthalene unit replaced by bicyclic heterocycles. Bioorg. Med. Chem. 2018, 26, 1797–1809. [Google Scholar] [CrossRef]
- Blaser, A.; Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Conole, D.; Franzblau, S.G.; Cooper, C.B.; Upton, A.M.; Lotlikar, M.; Denny, W.A.; et al. Structure-activity relationships for unit C pyridyl analogues of the tuberculosis drug bedaquiline. Bioorg. Med. Chem. 2019, 27, 1283–1291. [Google Scholar] [CrossRef]
- Sutherland, H.S.; Tong, A.S.T.; Choi, P.J.; Blaser, A.; Conole, D.; Franzblau, S.G.; Lotlikar, M.U.; Cooper, C.B.; Upton, A.M.; Denny, W.A.; et al. 3,5-Dialkoxypyridine analogues of bedaquiline are potent antituberculosis agents with minimal inhibition of the hERG channel. Bioorg. Med. Chem. 2019, 27, 1292–1307. [Google Scholar] [CrossRef]
- Sarathy, J.P.; Ragunathan, P.; Shin, J.; Cooper, C.B.; Upton, A.M.; Grüber, G.; Dick, T. TBAJ-876 retains bedaquiline’s activity against subunits c and ε of mycobacterium tuberculosis F-ATP synthase. Antimicrob. Agents Chemother. 2019, 63, e01191-19. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Mehra, R.; Sharma, S.; Bokolia, N.P.; Raina, D.; Nargotra, A.; Singh, P.P.; Khan, I.A. Screening of antitubercular compound library identifies novel ATP synthase inhibitors of mycobacterium tuberculosis. Tuberc. Edinb. Scotl. 2018, 108, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.; Haagsma, A.C.; Pham, H.; Maaskant, J.J.; Mol, S.; Lill, H.; Bald, D. Pyrazinoic acid decreases the proton motive force, respiratory ATP synthesis activity, and cellular ATP levels. Antimicrob. Agents Chemother. 2011, 55, 5354–5357. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Zhu, W.; Schurig-Briccio, L.A.; Lindert, S.; Shoen, C.; Hitchings, R.; Li, J.; Wang, Y.; Baig, N.; Zhou, T.; et al. Antiinfectives targeting enzymes and the proton motive force. Proc. Natl. Acad. Sci. USA 2015, 112, E7073–E7082. [Google Scholar] [CrossRef] [PubMed]
- Jeon, A.B.; Ackart, D.F.; Li, W.; Jackson, M.; Melander, R.J.; Melander, C.; Abramovitch, R.B.; Chicco, A.J.; Basaraba, R.J.; Obregón-Henao, A. 2-aminoimidazoles collapse mycobacterial proton motive force and block the electron transport chain. Sci. Rep. 2019, 9, 1513. [Google Scholar] [CrossRef] [PubMed]
- Jeon, A.B.; Obregón-Henao, A.; Ackart, D.F.; Podell, B.K.; Belardinelli, J.M.; Jackson, M.; Nguyen, T.V.; Blackledge, M.S.; Melander, R.J.; Melander, C.; et al. 2-aminoimidazoles potentiate ß-lactam antimicrobial activity against Mycobacterium tuberculosis by reducing ß-lactamase secretion and increasing cell envelope permeability. PLoS ONE 2017, 12, e0180925. [Google Scholar] [CrossRef]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [Google Scholar] [CrossRef]
- Zhang, Y.; Wade, M.M.; Scorpio, A.; Zhang, H.; Sun, Z. Mode of action of pyrazinamide: Disruption of Mycobacterium tuberculosis membrane transport and energetics by pyrazinoic acid. J. Antimicrob. Chemother. 2003, 52, 790–795. [Google Scholar] [CrossRef]
- Yang, C.-S.; Yuk, J.-M.; Jo, E.-K. The role of nitric oxide in mycobacterial infections. Immune Netw. 2009, 9, 46–52. [Google Scholar] [CrossRef]
- Manjunatha, U.; Boshoff, H.I.; Barry, C.E. The mechanism of action of PA-824. Commun. Integr. Biol. 2009, 2, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef]
- Van den Bossche, A.; Varet, H.; Sury, A.; Sismeiro, O.; Legendre, R.; Coppee, J.-Y.; Mathys, V.; Ceyssens, P.-J. Transcriptional profiling of a laboratory and clinical Mycobacterium tuberculosis strain suggests respiratory poisoning upon exposure to delamanid. Tuberc. Edinb. Scotl. 2019, 117, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Developing New TB Regimens. Available online: https://www.tballiance.org/rd/developing-new-regimens (accessed on 23 December 2019).
- Berube, B.J.; Parish, T. Combinations of respiratory chain inhibitors have enhanced bactericidal activity against mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e01677-17. [Google Scholar] [CrossRef] [PubMed]
- Moraski, G.C.; Cheng, Y.; Cho, S.; Cramer, J.W.; Godfrey, A.; Masquelin, T.; Franzblau, S.G.; Miller, M.J.; Schorey, J. Imidazo[1,2-a]Pyridine-3-Carboxamides are active antimicrobial agents against mycobacterium avium infection in vivo. Antimicrob. Agents Chemother. 2016, 60, 5018–5022. [Google Scholar] [CrossRef]
- Heinrich, N.; Dawson, R.; du Bois, J.; Narunsky, K.; Horwith, G.; Phipps, A.J.; Nacy, C.A.; Aarnoutse, R.E.; Boeree, M.J.; Gillespie, S.H.; et al. Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 2015, 70, 1558–1566. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhu, H.; Fu, L.; Wang, B.; Guo, S.; Chen, X.; Liu, Z.; Huang, H.; Yang, T.; Lu, Y. Identifying regimens containing TBI-166, a new drug candidate against mycobacterium tuberculosis in vitro and in vivo. Antimicrob. Agents Chemother. 2019, 63, e02496-18. [Google Scholar] [CrossRef]
- Converse, P.J.; Almeida, D.V.; Tyagi, S.; Xu, J.; Nuermberger, E.L. Shortening buruli ulcer treatment with combination therapy targeting the respiratory chain and exploiting mycobacterium ulcerans gene decay. Antimicrob. Agents Chemother. 2019, 63, e00426-19. [Google Scholar] [CrossRef]


| Chemical Class | Represented by | Structure | References |
|---|---|---|---|
| Riminophenazines | Clofazimine |  | [113] |
| TBI-166 |  | [114] | |
| Phenothiazines | Chlorpromazine |  | [99] |
| Thioridazine |  | ||
| Trifluoperazine |  | ||
| Quinolinyl pyrimidines | 13a |  | [106] |
| Thioquinazoline | CBR-1825 |  | [29] |
| Tetrahydroindazole | CBR-4032 |  | [29] |
| 2-mercapto-quinazolinone | 1 |  | [30] |
| Quinolinequinones | QQ8c |  | [107,111] |
| 16b |  | [112] |
| Chemical Class | Represented by | Structure | Ref |
|---|---|---|---|
| MenD inhibitors | |||
| methyl succinylphosphonate | 2 |  | [115] |
| succinylphosphonate | 1 |  | [115] |
| MenE inhibitors | |||
| vinyl sulphonamide MeOSB-AVSN | 3 |  | [50] |
| MenA inhibitors | |||
| benzophenone O-methyl oxime derivatives | (R)-13 |  | [49] |
| 4-bromophenyl)[2-fluoro-4-[[6-(methyl-2-propenylamino)hexyl]oxy]phenyl]-methanone | Ro-48-8071 |  | [46] |
| aminoalkoxydiphenylmethane | CSU-20 |  | [46,116] |
| bicyclic inhibitors | NM-4 |  | [117] |
| MenG inhibitors | |||
| diphenylborinic acid quinoline esters | 4b |  | [118] |
| biphenyl amide | DG70 |  | [51] |
| Chemical Class | Represented by | Structure | Ref |
|---|---|---|---|
| Cytochrome bcc inhibitors | |||
| Imidazopyridines | Q203 (Telacebec) |  | [12] |
| Imidazo[2,1-b]thiazole-5-carboxamide | ND-11543 |  | [126] |
| Imidazopyridine ethers | 19e |  | [127] |
| Pyrazolo[1,5-a]pyridine-3-carboxamide | TB47 |  | [128] |
| Lansoprazole sulphide (LPZS) |  | [129] | |
| Phenoxyalkylbenzimidazole | 54 |  | [130] |
| Pyrrolo[3,4-c]pyridine(2H)-dione | 5h |  | [131] |
| 2-(quinolin-4-yloxy)acetamides | 5s |  | [132] |
| 12 n |  | [133] | |
| Arylvinylpiperazine amides | AX-35 |  | [76] |
| Morpholino thiophenes | 37 |  | [134] |
| 4-Amino-thieno[2,3-d]pyrimidines | CWHM-1023 |  | [135] |
| 2-Ethylthio-4-methylaminoquinazolines | 11726148 |  | [77] |
| Cyt-bd inhibitor | |||
| Aurachin D |  | [136] | |









{kind=link}
{kind=link}
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foo, C.S.-Y.; Pethe, K.; Lupien, A. Oxidative Phosphorylation—an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis. Appl. Sci. 2020, 10, 2339. https://doi.org/10.3390/app10072339
Foo CS-Y, Pethe K, Lupien A. Oxidative Phosphorylation—an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis. Applied Sciences. 2020; 10(7):2339. https://doi.org/10.3390/app10072339
Chicago/Turabian StyleFoo, Caroline Shi-Yan, Kevin Pethe, and Andréanne Lupien. 2020. "Oxidative Phosphorylation—an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis" Applied Sciences 10, no. 7: 2339. https://doi.org/10.3390/app10072339
APA StyleFoo, C. S.-Y., Pethe, K., & Lupien, A. (2020). Oxidative Phosphorylation—an Update on a New, Essential Target Space for Drug Discovery in Mycobacterium tuberculosis. Applied Sciences, 10(7), 2339. https://doi.org/10.3390/app10072339