Ion Pairs of 1-Butyl-3-Methylimidazolium Triflate Do Not Dissociate in Propan-1-ol: A Vibrational Spectroscopic Viewpoint



Abstract
Featured Application
Abstract
{kind=link}
{kind=link}
{kind=link}
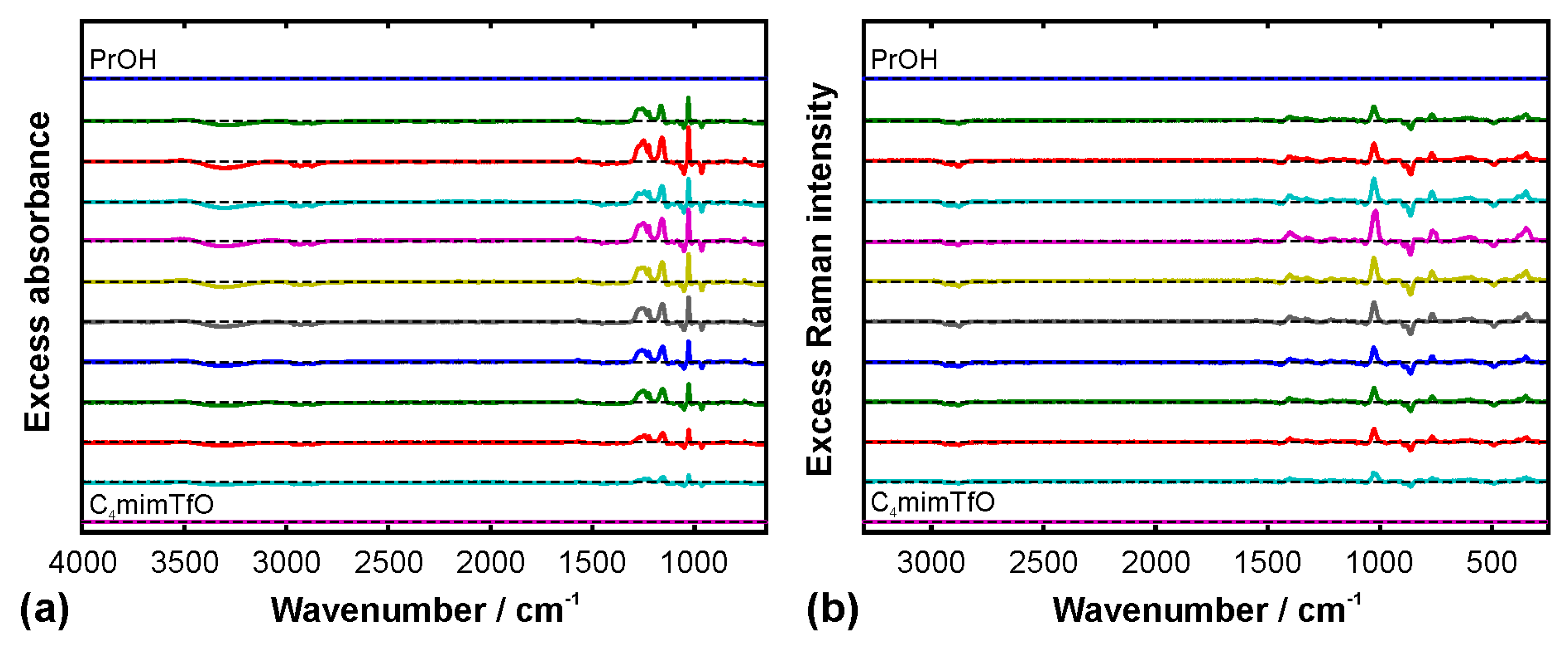{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Raman Spectroscopy
2.3. Infrared Spectroscopy
2.4. Excess Spectra
2.5. Decomposition of Spectra
3. Results
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Turner, E.A.; Pye, C.C.; Singer, R.D. Use of ab initio calculations toward the rational design of room temperature ionic liquids. J. Phys. Chem. A 2003, 107, 2277–2288. [Google Scholar] [CrossRef]
- Katsyuba, S.; Zvereva, E.; Vidis, A.; Dyson, P. Application of density functional theory and vibrational spectroscopy toward the rational design of ionic liquids. J. Phys. Chem. A 2007, 111, 352–370. [Google Scholar] [CrossRef]
- Cammarata, L.; Kazarian, S.G.; Salter, P.A.; Welton, T. Molecular states of water in room temperature ionic liquids. Phys. Chem. Chem. Phys. 2001, 3, 5192–5200. [Google Scholar] [CrossRef]
- Cha, S.; Ao, M.; Sung, W.; Moon, B.; Ahlström, B.; Johansson, P.; Ouchi, Y.; Kim, D. Structures of ionic liquid-water mixtures investigated by IR and NMR spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 9591–9601. [Google Scholar] [CrossRef]
- Jaeger, V.W.; Pfaendtner, J. Structure, dynamics, and activity of xylanase solvated in binary mixtures of ionic liquid and water. ACS Chem. Biol. 2013, 8, 1179–1186. [Google Scholar] [CrossRef]
- Köddermann, T.; Wertz, C.; Heintz, A.; Ludwig, R. The association of water in ionic liquids: A reliable measure of polarity. Angew. Chem. Int. Ed. 2006, 45, 3697–3702. [Google Scholar] [CrossRef]
- Zhang, Q.-G.; Wang, N.-N.; Yu, Z.W. The hydrogen bonding interactions between the ionic liquid 1-ethyl-3-methylimidazolium ethyl sulfate and water. J. Phys. Chem. B 2010, 114, 4747–4754. [Google Scholar] [CrossRef]
- Noack, K.; Leipertz, A.; Kiefer, J. Molecular interactions and macroscopic effects in binary mixtures of an imidazolium ionic liquid with water, methanol, and ethanol. J. Mol. Struct. 2012, 1018, 45–53. [Google Scholar] [CrossRef]
- Chang, H.; Jiang, J.; Liou, Y.; Hung, C.; Lai, T.; Lin, S. Effects of water and methanol on the molecular organization of 1-butyl-3-methylimidazolium tetrafluoroborate as functions of pressure and concentration. J. Chem. Phys. 2008, 129, 04452. [Google Scholar] [CrossRef]
- Vale, V.R.; Will, S.; Schröer, W.; Rathke, B. The general phase behavior of mixtures of 1-alkyl-3-methylimidazolium bis[(trifluoromethyl)sulfonyl]amide ionic liquids with n-alkyl alcohols. ChemPhysChem 2012, 13, 1860–1867. [Google Scholar] [CrossRef]
- Stuckenholz, M.; Crespo, E.A.; Vega, L.F.; Carvalho, P.J.; Coutinho, J.A.P.; Schröer, W.; Kiefer, J.; Rathke, B. Vapor Liquid Equilibria of Binary Mixtures of 1-Butyl-3-methylimidazolium Triflate (C4mimTfO) and Molecular Solvents: N-Alkyl Alcohols and Water. J. Phys. Chem. B 2018, 122, 6017–6032. [Google Scholar] [CrossRef] [PubMed]
- Chouireb, N.; Khan, I.; Crespo, E.A.; Oliveira, M.B.; Llovell, F.; Vega, L.F.; Tafat-Igoudjilene, O.; Kaci, A.A.; Santos, L.M.N.B.F.; Carvalho, P.J.; et al. Evaluation of the Solvent Structural Effect Upon the Vapor-Liquid Equilibrium of [C4C1im][Cl] + Alcohols. Fluid Phase Equilibria 2017, 440, 36–44. [Google Scholar] [CrossRef]
- Verevkin, S.P.; Safarov, J.; Bich, E.; Hassel, E.; Heintz, A. Thermodynamic Properties of Mixtures Containing Ionic Liquids Vapor Pressures and Activity Coefficients of n-Alcohols and Benzene in Binary Mixtures with 1-Methyl-3-butyl-imidazolium Bis(trifluoromethyl-sulfonyl)imide. Fluid Phase Equilibria 2005, 236, 222–228. [Google Scholar] [CrossRef]
- Kiefer, J.; Stuckenholz, M.; Rathke, B. Influence of the alkyl chain on the vibrational structure and interionic interactions in 1-alkyl-3-methylimidazolium trifluoromethanesulfonate ionic liquids. J. Mol. Liquids 2018, 255, 413–418. [Google Scholar] [CrossRef]
- Herstedt, M.; Smirnov, M.; Johansson, P.; Chami, M.; Grondin, J.; Servant, L.; Lassègues, J.C. Spectroscopic characterization of the conformational states of the bis(trifluoromethanesulfonyl)imide anion (TFSI−). J. Raman Spectrosc. 2005, 36, 762–770. [Google Scholar] [CrossRef]
- Zhou, Y.; Gong, S.; Xu, X.; Yu, Z.; Kiefer, J.; Wang, Z. The interactions between polar solvents (methanol, acetonitrile, dimethylsulfoxide) and the ionic liquid 1-ethyl-3-methylimidazolium bis(fluorosulfonyl)imide. J. Mol. Liquids 2020, 299, 112159. [Google Scholar] [CrossRef]
- Zehentbauer, F.M.; Kiefer, J. Molecular solution behaviour of an intermediate biofuel feedstock: Acetone-butanol-ethanol (ABE). ChemPhysChem 2015, 16, 3846–3858. [Google Scholar] [CrossRef]
- Kiefer, J.; Wagenfeld, S.; Kerle, D. Chain length effects on the vibrational structure and molecular interactions in the liquid normal alkyl alcohols. Spectrochim. Acta A 2018, 189, 57–65. [Google Scholar] [CrossRef]
- Singh, D.K.; Rathke, B.; Kiefer, J.; Materny, A. Molecular Structure and Interactions in the Ionic Liquid 1-Ethyl-3-methylimidazolium Trifluoromethanesulfonate. J. Phys. Chem. A 2016, 120, 6274–6286. [Google Scholar] [CrossRef]
- Noack, K.; Schulz, P.S.; Paape, N.; Kiefer, J.; Wasserscheid, P.; Leipertz, A. The role of the C2 position in interionic interactions of imidazolium based ionic liquids: A vibrational and NMR spectroscopic study. Phys. Chem. Chem. Phys. 2010, 12, 14153–14161. [Google Scholar] [CrossRef]
- Michniewicz, N.; Muszyński, A.S.; Wrzeszcz, W.; Czarnecki, M.A.; Golec, B.; Hawranek, J.P.; Mielke, Z. Vibrational spectra of liquid 1-propanol. J. Mol. Struct. 2008, 887, 180–186. [Google Scholar] [CrossRef]
- Plyler, E.K. Infrared spectra of methanol, ethanol, and n-propanol. J. Natl. Bur. Stand. 1952, 48, 281–286. [Google Scholar] [CrossRef]
- Tomsic, M.; Jamnik, A.; Fritz-Popovski, G.; Glatter, O.; Vlcek, L. Structural properties of pure simple alcohols from ethanol, propanol, butanol, pentanol, to hexanol: Comparing Monte Carlo simulations with experimental SAXS data. J. Phys. Chem. B 2007, 111, 1738–1751. [Google Scholar] [CrossRef] [PubMed]
- Ozawa, R.; Hayashi, S.; Saha, S.; Kobayashi, A.; Hamaguchi, H. Rotational isomerism and structure of the 1-butyl-3-methylimidazolium cation in the ionic liquid state. Chem. Lett. 2003, 32, 948–949. [Google Scholar] [CrossRef]
- Garaga, M.N.; Nayeri, M.; Martinelli, A. Effect of the alkyl chain length in 1-alkyl-3-methylimidazolium ionic liquids on inter-molecular interactions and rotational dynamics: A combined vibrational and NMR spectroscopic study. J. Mol. Liquids 2015, 210, 169–177. [Google Scholar] [CrossRef]
- Holomb, R.; Martinelli, A.; Albinsson, I.; Lassègues, J.C.; Johansson, P.; Jacobsson, P. Ionic liquid structure: The conformational isomerism in 1-butyl-3-methyl-imidazolium tetrafluoroborate ([bmim][BF4]). J. Raman Spectrosc. 2008, 39, 793–805. [Google Scholar] [CrossRef]
- Kiefer, J.; Eisen, K. Unsupervised screening of vibrational spectra by principal component analysis for identifying molecular clusters. ChemPhysChem 2018, 19, 795–800. [Google Scholar] [CrossRef]
- Li, Q.; Wang, N.; Zhou, Q.; Sun, S.; Yu, Z. Excess infrared absorption spectroscopy and its applications in the studies of hydrogen bonds in alcohol-containing binary mixtures. Appl. Spectrosc. 2008, 62, 166–170. [Google Scholar] [CrossRef]
- Wang, N.-N.; Zhang, Q.-G.; Wu, F.-G.; Li, Q.-Z.; Yu, Z.-W. Hydrogen bonding interactions between a representative pyridinium-based ionic liquid [BuPy][BF4] and water/dimethyl sulfoxide. J. Phys. Chem. B 2010, 114, 8689–8700. [Google Scholar] [CrossRef]
- Kiefer, J.; Molina Martinez, M.; Noack, K. The peculiar nature of molecular interactions between an imidazolium ionic liquid and acetone. ChemPhysChem 2012, 13, 1213–1220. [Google Scholar] [CrossRef]
- Joseph, J.; Jemmis, E.D. Red-, blue-, or no-shift in hydrogen bonds: A unified explanation. J. Am. Chem. Soc. 2007, 129, 4620–4632. [Google Scholar] [CrossRef] [PubMed]
- Serra, P.B.P.; Rocha, M.A.A.; Rathke, B.; Ruzicka, K.; Fulem, M.; Kiefer, J. Infrared spectroscopy of the symmetric branched isomers of n-heptanol. J. Mol. Liquids 2017, 244, 528–532. [Google Scholar] [CrossRef]
- Wang, H.; Wang, J.; Zhang, S.; Pei, Y.; Zhuo, K. Ionic Association of the Ionic Liquids [C4mim][BF4], [C4mim][PF6], and [Cnmim]Br in Molecular Solvents. ChemPhysChem 2009, 10, 2516–2523. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiefer, J.; Stuckenholz, M.; Rullich, C.C.; Rathke, B. Ion Pairs of 1-Butyl-3-Methylimidazolium Triflate Do Not Dissociate in Propan-1-ol: A Vibrational Spectroscopic Viewpoint. Appl. Sci. 2020, 10, 1620. https://doi.org/10.3390/app10051620
Kiefer J, Stuckenholz M, Rullich CC, Rathke B. Ion Pairs of 1-Butyl-3-Methylimidazolium Triflate Do Not Dissociate in Propan-1-ol: A Vibrational Spectroscopic Viewpoint. Applied Sciences. 2020; 10(5):1620. https://doi.org/10.3390/app10051620
Chicago/Turabian StyleKiefer, Johannes, Marcus Stuckenholz, Claudia Caroline Rullich, and Bernd Rathke. 2020. "Ion Pairs of 1-Butyl-3-Methylimidazolium Triflate Do Not Dissociate in Propan-1-ol: A Vibrational Spectroscopic Viewpoint" Applied Sciences 10, no. 5: 1620. https://doi.org/10.3390/app10051620
APA StyleKiefer, J., Stuckenholz, M., Rullich, C. C., & Rathke, B. (2020). Ion Pairs of 1-Butyl-3-Methylimidazolium Triflate Do Not Dissociate in Propan-1-ol: A Vibrational Spectroscopic Viewpoint. Applied Sciences, 10(5), 1620. https://doi.org/10.3390/app10051620