1. Introduction
The present work addresses the structure of the active site of recombinant [NiFeSe] hydrogenase isolated from
Desulfovibrio vulgaris Hildenborough [
1]. The [NiFeSe] hydrogenase represents, in addition to the [NiFeS] hydrogenase, one of the two subclasses of [NiFe] hydrogenases. Hydrogenases are metalloenzymes with an active site that contains, under a reducing atmosphere, iron, nickel and sulfur as well as diatomic ligands like CN and CO, which catalyze the reversible oxidation of molecular hydrogen,
Unlike [FeFe] hydrogenases, which are irreversibly inactivated after exposure to O
2, [NiFe] hydrogenases can be reductively reactivated after exposure to O
2 [
1]. Due to their potential of producing hydrogen, these enzymes represent a design template for molecular catalysts being applied in electrolyzers, which use electricity to split water into hydrogen and oxygen, as well as in fuel cells [
2]. Nonetheless, as O
2 acts as an oxidant that competes with protons, the hydrogenase’s protein scaffold protects the redox centers from high O
2 concentrations that would inhibit H
2 production at the active site [
2].
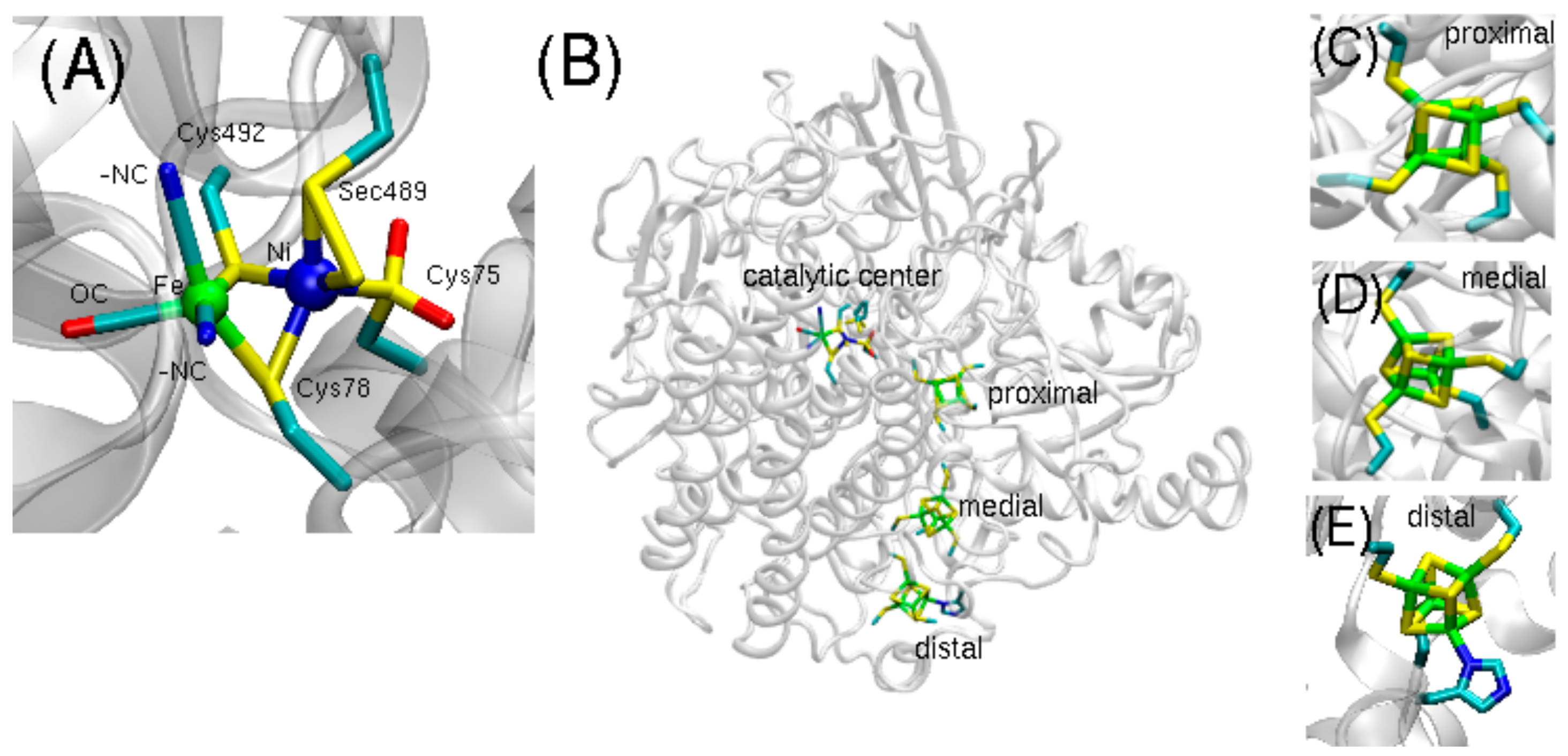
Representing the predominant form, the [NiFeS] hydrogenase contains the catalytic Ni-Fe center, bound by two bridging cysteines and surrounded by two additional cysteine ligands on the Ni site and two CN- and one CO ligands coordinated to the Fe site. The [NiFeSe] hydrogenase is structured analogously, with the exception that a selenocysteine (Sec489) replaces a terminal cysteine (
Figure 1A). Three iron-sulfur clusters (proximal, medial, and distal) form an electron transfer path connecting the catalytic center to the protein surface (
Figure 1B–E).
In general, the activity of selenoproteins is usually higher than that of sulfur-containing protein homologues due to the lower pKa and higher nucleophilicity of the selenol group compared to that of the thiol group [
3,
4]. Selenium-containing cysteine (Se-Cys) is more acidic than sulfur-containing cysteine (S-Cys), and the high nucleophilicity of selenium leads to a higher O
2 tolerance of [NiFeSe] hydrogenase. The insertion of selenocysteine requires intricate biosynthetic machinery and is energetically more costly than cysteine insertion, so [NiFeSe] hydrogenases have evolved in only a few microorganisms, such as in
Desulfovibrio vulgaris and
Methanococcus voltae [
2].
The enzymes belonging to the subfamily of [NiFeSe] hydrogenases are special because they have a fast H
2 production rate [
5,
6,
7]. Compared to [NiFeS] hydrogenases, the Se enzyme records a higher H
2 production activity [
8]. Indeed, if Se is available to the microorganism, a preferred expression of NiFeSe over NiFeS hydrogenase is observed [
8]. Importantly, the recombinant expression system developed by Marques et al. is a step toward engineering large quantities of these valuable enzymes [
1].
An earlier, lower-resolution (2.04 Å) structure of aerobically crystallized [NiFeSe] hydrogenase revealed the oxidized, “as-isolated” active site that lacks an oxide bridging ligand [
9]. More recently, high-resolution (0.95–1.4 Å) crystal structures of [NiFeSe] hydrogenase have revealed three conformers of the active site that differ not only in the arrangement, but also in the number of sulfur atoms (
Figure 2).
In the recent, high-resolution structures, conformers I and II contain a sulfur atom bound between the nickel atom and the selenocysteine unit (
Figure 2A,B), while conformer III does not contain this additional sulfur atom (
Figure 2C). In contrast, the nickel atom of conformer III is only bound to a selenocysteine. Conformer I and II, in turn, differ in their binding geometries of the Ni-S-Se-Cys unit. While in conformer II, the additional sulfur atom is directly inserted between the Ni-Se-Cys bond compared to conformer III, and in conformer I, the nickel atom forms a direct bond to the additional sulfur atom as well as to the selenocysteine unit. In addition, the sulfur and selenium atoms are also directly bound to each other.
The structural and mechanistic details of [NiFeSe] hydrogenase have been investigated using a range of spectroscopic methods including X-ray diffraction (XRD) [
2], Fourier transform infrared spectroscopy (FTIR) [
2,
10], resonance Raman (RR) [
2], electron paramagnetic resonance (EPR) [
1], circular dichroism (CD), UV/Vis [
5], and gas chromatography-mass spectrometry GC-MS [
1]. RR experiments indicate various redox dependent states, giving insight into hydrogen production as well as oxygen tolerance [
2]. The UV/Vis spectrum of the [NiFeSe] hydrogenase is very similar to that of the [NiFe] hydrogenase, showing the characteristic broad absorption centered at 400 nm due to the oxidized FeS centers [
5]. [NiFeSe] hydrogenase is nickel-EPR silent in the oxidized state [
1], indicating that the enzyme is in a Ni(II) state that is contrary to most oxidized [NiFe] hydrogenases, in which nickel is in a paramagnetic Ni(III) spin state. GC experiments [
1] indicate that H
2 production of [NiFeSe] hydrogenases is higher than that of [NiFeS] hydrogenases [
5].
Particularly, FTIR spectroscopy has provided insight into the electronic structure and coordination pattern of the catalytic metal ions of hydrogenases. As the frequencies of the nonproteic ligands (CO, CN-) are highly sensitive to the changes in electronic environment, particularly at the Fe locus, FTIR spectra can help elucidate the electronic states governing the catalytic events. In general, for a given redox state, the frequencies of CO bands of [NiFeSe] are lower than those of equivalent redox states of standard sulfur-Ni-Fe hydrogenases [
11]. This behavior is rationalized by the lower electronegativity of the Se atom relative to the S atom present in standard Ni-Fe hydrogenases; an increased π-donating effect from the
d orbitals of Se to the 2pπ* orbital of the CO ligand is also present [
12]. Also, the FTIR spectra of [NiFeSe] show CN-bands that are comparable in intensity with CO bands, suggesting a high π-mobility in the CN-ligands due to hydrogen bond coordination with atoms of surrounding amino acids [
10].
In addition to a range of experimental studies, computational studies have also attempted to shed light on the structure and chemistry of [NiFe] hydrogenases with FeS active sites [
13,
14,
15]. Hybrid quantum mechanical and molecular mechanical (QM/MM) applications have been particularly invaluable for probing the connection between the electronic structure and function in these complex enzymatic systems [
16,
17,
18]. The combination of QM methodology, which describes the chemically active region involving charge-transfers or bond-formation, and MM methods to treat the bulk protein and solvent environment is a computationally efficient method for describing large biomolecules [
16]. In the case of hydrogenase, this QM/MM approach has been shown to accurately describe experimentally measured vibrational frequencies [
13,
14,
15].
The present work reports on QM/MM structural and vibrational frequency computations to analyze the three proposed [NiFeSe] conformers from the structures of Marques et al. [
1]. For each of the three conformers, both Fe
2+ and Fe
3+ oxidization states have been investigated, providing for a total of six models. For the QM/MM optimized geometries of the six species, the IR spectra have been calculated and the vibrational frequencies for the CO and CN-ligands have been analyzed and compared to the experimental values. In the next section, the preparation of the computational models and the QM/MM methodology will be described, and then they will be followed by a discussion of the QM/MM optimized geometries of each conformer. Finally, computed vibrational frequencies based on the QM/MM optimized geometries will be presented and analyzed.
2. Methods
Three conformers of [NiFeSe] hydrogenase from
D. vulgaris Hildenborough were observed in the crystallographic data from PDB ID: 5JSH based on an oxidized species [
1]. A model was therefore built for each of the three conformers by selecting the coordinates of the active site of each of the three conformers, which will henceforth be referred to as conformer I, II, and III (
Figure 2 and
Figure 3). These conformers differ not only in the number of sulfur atoms and their bonding patterns at the Ni site, but also in the geometry of the selenocysteine.
Hydrogen atoms were modeled using CHARMM with the charmm36 force field [
19,
20]. His82 was protonated at position Nε due to possible interactions with the Fe[CN]
2CO unit. Unless otherwise specified, protonation of amino acid side chains was according to the standard assignment for pH 7. Missing atoms were modeled with CHARMM using the charmm36 force field [
19,
20].
The entire protein, including the catalytic center and three [Fe
4S
4] clusters, and crystal water molecules were then solvated in a water box with sodium chloride ions (0.1 M). Partial charges for the three [Fe
4S
4] clusters were adapted from previous work [
13,
14,
15]. Energy minimizations were performed to optimize the hydrogen bond networks at the protein-solvent interface. In a final step, all protein atoms, crystal water, solvent water, and salt ions within a radius of 40 Å centered on the Ni atom were selected to comprise the complete model (
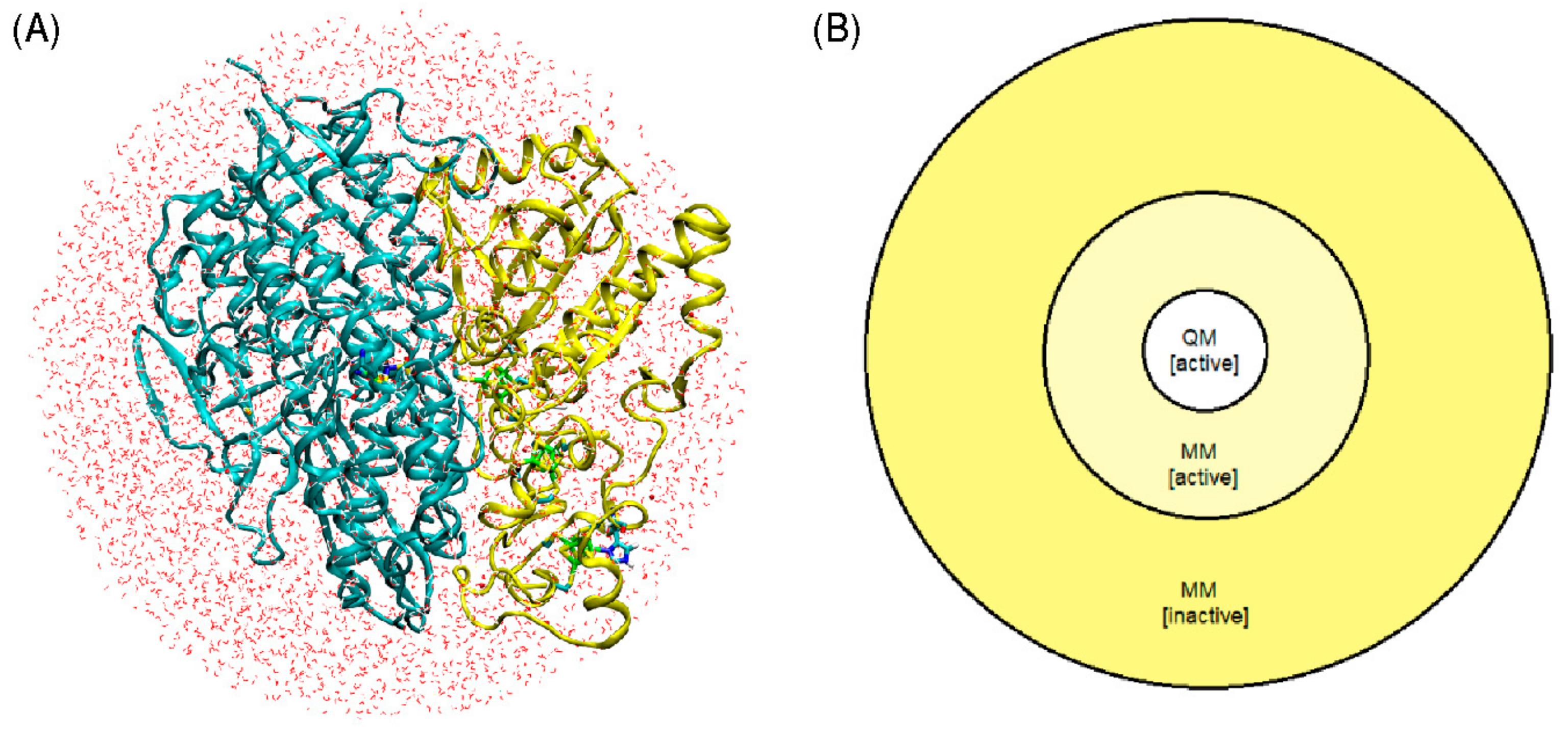
Figure 4A).
The QM/MM geometry optimization of each of the six models was carried out using a model that divides the enzyme into three regions: QM, MM active, and MM inactive (
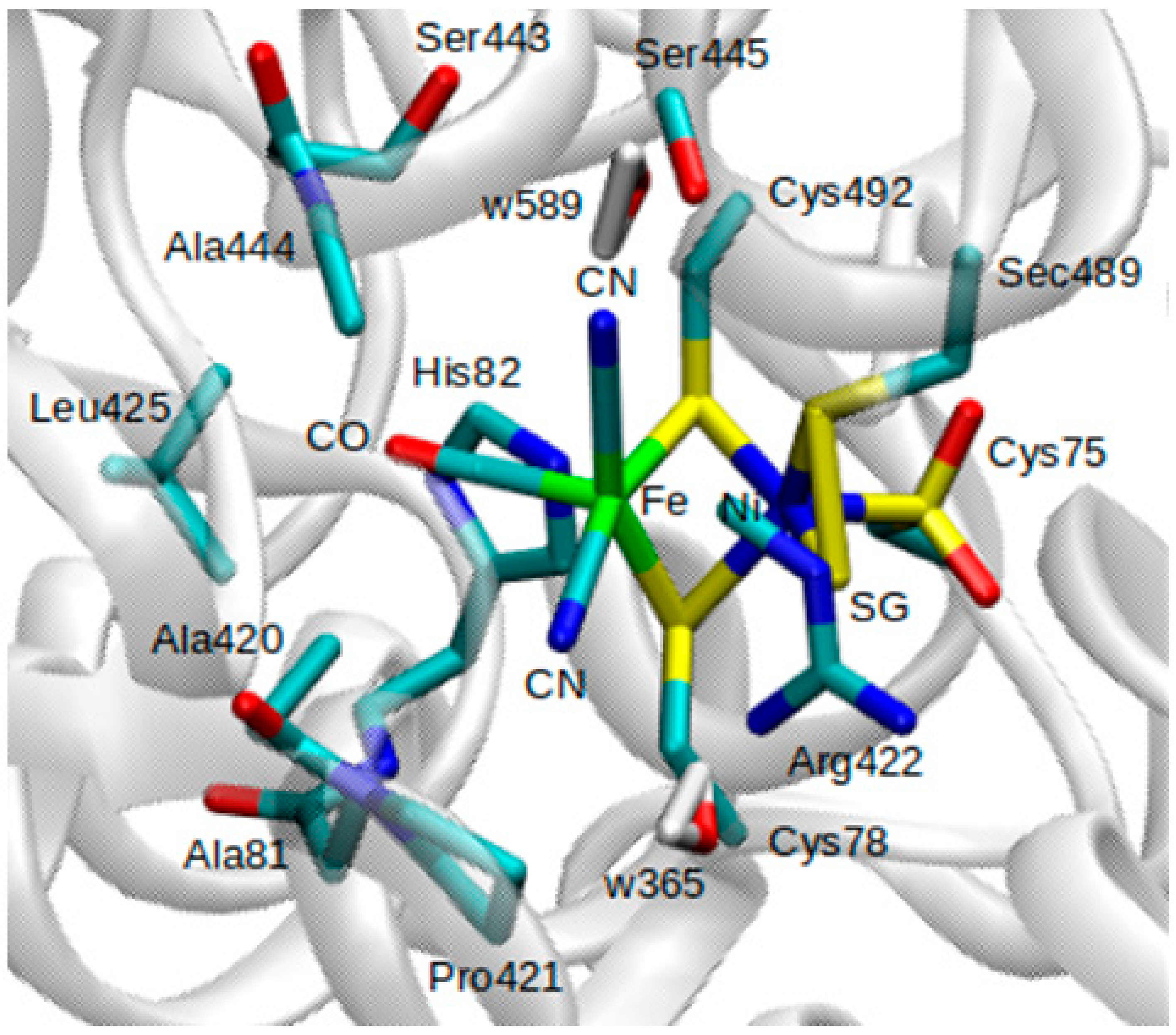
Figure 4B). Only the positions of atoms within the QM and MM active regions were optimized, whereas atoms in the MM inactive region were held fixed. Atoms included in the QM region are all atoms involved in the Fe-Ni catalytic center, as well as atoms belonging to amino acids Ala81, Ala420, Ala444, Arg422, His82, and Leu425.
Pro421, Ser443, and Ser445, two water molecules (w365 and w589) from the crystal structure, were also included in the QM region (
Figure 3). Atoms belonging to protein and crystal water (oxygen) within a 15 Å radius of the Ni atom were considered as part of the MM active region.
Atoms in the QM region were treated with the Density Functional Theory (DFT) and the BP86 functional with the resolution of identity (RI) technique, as implemented in Turbomole6.2 [
21,
22,
23]. A mixed basis set of def2-TZVP (Ni, Fe, S, and Se atoms) and 6-31G* (C, N, O, and H atoms) was used; this combination of basis sets has been tested and shown to be appropriate for geometry optimizations and vibrational frequency analyses on such enzymes [
14].
Based on EPR studies indicating that [NiFeSe] hydrogenase is nickel-EPR silent in the oxidized state [
1], the computations presented here treat Ni in the 2+ oxidation state. Furthermore, a common feature of hydrogenases is the presence in the active site of a low-spin Fe coordinated by CO- and CN-ligands [
24,
25]. Therefore, the electronic states considered in the models are based on [Ni
2+Fe
2+] and [Ni
2+Fe
3+] configurations. For Fe
2+, the total charge of the QM system is −1 for all three conformers and the total spin is S = 0 (multiplicity 1). For Fe
3+, the total charge of the system is 0 for all three conformers, giving a total spin of S = 1/2 and a spin multiplicity of 2.
The energetic coupling between the first and second layers (QM and mobile MM) was treated using electrostatic embedding with a charge-shift scheme as implemented in ChemShell [
26,
27,
28]. Within the MM region, the charmm36 force field was used to describe all bonding and non-bonding interactions [
19]. Any covalent bonds traversing the QM-MM border were cut and hydrogen link-atoms were used to satisfy valency. Vibrational frequency analyses and IR spectra were carried out with Turbomole6.2 [
22,
23] using the normal mode analysis approximation [
29].
3. Results and Discussion
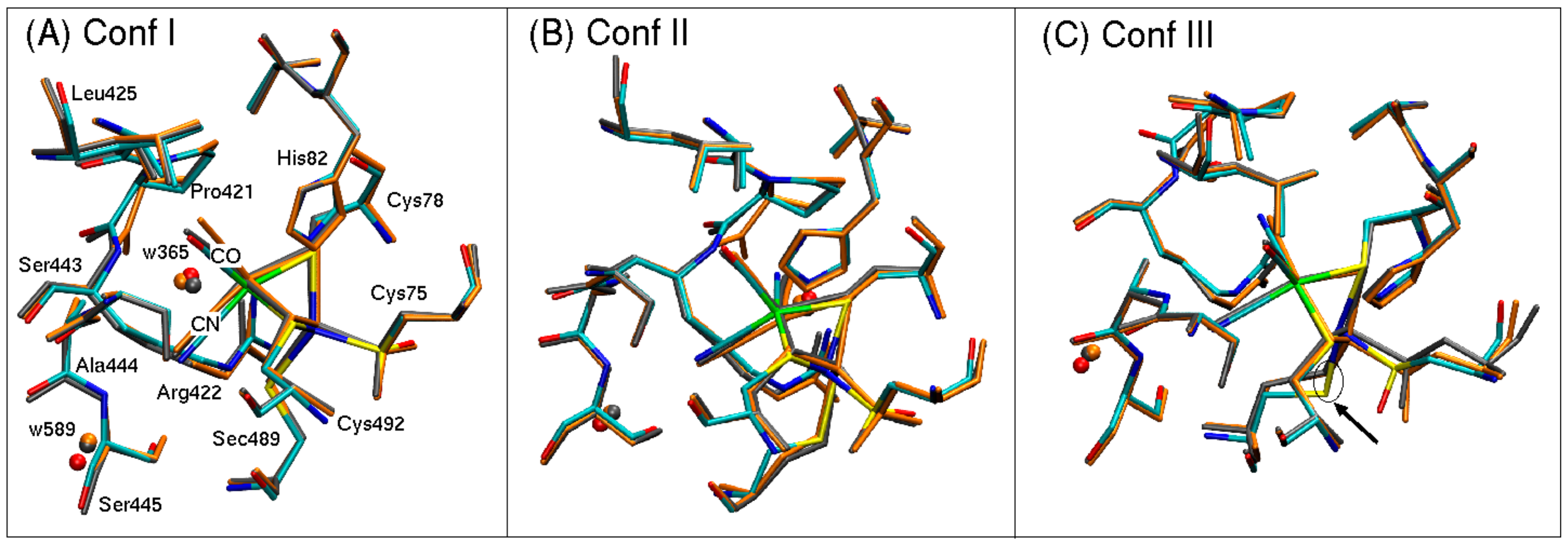
The QM/MM optimized geometries of each conformer are shown in
Figure 5 for both Fe
2+ (gray) and Fe
3+ (orange) superimposed on the oxidized structure determined with X-ray crystallography at 1.3 Å resolution [
1].
In general, the QM/MM optimized geometries reproduce well the structures resolved with X-ray crystallography [
1]. Bond lengths and relative orientations of atoms are well preserved. The positions of oxygen atoms from crystal water w365 and w589 are also preserved in each model, suggesting that the methodology is suited to describe the overall electronic environment. Bond lengths of atoms coordinated to a nickel or iron atom are reported in
Table 1 and
Table 2 for [Ni
2+Fe
2+Se] and [Ni
2+Fe
3+Se], respectively; for comparison, the bond lengths obtained from the crystal structure (PDB ID: 5JSH from Marques et al. [
1]) are listed in bold print. Ni-S lengths for the coordinated cysteine thiolates are well reproduced, but Fe-Ni distances in all three conformers are overestimated by 0.1–0.2 Å compared to the crystal structure. Particularly, conformer III demonstrates an overestimation in the Fe-Ni bond length for both Fe
2+ and Fe
3+ oxidation states (2.61 Å and 2.76 Å, respectively, versus the experimental value of 2.41 Å). This Fe-Ni separation is accompanied by a rotation of the side chain of Sec489, which leads to a 1 Å shift in the position of the selenium atom (see arrow pointing to Sec489 in
Figure 5C). This overestimation in bond lengths is also observed for Ni-S and Ni-Se bonds for conformers I and II. For the oxidized species of [Ni
2+Fe
3+Se], reasonable agreement is observed between the computed Fe-Ni bond length of conformer II (2.45 Å) and the experimentally reported value of 2.41 Å (
Table 2). Conformers I and III overestimate the Fe-Ni distance by more than 0.1 Å and 0.34 Å, respectively, compared to the experimentally measured values [
1].
The discrepancies between the computed and measured bond lengths suggest that either the assumed electronic state of the catalytic site may not accurately reflect the state what is captured in the crystal structure or that the assignment of atomic positions may be ambiguous due to insufficient experimental resolution. Here, we have treated the electronic state of nickel as 2+ since [NiFeSe] hydrogenases are known to be EPR silent, in contrast to standard Ni-Fe hydrogenases [
8]. Another factor that may lead to discrepancy between computed and experimental geometries may be the existence of a bridging ligand (such as a hydride) that may not be resolved from electron density.
The bond lengths between nonproteic ligands (CO, CN1, and CN2) coordinated to Fe show reasonable agreement (0.05–0.14 Å). In our models, we have adopted the assignment of positions from the crystal structure of Marques et al. [
1], namely CO pointing toward Leu425 and CN-ligands pointing toward Pro421 and Ser445. Nonetheless, the experimental resolution (1.3 Å) does not allow for unambiguous assignment of ligand position. In other words, if the CO ligand were exchanged with either of the CN-ligands, the electronic structure of the hydrogen bonding network to neighboring amino acid side chains and crystal water molecules would change (see
Figure 3 for up-close view of active site). These alternate two scenarios have not been tested yet but would be worthwhile extensions of the current investigation.
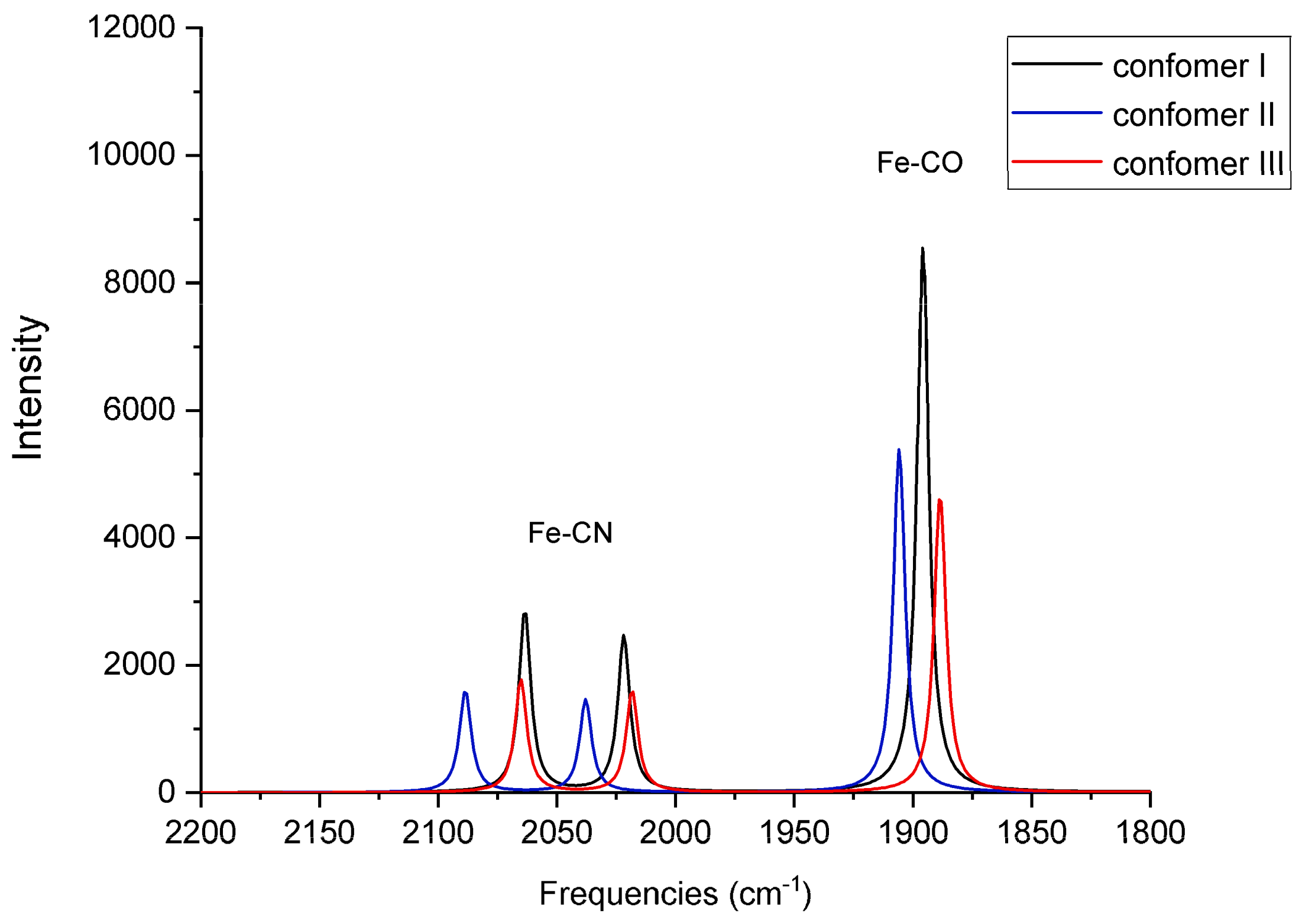
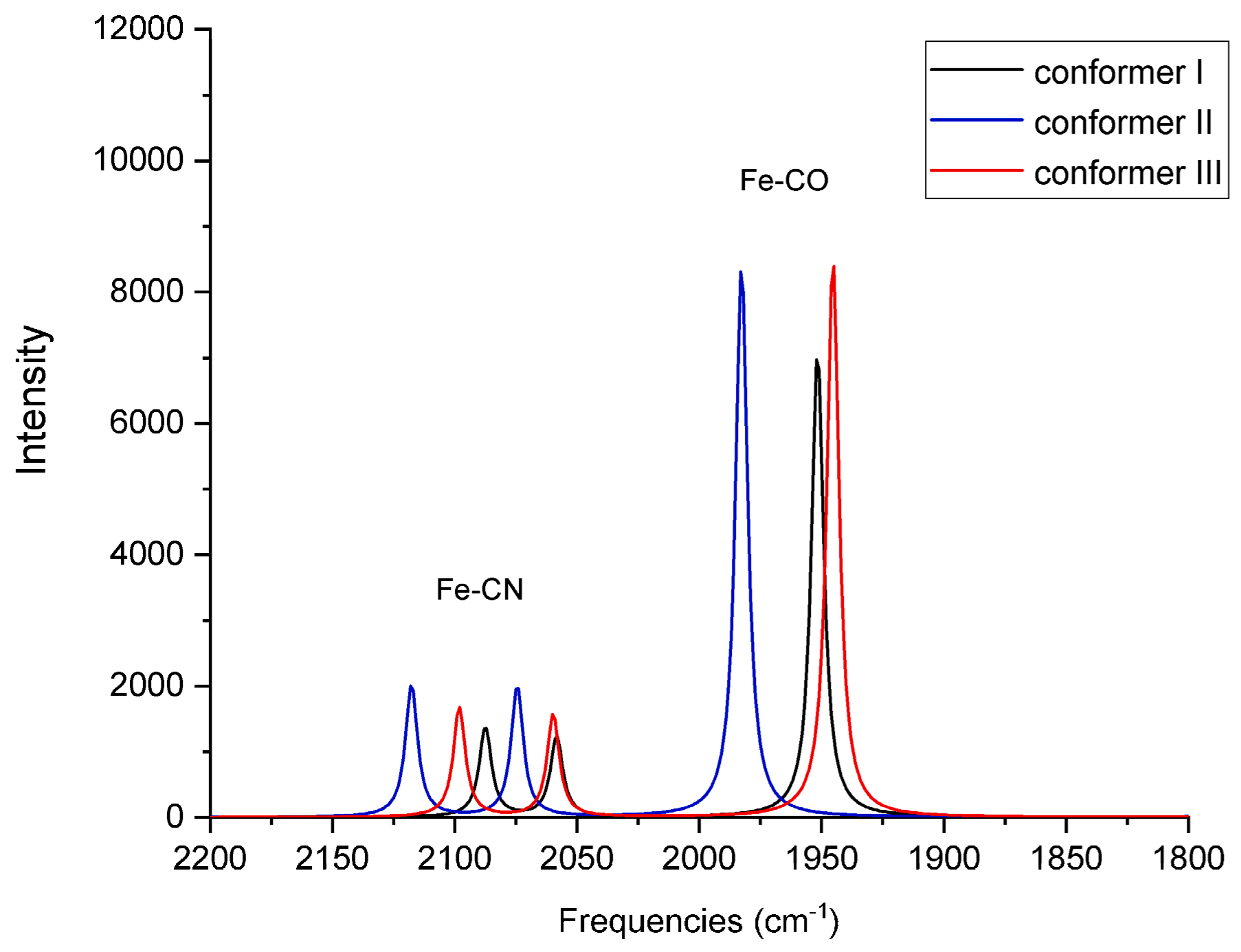
For each of the six QM/MM optimized structures, vibrational frequencies and IR intensities were computed; the results for Fe
2+ and Fe
3+ are listed in
Table 3, respectively, and the spectra are shown in
Figure 6 and
Figure 7, respectively. The results are compared with the FTIR spectra obtained from the oxidized form of native, wild-type (WT) [NiFeSe] hydrogenase as-isolated from
D. vulgaris [
10]. In their experiment, DeLacey et al. observed two sets of CO bands for different batches of purified enzymes [
10]. These two bands were assigned to two different conformations, or isomers (IS), of the active site and termed Ni-IS
I and Ni-IS
II; Ni-IS
I is the conformer that shows a more intense CO band at 1904 cm
−1 [
10]. For sake of reference, the frequencies obtained from the [NiFeSe] species from the recombinant enzyme (r[NiFeSe]) of Marques et al., as-isolated (as is) and reduced (red) with H
2, are listed as well. An important note: the experimental values of DeLacey et al. listed in
Table 3 were obtained from experiments that are not based on the same crystallized protein that is the basis of our computational study [
1]. Nonetheless, as these experimental frequencies are for the native WT [NiFeSe] hydrogenase isolated from
D. vulgaris under non-reducing conditions, we will refer to them as a reference with which to compare our calculated frequencies based on the oxidized, recombinant enzyme in Ref. [
1].
Comparison with the frequencies measured from an oxidized species isolated from the native, WT enzyme indicates that the computed frequencies are reasonable and are representative of the QM/MM optimized structures. Conformer II in the Fe2+ species demonstrates CO and symmetric CN-stretching frequencies that are in agreement with those of ISI (compare 1906 and 2089 cm−1 with 1904 and 2085 cm−1, respectively). This agreement suggests the assumed Fe2+ oxidation state may be valid for the species that were measured in the experiments. Compared to the experimental antisymmetric CN-stretching frequencies for as-isolated (~2076–2079 cm−1) or reduced (~2059–2063 cm−1) species, the calculated antisymmetric CN-stretching frequencies for the Fe2+ species in all three conformers are red-shifted (2022 cm−1, 2038 cm−1, and 2018 cm−1). Nonetheless, the antisymmetric CN-stretching frequency for conformer II (2038 cm−1) is closest to the experimental values.
The discrepancy between computed and measured CN-antisymmetric stretching frequencies indicates that, although conformer II from Marques et al. may be present in the mixture of the oxidized species of the WT native enzyme measured in the experiment of DeLacey et al. [
10], the antisymmetric stretching vibration may be sensitive to the protein environment in the model. A similar systematic red-shift in the computed CN-antisymmetric stretching frequency has been observed for membrane-bound [NiFe] hydrogenase from
Ralstonia eutropha [
13].
Based on the intense band that they observe at 1940 cm
−1, Marques et al. believe their as-isolated sample is more than 80% in the Ni-IS
II state [
1]; the computed vibrational frequencies for conformers I and III for Fe
3+ (1952 cm
−1 and 1945 cm
−1, respectively) are in best agreement with this frequency. Interestingly, based on electron density occupancy fitting, Marques et al. believe conformer I is dominant (51%), followed by conformer III (29%) and conformer II (20%) [
1].
The isolated species of DeLacey et al. are believed to be diamagnetic Ni
2+ states with one, or more than one, oxygen species in the active site [
10]. An interesting suggestion has been made that the side chains of either cysteine or selenocysteine residues can be oxidized to sulfenates; this phenomenon has been used to explain data from X-ray absorption experiments [
27]. In their experiments involving the Ni-Fe cofactor of the NAD-reducing soluble hydrogenase, Burgdorf et al. attribute the long measured Ni-S distance (3.6 Å) to thiol groups that may be in an oxidized form (Cys-SOH) [
30]. Similar chemical modifications of Cys residues have been detected in a range of proteins [
31]. DeLacey et al. also speculate that oxidation of their Ni-IS species leads to a Ni-OX species in which selenocysteine or cysteine ligands are oxidized [
10].
In the case of Fe
3+ (
Table 3 and
Figure 7), the computed CO and CN-symmetric stretching frequencies for conformers I (1952 cm
−1 and 2087 cm
−1) and III (1945 cm
−1 and 2098 cm
−1) agree well with the frequencies measured for both WT (1939 cm
−1 and 2094 cm
−1) and recombinant enzymes (1940 cm
−1, 2085 cm
−1, and 2094 cm
−1), respectively. The computed CN-antisymmetric stretching frequencies for conformers I and III (2058 cm
−1 and 2060 cm
−1, respectively) are red-shifted compared to the experimental frequencies, behavior that was also observed in the case of Fe
2+.
4. Conclusions and Outlook
Here we report for the first time a QM/MM computational investigation of the active site geometry of the [NiFeSe] hydrogenase based on the available structural data from X-ray crystallographic data collected at 1.3 Å resolution [
1]. The IR vibrational frequencies of CN- and CO ligands coordinated to the Fe ion have been computed using a normal-mode analysis based on the QM/MM optimized geometries. Overall, the computed QM/MM optimized geometries are in good agreement with the experimentally resolved geometries, such that the computed vibrational frequencies may be assumed to reflect accurately the underlying structures. In comparison with experimental FTIR frequencies, the computed frequencies suggest that a mixture of structural isomers, as well as Fe oxidation states, may be present in a given protein sample, thus leading to the pattern of CO and CN-vibrational stretching bands observed. Based on the CO and CN-symmetric stretching frequencies, the Ni-IS
I state can be assigned to conformer II in the case of Fe
2+, while the Ni-IS
II state shows best agreement with conformers I and III in the case of Fe
3+. This pattern suggests that iron oxidation may be favored in conformers I and III. Independent of the iron oxidation state, conformers I and III show similar vibrational behavior, an interesting correlation that requires additional investigation. A systematic red-shift of 20–40 cm
−1 in the antisymmetric stretching frequencies for both Fe
2+ and Fe
3+ is observed.
Here, the electronic state of nickel has been treated as 2+ as the oxidized states of [NiFeSe] hydrogenases are known to be EPR silent. Nonetheless, a worthwhile computational investigation would be a comparison of the optimized geometries considering additional ligand oxidization states, such as cysteines or selenocysteines oxidized to sulfenates. In addition, modeling bridging ligands at the Fe-Ni locus would provide some insight into the enzyme’s catalytic mechanisms. Furthermore, as the assignment of CN- and CO ligands in the crystal structure may not necessarily be accurate, the present calculations should be extended to test for alternate assignments. Also, in the structure of Marques et al., the total Ni occupancy of the catalytic site could only be refined to 85%, while a minor Ni site could not be clearly resolved. Therefore, future models may also wish to address variable Ni positions.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}