Featured Application
The potential-controlled sorption of charged molecules using multi-walled carbon nanotubes as electrodes is an innovative step to economizing chromatographic processes. However, any process design requires understanding the basic operators, such as material properties and effects triggered by a potential at the solid-liquid interface, to define the framework for future applications.
Abstract
Triggering the interaction of nanomaterials with molecules by means of electrical potentials in aqueous media remains challenging, especially if 3D through-flow systems are used as electrodes, as in potential-controlled liquid chromatography (PCC). In this paper, multi-walled carbon nanotubes (MWCNTs) function as a particulate packed bed electrode in order to study the system’s response to various applied potentials and electrolyte compositions. The process principle was analyzed using chronoamperometry and cyclic voltammetry. Applying an electrical potential to the hydrophilic MWCNTs induces the presence of both capacitive and faradaic currents. This leads, over time, to a degradation of the electrode due to structural changes of the MWCNT matrix and an increase in redox reactions on the surface. The role of the electrochemical double layer (EDL) is highlighted as a main player in the process, directly influencing the adsorption capability of the electrode. The EDL rearrangement time and coverage radius depend on the composition of the mobile phase and on the potential applied. The capacity of the electrode for the target (maleic acid) increases at high positive potentials (+800 mV vs. Ag/AgCl), while the presence of electrolytes leads to a capacity decrease. Our research enhances the understanding of capacitive through-flow cells.
1. Introduction
Chromatography, and ion exchange chromatography in particular, is a widely used separation and purification technique at the biotechnological industrial scale. However, the process consumes a large amount of salt for elution and requires specifically functionalized resins. An alternative which circumvents these disadvantages is potential-controlled chromatography (PCC) or electrochemically modulated liquid chromatography (EMLC), both of which have been known to scientists for several decades [1,2,3,4,5,6,7,8]. Here, the surface charge of a conductive resin is modified by an applied electrical potential. Consequently, a selective interaction between charged or polarized molecules and the charged particulate packed bed surface takes place because of electrosorption. In a subsequent step, the adsorbed molecules are forced to desorb by a change in the potential direction without any further additives. With this advantage, PCC is a promising and flexible technique, which is inexpensive and seems suitable for all types of charged or polarized molecules.
Initially addressed in the second half of the 20th century by researchers like Okazaki [8] and Fujinaga et al. [2], the process concept of EMLC was developed as an analytical methodology, used to separate metal ions through the influence of electric potential gradients. Other researchers followed the concept and investigated the separation of inorganic [9,10,11] and organic compounds [12,13,14]. The Porter group [15,16,17,18,19,20,21], however, carried out a major part of the process’s development, studying the retention time of different molecules combined with several resin materials [22,23] and mobile phases [24,25]. Knizia et al. [26] and Kocak et al. [27] pushed the development of PCC further by extending the process from the analytical to the preparative scale. They employed a packed bed electrode of spherical, non-functionalized glassy carbon particles which had a relatively small specific surface area (<10 m2/g) [26,27]. Recently, Brammen et al. [28] implemented multi-walled carbon nanotubes (MWCNTs; Baytubes C150P) as an innovative packed bed electrode material with high specific surface area (≈200 m2/g).
Indeed, MWCNTs have already been introduced as a resin for typical gas or liquid chromatography applications [29,30,31,32,33], and as electrodes material in batteries and in sensor science [34,35,36,37]. Nevertheless, the nanotubes agglomerate in complex morphology, which depends on the setup geometry, the mobile phase and numerous external parameters. Hence, the interactions of this carbon material with liquid media are still hotly debated in the literature and are not yet fully understood. Furthermore, predicting the adsorption behavior on MWCNTs under potential is challenging, and the effects inside the MWCNT electrode matrix are largely unknown. Even if the innovative material is already broadly applied [38,39,40,41,42] and has decisive advantages compared to other conductive stationary phases, its use for PCC is still in its infancy. Descriptions of the behavior of the particulate MWCNT matrix under the influence of an electrical potential are required and multiscale concepts for analyzing the relation between electrical current, mobile phase and column capacity are essential for developing a process convertible to the technical scale [43,44]. To fill this gap, we present here a characterization of the particulate MWCNT electrode in an aqueous system. We describe the interaction of the electrode with charged molecules and the process performance under electrical potential. Based on the work of Brammen et al. [28], and due to the fact that Baytubes C150P (Bayer Materials Science AG, Germany) are no longer commercially available, we establish a new type of non-functionalized, low-cost and easily accessible MWCNT (CNT-K, Future Carbon GmbH, Germany) as a resin and closely examine several processing features. Accordingly, the specific material properties of the CNT-K are analyzed, and the particulate electrode is described using a combination of chromatographic and electrochemical methods. In this context, chronoamperometry and cyclic voltammetry are introduced as characterization methods, to detect a correlation between electrical potential, electrical current and adsorption capacity, as well as between the current, electrode structure and the ionic strength of the mobile phase.
2. Materials and Methods
The experimental work is divided into four sections, beginning with a material characterization, and followed by the introduction of electrochemistry and then static and dynamic chromatography techniques.
2.1. Materials and Instrumentation
The work presented here was carried out entirely in a self-designed PCC column based on the example of Brammen et al. [28]. As stationary phase material, MWCNTs (CNT-K, Future Carbon GmbH, 95448 Bayreuth, Germany) with an outer diameter of 20–40 nm and a density of 2.1 g/cm3 were packed in a curled, strong cation exchange membrane (CMI-7000, Membranes International Inc., Ringwood, NJ 07456, USA). Before the column was packed with the particles, the MWCNTs were treated with 1 M HCl at 80 °C for more than 4 h to reduce the catalystic residues and other contaminants. Afterwards, they were neutralized in DI-water and stored as a MWCNT-DI-water suspension. As mobile phases, either pure DI-water with a conductivity below 0.02 mS/cm (pH = 6.2) or 10 mM phosphate buffer (pH = 7.2, 1.7 mS/cm) were used. The buffer was prepared with sodium dihydrogen phosphate, (absolute, Ph. Eur. AppliChem GmbH, 64291 Darmstadt, Germany) and di-sodium hydrogen phosphate (≥98%, Ph. Eur., water free, Carl Roth GmbH & Co. KG, 76185 Karlsruhe, Germany). When necessary, the pH value was regulated with hydrochloric acid (37%, VWR International GmbH, 64295 Darmstadt, Germany) and sodium hydroxide (≥99% p.a., ISO, Carl Roth GmbH & Co. KG, 76185 Karlsruhe, Germany). Maleic acid (absolute, Ph. Eur, AppliChem GmbH, 64291 Darmstadt, Germany) was used as model adsorbate for potential-step experiments; sodium chloride (≥98%, Ph. Eur., Carl Roth GmbH & Co. KG, 76185 Karlsruhe, Germany) was used as tracer to quantify the homogenity of the packed bed. All PCC experiments were performed with an ÄktaTM pure system (ÄktaTM pure 25 M, GE Healthcare GmbH, 79111 Freiburg, Germany). As analytical tools, UV–Vis spectroscopy was used for molecule detection at 216 nm, and a conductivity sensor and a pH sensor were applied at the column outlet. Furthermore, we used a Gamry G750 potentiostat (Gamry Instruments, Warminster, PA 18974, USA) connected to the column for electrochemical application and evaluation. For static binding experiments, an Agilent HPLC system (Agilent 1100 Series HPLC Value System, Agilent Technologies, Inc., Santa Clara, CA 95051, USA) was employed, including a UV–Vis spectrophometer at 216 and 280 nm. Adsorbates for the static binding experiments were L-alanine (Ala, Serva Electrophoresis GmbH, 69115 Heidelberg, Germany), L-histidine (His, ≥99% Cellpure, Carl Roth GmbH & Co. KG, 76185 Karlsruhe, Germany), L-phenylalanine (Phe, Merck KGaA, 64293 Darmstadt, Germany), L-tryptophan (Trp, Serva Electrophoresis GmbH, 69115 Heidelberg, Germany), maleic acid (absolute, Ph. Eur, AppliChem GmbH, 64291 Darmstadt, Germany) and fumaric acid (FA, AppliChem GmbH, 64291 Darmstadt, Germany). The specific surface area was determined using the BET method with nitrogen in a Gemini VII system (Micromeritics Instrument Corp., Norcross, GA 30093-2901, USA), while the particle agglomerate size distribution was measured in suspension with a Microtrac SYNC (Microtrac Retsch GmbH, 42781 Haan, Germany). Transmission electron spectroscopy (TEM; JEM 100-CX, JEOL GmbH, 85356 Freising, Germany), scanning electron microscopy (SEM; CamScan 4, Oxford Instruments/ JSM 5900 LV, JEOL GmbH, 85356 Freising, Germany) and X-ray photoelectron spectroscopy measurements (XPS; LHS 10 XPS with MgKα X-ray source (hυ = 1253.6 eV), Leybold Heraeus GmbH, 50968 Köln, Germany) were carried out additionally.
2.2. Electrochemical Experiments
All electrochemically-based experiments were performed in the PCC column with a 3-electrode setup. The reference electrode was a screwable Ag/AgCl electrode (RE-3VT, C3 Prozess- und Analysentechnik GmbH, 85540 Haar, Germany). The volume flow was 1 mL/min in all experiments. The aqueous media applied were DI-water or 10 mM phosphate buffer. The Gamry G750 potentiostat was operated in surface mode. Open circuit measurements were taken for up to 300 s; chronoamperometry experiments for up to 360 s in ±100 mV steps; and cyclic voltammetry was conducted three times at rates of 0.1 and 1.0 mV/s. The operation range was set between −800 mV to +800 mV to prevent water decomposition, which can occur in the column at higher electrical potentials (positive or negative).
2.3. Static Binding Experiments
Static binding capacities (SBC) were determined outside the column and without external potential application at room temperature, 24 h, 500 rpm. Therefore, 2.0 g of wet MWCNT mud was incubated with 10 mL adsorbate solution and stored in 15 mL roll edge glasses. Besides, 0.3 g to 0.4 g of wet MWCNT mud was incubated with 0.7 mL of an adsorbate solution, and stored in a 96 well plate. The CNT mud used was generated by centrifuge dispersed MWCNT in 50 mL falcon at 3200× g for 3 min; the DI-water supernatant was rejected afterwards. The adsorbate was dissolved in DI-water at different concentrations, amino acids at their pI value and carbon acids at neutral pH. The supernatant concentration was measured three times, each taken from two to three technical replicates.
2.4. Chromatographic Experiments
All chromatographic experiments were performed under flow in the PCC column, both with and without applied potential. DI-water and 10 mM phosphate buffer (pH = 7.2) were used as mobile phases with a flow rate of 1 mL/min. The potential-step experiments were executed with 50 μL of neutralized 2 g/L or 5 g/L maleic acid dissolved in DI-water, which was injected at a selected applied potential (adsorption phase) and eluted at −800 mV (elution/desorption phase). The potential switch was induced after 30 mL or 40 mL. To determine the packed bed asymmetry and the HETP value, tracer experiments were conducted without an applied potential using 1 M NaCl (50 μL, 1 mL/min). The results presented were taken from four different but identically prepared columns, which were packed with 0.5 g of MWCNT powder (purified and suspended in DI-water first) under a DI-water flow of 1 mL/min.
3. Results and Discussion
Even though the PCC concept has been verified several times in the literature (see Section 1), essential questions remain regarding the system response due to the application of the potential and due to the effects of the species in solution. Furthermore, there is little literature on aqueous systems. When nano powders are used as a resin, their physical properties are especially challenging; thus, understanding the process is clearly of great importance. For that reason, the column description is introduced first, as this is a key element of the process. A detailed characterization of the CNT-K material follows, which is necessary because the properties of the MWCNT depend on the manufacturing process and affect the process performance. Afterwards, the electrochemical analysis of the column response under flow is presented to evaluate the aging of the packed bed using chronoamperometry and cyclic voltammetry. The resultant relation to the EDL rearrangement forms the basis for the discussion of the relationship between potential-dependent binding capacity and the ion concentration of the mobile phase. The description of the PCC operating principle can be found in the Supplementary Material, Figure S1.
3.1. The Setup
Brammen et al. [28] developed a three-electrode column setup, with a bed volume of approximately 10 mL, and used Baytubes C150P carbon nanotubes as a stationary phase material. They confirmed that these MWCNTs, with excellent electrical, chemical and mechanical properties, are suitable and offer decisive advantages for PCC. In this work, the process fundamentals are investigated using the readily available and low-cost MWCNT material CNT-K. Only 0.5 g of CNT-K is required to fill one column (material cost about 50 ct). However, a significant challenge of working with a nanopowder is to obtain a homogeneous packed bed as the basis for reproducible and consistent experiments. Therefore, an improvement of Brammen’s column design was necessary and the first task to solve here (see Figure S2). The stamp was then installed in self-printed column clamps (rapid manufacturing), leading to a homogeneous force transmission and facilitating the particulate electrode packing process. The packed bed electrode could then withstand more than 20 bar and be used for at least 320 working hours. The twisted contact wire was improved in a computational fluid dynamic simulation study (radius, number of twists, distance between them). In the end, the optimized column setup together with the improved wire should ensure that the entire particulate electrode is uniformly charged, by a radial potential distribution. Thus, the particles accordingly act simultaneously as a working electrode and as a stationary phase material.
3.2. Specific Properties of the Multi-Walled CNT-K
MWCNTs are a very versatile graphene-based material with sp2 character, forming single-walled or multi-walled tubes of different conformations and with a distinct delocalized π-electron system. Depending on how they are manufactured (e.g., a chemical vapor deposition with an iron-based catalyst), the resulting particles will have different properties, generally of high complexity. Therefore, it is important to characterize the nanotubes used here to be able to understand their role in the potential-controlled process and to identify changes in the MWCNT matrix due to environmental and electrical impacts. The morphological description of the CNT-K (Future Carbon GmbH, Germany) used here is provided on different scales (see Figure 1) and shows the altered configuration of the particle network in solution: CNT-K form spongy agglomerates at the macro level or a fibril network at the nano level. Moreover, the CNT-K stick together under pressure (like in the column) and create a uniform matrix (MWCNT matrix). Thus, a highly porous monolithic structure and a homogeneous particulate packed bed are present in the chromatographic column. Characteristic numbers are an asymmetry factor of 1.1 (describes the homogeneity of the packed bed); a HETP of 0.04 cm (represents the separation efficiency, see Equation S1); a column porosity of 0.4; and a total column porosity of 0.8 (see Figure S3 and Table S1). The description of the packed bed is extended using electrochemical methods, such as open circuit potential (ocp) measurements, which provide about +300 mV to +500 mV at neutral pH, depending on the arrangement of the CNT (of the matrix structure). This value is comparable to others in the literature [45,46]. Thus, a lack of electrons exists in the passive state, and the particulate electrode turns out to be positively charged. In the presence of ions (10 mM phosphate buffer, pH = 7.2) the ocp measurement yields even higher positive potentials, showing a more pronounced positive charge for the CNT matrix.

Figure 1.
Configuration of the MWCNT “CNT-K” (Future Carbon GmbH, Germany) at different scales. From left: (a) CNT-K agglomerate at macro scale (SEM, 2020×, Marco Breidinger); (b) surface of CNT-K agglomerates (SEM; 5000×, Katia Rodewald); (c) single CNT-K network at nano scale (TEM, 50,000×, Dr. Sebastian Schwaminger)
The single CNT-K tubes have an outer diameter between 20 and 40 nm, with a length up to hundreds of μm (Figure S4). The agglomerates are between 10 and 500 μm (Figure S5) and have pore sizes in the macro range. The specific surface area of the particulate material is around 200 m2/g, which is low compared to the single-walled CNT, with up to 1000 m2/g [47,48]. However, the commercially available SWCNTs are 10 to 100 times more expensive than commercially available MWCNTs; moreover, no technology for a large-scale production of SWCNTs currently exists. The relatively small surface area of the MWCNT used may be caused by the agglomeration behavior of the CNT-K [47], limiting the access to the total CNT-K surface. Another possibility is that only the outer surface of a single nanotube is accessible, as around 85% of tubes are closed tubes (Figure S6). In literature, the values for the surface area of open or closed MWCNT are 300 m2/g or 150 m2/g [49] respectively, so the specific surface area of CNT-K with 200 m2/g is within an expected range. The fact that over 85% of the CNT-K tubes from the TEM pictures seem to be closed tubes leads to the conclusion that adsorption can take place exclusively on the outer MWCNT shell, which is important information for the adsorption capacity of static and dynamic adsorption experiments.
3.3. CNT-K Hydrophilicity and Surface Composition
The hydrophilicity or hydrophobicity of MWCNTs is a hot topic in literature and has been discussed from different perspectives [50,51,52,53,54,55]. Commonly, the hydrophilicity of the tubes is attributed to surface defects in the curled layers or to the functionalization of the surface. Both effects lead to the presence of surface charges and improve the interaction with water. If these effects are not present (and when the tubes are ideally shaped), they are described as hydrophobic and uncharged. In our case, the manufacturer refers to the CNT-K as a non-functionalized and rather hydrophobic material, but in the contact angle measurements of the CNT-K carried out by our group, an angle of 80° [54] or even lower was detected (depending on the density and homogeneity of the sample prepared for the Washburn method), which suggests a slight hydrophilicity. Moreover, the twisted configuration of the tubes leads us to expect defects on the surface, reinforcing the supposition of hydrophilicity (see Figure 1). However, detailed knowledge of the interaction of the MWCNT surface and water is lacking in literature. This knowledge is essential for understanding the impact of the properties of the packed material on the adsorption processes of charged molecules in aqueous media. Therefore, we determined the swelling behavior of the CNT-K in water and analyzed the water binding of dry CNT-K particles. The correlation between dry and wet CNT-K particles is , which means that 1 g of wet CNT mud (CNT-K suspended in DI-water, centrifuged at 3200× g, 3 min, decantation of CNT-free water) contains only 7% of CNT-K; or on the contrary: 1 g of wet CNT-K mud contains 93% water even after centrifugation. Due to this, the dry CNT-K agglomerates swell to 125% of their original volume, leading to an increase in pore sizes, confirming the existence of macro pores in the system [52].
The high affinity of the CNT-K to water suggests the existence of surface oxygen, which we confirmed in XPS measurements (see Table 1). The XPS results showed that a small amount of surface oxygen is already present in untreated CNT-K powder. Conditioned CNT-K (acidic treatment to remove the catalyst residue, washed afterwards to neutral conditions and stored in DI-water) showed a similar quantity of oxygen on the surface. Ndunda et al. [56] and Zajíčková et al. [57] reported comparable values to ours and suggested that these confirm the oxidation of the surface. Samples of CNT-K used as an electrode and treated several times in potential-controlled experiments showed an increase of the surface oxygen content (see Figures S7 and S8), due to redox reactions occurring on the surface, or other electrostatic or covalent molecule-surface interactions appearing during the experiments [58,59]. These effects will be further discussed in the following sections.

Table 1.
Determination of surface oxygen content using X-ray photoelectron spectroscopy for untreated (as provided by the producer), conditioned (acidic treatment, washed, stored as DI-water slurry and dried) and used CNT-K (applied in different packed beds). The spectra and further information are found in Figures S7 and S8. The material was washed several times with DI-water to avoid adsorbate residues.
3.4. Static Binding Capacity of CNT-K for Small Molecules
Before the interaction between the mobile phase, target adsorbates and CNT-K were studied inside the chromatographic column (dynamic processing), it is helpful to analyze the adsorption behavior of different model molecules on the particles under static conditions; i.e., to quantify this behavior. These so-called static binding capacity experiments were performed with maleic acid (MA) as the standard adsorbate and fumaric acid (FA) for comparison due to the similarity of its chemical structure (Figure 2a). Both carboxylic acids carry one or two negative charges in DI-water at neutral pH. Incubation of 24 h leads to an adsorption amount higher than 100 mg/g, probably caused by electrostatic and polar interactions between the negative molecules and the positive, hydrated nanotube’s outer surface. Moreover, both the capillarity effect on the spongy agglomerates and the possibility of precipitation and crystallization of the organic molecules on the nanotubes further explain the high adsorption capacity [60]. In addition, π-π interactions between the carbon acids and CNT-K can occur. To gain further insight into the interaction of the CNT-K with small molecules, several other adsorbates with different properties were analyzed. The amino acids histidine (His), phenylalanine (Phe) and tryptophan (Trp) were selected as aromatic representatives, which suggest dominating π-π interactions, while alanine (Ala) was selected due to its aliphatic structure. All molecules were dissolved at their pIs, leading to net charges in solution. The isotherms in Figure 2b show that only Trp follows a typical saturation trend with increasing equilibrium concentration and a maximum of up to 75 mg/g. His and Phe adsorb exponentially after an overload, which indicates weak π-π interactions, whereas Ala shows a typical multi-layer behavior. All amino acids reach similar adsorbed amounts to the ones of the carbon acids around the equilibrium concentrations of 5 g/L, although their adsorption behaviors are different. The exception is alanine, which reveals clearly higher adsorbed amounts. Hence, CNT-K exhibit not only a high interaction capacity with the molecule selected, but also have different interaction mechanisms, depending on the target molecule properties. The first attribute, the adsorption capability of the CNT-K, is a critical requirement for the profitability of the PCC process, while the second attribute, the existence of various interaction mechanisms, is also an important feature necessary for triggering molecule adsorption and separation in the through-flow system. Thus, both points indicate that the CNT-K material is suitable for the development of a potential-controlled separation technology.
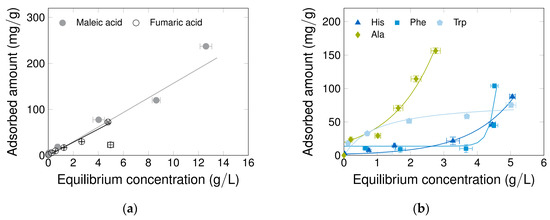
Figure 2.
Static binding capacities of (a) two carbon acids isomers at pH = 7 and (b) four amino acids at pI (neutral charged). The adsorbed amount is related to the dry mass of CNT-K. The experiment was performed at RT, 24 h, 500 rpm, with 2 or 3 technical replicates, triple determination at 216 or 280 nm.
3.5. Electrochemical Characterization of the Packed Bed
Chronoamperometry and cyclic voltammetry experiments were performed to study the electrochemical effects which occur inside the particulate fixed bed electrode, between the material surface and the mobile phase environment, when a potential is applied. Unlike conventional systems, which often operate in static conditions, the major goal here was to determine the operating quality by analyzing the response of the MWCNT packed bed directly in the flow cell. Accordingly, a constant potential was applied in the chronoamperometry measurements to first determine the potential-dependent current profile and its stabilization over time. The changes in the current profile led to a greater understanding of the electrochemical double layer (EDL) rearrangement, and later, the aging effects of the MWCNT matrix, which occur due to structural changes of the matrix or of the surface groups’ chemistry. As the through flow of the mobile phase is expected to play a role in the EDL, it was kept constant at 1 mL/min during all experiments.
Regarding the chronoamperometry experiments (Figure 3), immediate changes in the current strength are recognizable when a constant potential is applied to the particulate electrode over a defined process time. Simultaneously, the enforced changes in the electrode surface charge provoke a rearrangement of the ions in solution. The time needed to load the capacitor and to reach a stable current corresponds to the EDL rearrangement rate, whereas the mean current in this region after the exponential rearrangement represents the resistance in a non-ideal capacitor setup. As the profiles of the electric current converge to a constant value and do not achieve zero, which would be typical for an ideal capacitor, capacitive and faradaic current must exist in the PCC system. By comparing the constitutions of several packed CNT-K columns chromatographically with tracer experiments and electrochemically with chronoamperometry, we determined that the higher the current strength, the greater the CNT mass in the system. This correlates with the fact that more particle-particle contacts are available, as the MWCNT matrix is more homogeneous. The analysis of the response of various columns chronoamperometrically enables us not only to identify the constitution of the column, but also record the aging of the MWCNT matrix. This information on the life cycle of the particulate packed bed is directly connected to the quality of the particulate electrode and is extremely relevant for the consistency of results in a chromatographic, electrically driven process.
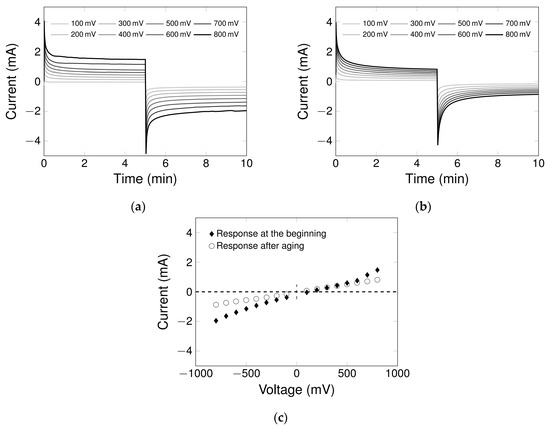
Figure 3.
Chronoamperometry experiments in DI-water (1 mL/min) to determine the constitution of the packed bed in a column A: (a) electrode response at the beginning, (b) electrode response after aging, (c) current/potential diagram showing mean current after electrochemical double layer (EDL) rearrangement. The chromatographic interpretation of changes in the column constitution can be found in Figure S9 (tracer experiment).
The chronoamperograms in Figure 3 demonstrated that the influence of the low volume flow rate significantly lengthens the EDL rearrangement time in comparison with static experiments, where it only takes a few seconds [61]. Figure 3a presents a chronoamperogram of a packed bed shortly after the packing process. The EDL rearrangement time is approximately 60 s for +800 mV and 120 s for −800 mV, and the current profiles maintain high mean currents after the exponential current decrease/increase. The response of one package looks different after repeated use and several processing cycles: aging leads to a longer EDL rearrangement time and a reduction of the mean current (Figure 3b). This change is interpreted as a degradation of the MWCNT matrix, which is also detectable with tracer experiments (for the chromatographic characterization see Figure S9). A possible reason for the modification of the MWCNT matrix is the mutual repulsion of the tubes due to charging the surface through the repeated application of an electrical potential [62]. This aging effect reduces the number of particle-particle contact points and changes the fluid pathway through the column. In addition to structural changes, the increase in the column resistance might be due to irreversible redox reactions on the surface [63], as confirmed by XPS results (see Section 3.3).
By plotting the mean current after EDL rearrangement over the applied potential, the relationship between current/potential and packed bed constitution is represented (see Figure 3c), showing higher mean current for freshly packed, more homogeneous MWCNT electrodes. In addition to the information on packing structure and aging effects, the slightly hyperbolic trend of the current, determined at higher potentials (positive and negative), indicates the presence of faradaic current, which points to redox reactions. This phenomenon appears at the outer measurement limit, between −500 and −800 mV, at which CNT material can oxidize. The linear regime, visible at lower applied potentials, represents dominating physicosorptive effects and capacitive current. The x-axis intersection is around +100 mV and corresponds to the point zero current of the total working electrode. The further the applied potential is from this point, the higher the net current is and the more likely it is that faradaic current occurs. The electrical charge of the particle electrode is potential-dependent and increases with higher applied potential. Thus, more than −500 mC can be obtained at the beginning of the column life (if −800 mV are applied), while the electric charge decreases with column aging (Figure S10).
Another characteristic factor calculated from the mean current of the chronoamperometry measurements is the specific resistance of the packed bed (see Equation S2), which is approximately 1.44 × 106 Ω mm2/m in the best case at ±800 mV (lowest resistance) (Figure 3a). This high resistance corresponds more to the resistance of tap water than to the expected MWCNT resistance (electrical conductivity, 1300 mS/cm [54] or even higher [64]). The high CNT-K column resistance is not surprising, as 93% of the MWCNT matrix contains DI-water, which has a poor conductivity (<0.02 mS/cm). Getting more than 0.5 g of particles into the system to increase the particle-particle contact points and to reduce the amount of water and the packed bed resistance is still a challenge because the CNT-K matrix swells in water and becomes tightly packed with high backpressure, thereby reducing the homogeneity of the packed bed and the flow profile quality.
3.6. Response of the System in Presence of Buffer Ions
To investigate the influence of the buffer ions on the electrochemical double layer and the column resistance, experiments with 10 mM phosphate buffer, conventionally employed in biotechnology, were carried out. Additionally, DI water experiments were run again for the direct comparison of results. These experiments confirmed the reproducibility of the current response behavior of the PCC column described in Section 3.5 as well as the aging effect, which is present for buffer and DI-water (after more than 250 working hours). The current/potential diagram in Figure 4 presents the relationship between the applied potential, the current, the electrode aging and the effect of the buffer ions. It shows that the mean current value increases in the phosphate buffer environment and the packed bed resistance decreases, caused by the higher ion concentration in the system. In addition, the EDL rearrangement time is longer (see Figure S11), and a higher concentration gradient is expected between the MWCNT surface and the bulk solution.
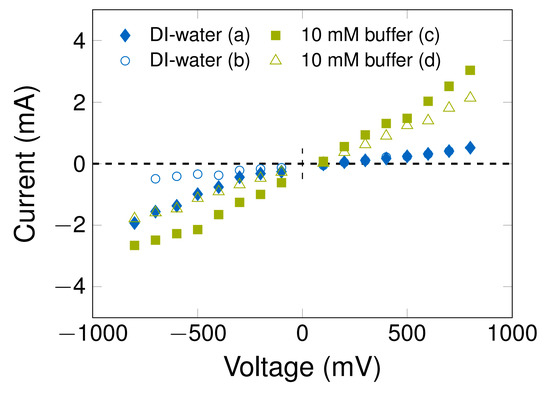
Figure 4.
Current/potential diagram to visualize the mean current after EDL rearrangement in a system with DI-water or 10 mM phosphate buffer as mobile phase (pH = 7.2, 1 mL/min). The MWCNT matrix was analyzed at an earlier stage (packed bed at initial condition) (a,c) and after long usage (after aging) (b,d). The experiments were performed with column B. The related chronoamperometry measurements and tracer experiments are illustrated in Figures S11 and S12. Information about the electric charge is given in Figures S13 and S14.
The aging of the electrode is caused by repeated use, which leads to structural changes in the CNT-K matrix (see Figure S12 for tracer experiments to demonstrate the aging of the MWCNT matrix). Consequently, the mean current decreases and the EDL rearrangement time changes. In the DI-water system, the mean current profile decreases, especially at high negative potentials when aging takes place, whereas this development is substantially less pronounced for the buffer. This non-linear behavior in the DI-water environment indicates that a detectable increase in electron transfer is present when fewer ions are available. Regarding the packed bed resistance, the ion rich buffer seems to be the more promising medium for an electrochemical cell, resulting in 6.47 × 105 mm2/m at the beginning and in 1.32 × 106 Ω mm2/m after aging. In comparison, the DI-water system reaches packed bed resistances between 1.26 × 106 Ω mm2/m and 1.09 × 109 Ω mm2/m. The electric charge of the working electrode correlates with the ion concentration (Figure S13): if more ions are available in the system, the electric charge is higher, up to ±1000 mC, while it is around ±500 mC in DI-water, independent of the potential-switch order of chronoamperometry experiments (Figure S14). A loss of electric charge occurs when aging effects occur, as caused by structural changes in the CNT matrix and redox reactions on the surface.
3.7. Sensitivity of the Electrochemical Process to the Environment
Along with chronoamperometry, in which the effect of the ion concentration on the current strength is determined, cyclic voltammetry (CV) is used to enhance the understanding of the electrochemical effects occurring in the particle based, through-flow electrode (Figure 5). To get more information about the potential-dependent adsorption of ions from the mobile phase, pH and conductivity are measured simultaneously to obtain the current values. The typical CV for a CNT-K column is presented in Figure 5a using DI-water as the mobile phase (1 mV/s). A reproducible current profile exists when a positive potential is applied, which represents reversible surface modification that includes the rearrangement of the EDL and the surface reactions. If negative potential gradients are applied, changes in the current profile are recognizable and an exponential current increase is detected. These changes are particularly pronounced between −500 and −800 mV and become less significant with each cycle, because the initial surface properties change more markedly at the beginning due to compensation from redox reactions, which take place on the electrode surface. This type of particle aging, discussed in literature [53,65], is consistent with our previous results (see Section 3.3 and Section 3.5) and helps to understand and confirm them. Regarding the shape of the CV, the flat and almost rectangular profile corresponds to a capacitor under flow [51,53,61,65,66,67] with current strength up to 200 μA [49,51,66]. Alongside the fluid dynamical effects, leakage current can shift the CV shape [68].
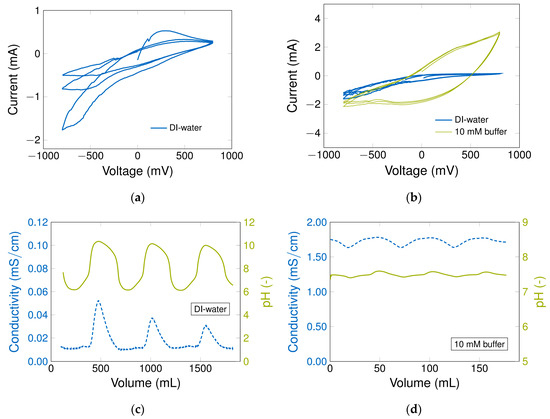
Figure 5.
Cyclic voltammogram to compare two columns and two different mobile phases (1 mL/min). (a) Aging effect in a CNT-K column used with DI-water as the mobile phase (1 mV/s); (b) the current differences of DI-water (0.1 mV/s) and 10 mM phosphate buffer (1 mV/s) determined in another column B; (c) the cyclic potential’s influence on pH and conductivity using DI-water in column B; (d) the cyclic potential’s influence on pH and conductivity using 10 mM phosphate buffer in column B. The potential gradient increases from 0 mV to +800 mV; then decreases to −800 mV; and increases again to +800 mV. The cycle is run three times and then stopped at 0 mV.
The CV in Figure 5b was run in another column with 0.1 mV/s to guarantee a complete picture of the voltage gradient influence on the current profile and pH value regarding a DI-water and buffer system. In the buffer system, the electrical current is higher and the CV shape is more pronounced, and the cathodic current shape is quite similar to anodic, suggesting a capacitor behavior and a reversible system [51]. The aging of the column due to redox reactions at the negative potential region is negligible in comparison to the CV with DI-water.
It is very interesting to follow pH and conductivity changes in the flow cell following potential application and to compare both systems, water and buffer. These effects increase understanding of what happens on the electrochemical double layer. The effect of the CV experiment in Figure 5b (increase from 0 mV to +800 mV, followed by a decrease to −800 mV and back) on the pH-value and the conductivity of the mobile phase are presented in Figure 5c,d. Even if the electrical current of DI water system is lower under applied potential due to the smaller number of ions in comparison to the buffer system, the pH value oscillates substantially upon applying a potential gradient. If the potential changes, the ion concentration in the bulk is disturbed, immediately influencing the pH value due to the adsorption or desorption of oxonium and hydroxyl ions inversely to the applied potential value (pH decrease at positive potential and pH increase at negative potential). At the same time, the low amount of salt ions in DI-water is responsible for the change in the mobile phase conductivity. The cation exchange membrane likely plays a role in the total system and interacts with the salt ions, too (for more details see Figure S15). However, the surface area of the membrane, and hence its number of binding sites, is less than the surface area of the electrode. If a buffer is used as a surrounding medium, the impact is clearly smaller and the signal output is more balanced because enough ions are available to dampen the concentration gradient of the mobile phase medium and the CNT-K surface (at least in the range of the chosen concentration profile). The regulation of the pH value is essential for biotechnological applications, the long-term target for our PCC process. Regarding this, the buffer system is more suitable for further application.
3.8. Potential-Controlled Interaction between Electrode and a Charged Molecule: Influence on Retention and Elution
The final goal of the PCC process is to separate molecules. To meet this objective, we studied the retention and elution of one model adsorbate, maleic acid, in the two media: DI-water and buffer. This molecule has two carboxyl groups and up to two negative charges at neutral pH. To analyze the potential influence on the column capacity, potential-step experiments were carried out. In the so-called adsorption phase, constant positive or negative potentials are applied to the CNT electrode, which induce a retention or a repulsion of the molecule. In the desorption phase, the potential is switched to -800 mV, which leads to a desorption of the previously adsorbed molecules. The injection concentration of maleic acid was chosen to be as high as the column overload, to underscore the potential effect on the adsorption and to induce a first breakthrough in the adsorption phase. Figure 6a represents typical chromatograms for maleic acid in a DI-water system. As expected, the twice negatively charged molecule does not adsorb at higher negative potential, slightly adsorbs if no potential is applied and almost completely adsorbs if a positive potential is applied (no peak appears here). If the elution is enforced by the potential switch to −800 mV, the amount of potential-dependent adsorbed maleic acid is visible in the elution peak, corresponding to the column capacity. The higher the applied positive potential, the more binding sites are on the CNT surface, and thus the higher the column capacity. However, due to the positive ocp of the CNT matrix, maleic acid is adsorbed without a potential being applied, and even −300 mV cannot interrupt the interaction of the CNT-K and the adsorbate. If a 10 mM phosphate buffer is selected as the mobile phase (see Figure 6b), the adsorption capacity for maleic acid decreases, because of the competition between the adsorbate and buffer ions for binding sites. Although the electrical conductivity is improved in the electrochemical experiments in the presence of a buffer, the concurring buffer ions occupy and somehow create a barrier between the MWCNT surface and the adsorbate [19,66]. Consequently, a buffer as a mobile phase, even if positive for the process, also has a limiting effect regarding the electrode capacity.
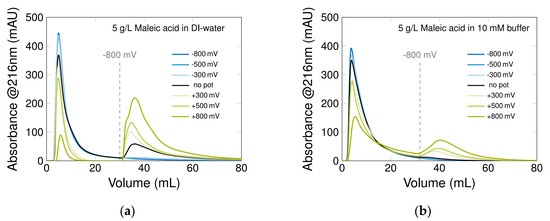
Figure 6.
Potential-step experiments to determine the retention of maleic acid in (a) DI-water and (b) 10 mM phosphate buffer (pH ≈ 7, 50 μL, 1 mL/min, 216 nm). The experiment was executed with different potentials applied during the adsorption phase (see legend); the elution was repeated each time with a potential switch to −800 mV (horizontal line): (a) injection of 5 g/L maleic acid in DI-water; (b) injection of 5 g/L maleic acid in 10 mM phosphate buffer. The experiment was performed in column B with a potential switch at 30 mL.
Figure 7 contains a plot of the percentages of the adsorbed maleic acid in DI-water and 10 mM phosphate buffer respectively, at different potentials. The use of an ionic mobile phase decreases the retention capacity for maleic acid. Thus, 92% of maleic acid is adsorbed with DI-water at +800 mV, while the hindrance due to the buffer ions reduces the value to just 43% maleic acid when a buffer is used. Furthermore, an exponential correlation between applied potential and column capacity is given within each mobile phase: 0.062 × e0.4x for DI-water and 0.005 × e0.6x for phosphate buffer.
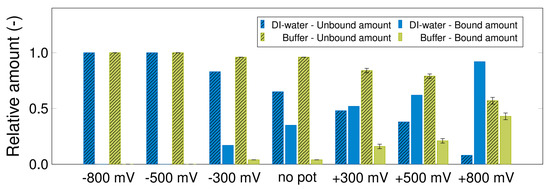
Figure 7.
Relative amounts of maleic acid in the adsorption phase (shaded bars) and in the desorption phase (bare bars). Potential-step experiment with 5 g/L maleic acid in DI-water (blue) and 10 mM phosphate buffer (green) (1 mL/min, 50 μL, 216 nm). The adsorption phase was executed by applying different potentials (x-axis); a potential switch to −800 mV induced the elution phase. The experiment was performed in a column B with a potential switch after 30 mL (experiments repeated twice).
Additional potential-step experiments in three independent PCC columns using DI-water and 5 g/L maleic acid demonstrate the reproducibility of the results (Figure S16). All experiments show that if more than the minimal saturation amount of maleic acid is injected (the column is overloaded), over 80% of the molecules are adsorbed at +800 mV and are eluted when the potential switch to −800 mV takes place. Reducing the injection concentration of maleic acid from 5 g/L to only 2 g/L (column C, Figures S17 and S18), 100% of the maleic acid can be bound at +800 mV in DI-water. These results confirm that PCC works when using CNT-K as a stationary phase and working electrode. They also highlight the differences between water and buffer as mobile phase and that water offers advantages for higher adsorption capacity.
4. Conclusions and Outlook
Carbon nanotubes are a material with exciting properties. For potential-controlled chromatography, they are primarily interesting due to their conductivity, inertness and high surface area. Our work demonstrates that CNT-K can be used as 3D packed bed electrode to electrically trigger retention and elution of maleic acid under flow in a chromatographic set up. Nevertheless, the nanotubes are also a challenging material. While our column optimization leads to a homogeneous and stable packed bed, the matrix structure suffers, and the packing loses homogeneity after approximately 300 working hours, as we have learned from chronoamperometry and cyclic voltammetry measurements. Along with the structural degradation, redox reactions take place on the carbon surface and change the electrode properties. Thus, aging causes a loss of consistency and efficiency of the potential-dependent retention process. This work focusses on understanding fundamental dependencies and effects in the PCC process to evaluate the impacts of individual components on the solid-liquid interface. The presence of capacitive and faradaic currents is confirmed; and column characteristics, such as the electric charge of the packing and the specific resistance, are determined at different applied potentials. As far as we know, there have been no similar analyses of the interaction of a multi-walled carbon nanotube solid phase with water in a through-flow cell, which, as we have demonstrated, induces a pH shift of the solution. Although the column resistance in water is high, the adsorption capacity for maleic acid is significantly higher (≈40%) than in 10 mM phosphate buffer, where ion hinderance is more relevant. In presence of a buffer, the faradaic current is reduced and the pH value stabilized. This knowledge is important for later applications in biotechnology or other life sciences, wherein many target molecules are sensitive to pH oscillations.
In this study, we demonstrated that the combination of electrochemistry and chromatography is a powerful strategy to evaluate quality and aging of the nanotube packed bed; to record and control the potential-controlled sorption process; and to estimate column performance. However, parameters such as the influence of the fluid dynamics and the effect of different electrolytes are also highly relevant and should be examined in future work. In addition, we continue to work toward identifying the processing framework for enhanced interaction profiles between charged molecules and MWCNTs.
Supplementary Materials
The following are available online at https://www.mdpi.com/2076-3417/10/3/1133/s1. Figure S1: Working principle of potential-controlled chromatography. Figure S2: Scheme of the adapted potential-controlled chromatographic (PCC) column. Figure S3: 1 M NaCl tracer to determine the quality of a CNT packing. Table S1: Long-term stability and quality of different particle packings. Figure S4: Minimum length, outer diameter and inner diameter of CNT-K. Figure S5: Particle size distribution of more than 2000 CNT-K agglomerates. Figure S6: Analysis of CNT-K ends. Figure S7: XPS analysis of the CNT functionalization based on the ratios between the surface oxygen/carbon. Figure S: Exemplary raw data of the XPS measurements. Figure S9: Tracer experiment to describe the packing quality at the beginning of the column life and after several experiments. Figure S10: The electric charge of a CNT-K electrode (column A) at different applied potentials. Figure S11: Additional chronoamperometry experiments to visualize the water and buffer influence on resistance and EDL rearrangement time. Figure S12: 1 M NaCl tracer experiment to analyze the quality of the packing chromatographically. Figure S13: The electric charge of a CNT-K electrode (column B). Figure S14: Electric charge from several chronoamperometry measurements. Figure S15: Scheme of a cyclic voltammetry experiment using DI-water as mobile phase. Figure S16: Relative amount of maleic acid in potential-step experiments using DI-water as mobile phase. Figure S17: Potential-step experiments to compare the absorbance values at the column outlet for different initial concentrations of maleic acid in DI-water (column C). Figure S18: Relative amount of maleic acid in potential-step experiments using DI-water as mobile phase (column C).
Author Contributions
T.T., P.F.-G. and S.B. designed the project together. T.T., T.S. and M.P. performed the experiments. T.T. and P.F.-G. wrote the manuscript. All authors assisted in evaluating the results, writing the discussion and editing the manuscript. Financial acquisition by S.B. and P.F.-G. All authors have read and agreed to the published version of the manuscript.
Funding
The research was funded by the Federal Ministry of Education and Research for their subsidy (grant number 13XP5038A). This work was supported by the German Research Foundation (DFG) and the Technical University of Munich (TUM) in the framework of the Open Access Publishing Program.
Acknowledgments
We express our gratitude to our partners of the Karlsruhe Institute for Technology, the groups of Matthias Franzreb and Wolfgang Wenzel. Furthermore, we thank our colleagues Andrés Martinez and Markus Brammen for their support in process specific questions, and the students Christian Weber, Stefan Rauwolf, Yannik Widmaier, Melanie Porzenheim, Timothy Stummvoll and Leonie Wittmann for their assistance in the laboratory. We thank Sebastian Schwaminger for his support and advice with the XPS measurements. For their support and the use of their equipment, we thank several groups at the Technical University of Munich: the Associate Professorship of Electron Microscopy (TEM, Carsten Peters), the Wacker-Chair of Macromolecular Chemistry (SEM, Katia Rodewald) and the Chair of Machine Elements (SEM, Marco Breidinger), as well as the Associate Professorship of Physical Chemistry with Focus on Catalysis (XPS, Sebastian Günther). Finally, we thank Thomas Benen from Microtrac Inc. for the analysis of the MWCNT agglomerate’s size and shape and Christian Schrader from C3 Prozess- und Analysentechnik GmbH for his comprehensive support.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fujinaga, T. Electrolytic chromatography and coulometric detection with the column electrode. J. Pure Appl. Chem. 1971, 25, 709–726. [Google Scholar] [CrossRef][Green Version]
- Fujinaga, T.; Kihara, S.; Bruckenstein, S. Electrolytic chromatography and coulopotentiography—A rapid electrolysis at the column electrode used for the preparation, separation, concentration, and estimation of trace and/or unstable substances. Crit. Rev. Anal. Chem. 1977, 6, 223–254. [Google Scholar] [CrossRef]
- Strohl, J.H.; Dunlap, K.L. Electrosorption and separation of quinones on a column of graphite particles. Anal. Chem. 1972, 44, 2166–2170. [Google Scholar] [CrossRef]
- Deinhammer, R.S. The development of novel chromatographic systems utilizing charge-controllable stationary phases for optimization of separation in liquid chromatography. Ph.D. Thesis, Iowa State University, Ames, Iowa, 1994. [Google Scholar]
- Nagaoka, T.; Fujimoto, M.; Nakao, H.; Kakuno, K.; Yano, J.; Ogura, K. Electrochemical separation of ionic compounds using a conductive stationary phase coated with polyaniline or polypyrrole film, and ion exchange properties of conductive polymers. J. Electroanal. Chem. 1994, 364, 179–188. [Google Scholar] [CrossRef]
- Ponton, L.M.; Porter, M.D. Electrochemically modulated liquid chromatographic separations of inorganic anions. J. Chrom. A 2004, 1059, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Yakes, B.J.; Keller, D.W.; Porter, M.D. Electrochemically modulated liquid chromatographic separation of triazines and the effect of pH on retention. J. Chrom. A 2010, 4395–4401. [Google Scholar] [CrossRef]
- Okazaki, S. Fundamental studies on the electrolysis with the glassy carbon column electrode. Rev. Polarogr. 1968, 15, 154–167. [Google Scholar] [CrossRef]
- Tsuda, T. Electrochromatography using high applied voltage. Anal. Chem. 1987, 59, 521–523. [Google Scholar] [CrossRef]
- Ting, E.-Y.; Porter, M.D. Separations of benzodiazepines using electrochemically modulated liquid chromatography. J. Chrom. A 1998, 793, 204–208. [Google Scholar] [CrossRef]
- Ting, E.-Y.; Porter, M.D. Separations of corticosteroids using electrochemically modulated liquid chromatography: Selectivity enhancements at a porous graphitic carbon stationary phase. Anal. Chem. 1997, 69, 675–678. [Google Scholar] [CrossRef]
- Antrim, R.F.; Scherrer, R.A.; Yacynych, A.M. Electrochromatography—a preliminary study of the effect of applied potential on a carbonaceous chromatographic column. Anal. Chim. Acta 1984, 164, 283–286. [Google Scholar] [CrossRef]
- Antrim, R.F.; Yacynych, A.M. The effect of supporting electrolytes in electrochromatography. Anal. Lett. 1988, 21, 1085–1096. [Google Scholar] [CrossRef]
- Ge, H.; Wallace, G.G. Characterization of novel conducting polymeric stationary phases and electrochemically controlled high-performance liquid chromatography. Anal. Chem. 1989, 61, 2391–2394. [Google Scholar] [CrossRef]
- Porter, M.D.; Takano, H. Electrochemically modulated liquid chromatography. Chromatography II 2000, 636–646. [Google Scholar]
- Deinhammer, R.S.; Ting, E.-Y.; Porter, M.D. Electrochemically modulated liquid chromatography (EMLC): A new approach to gradient elution separations. J. Electroanal. Chem. 1993, 362, 295–299. [Google Scholar] [CrossRef]
- Ting, E.-Y.; Porter, M.D. Electrochemically modulated liquid chromatography (EMLC) as a probe of the adsorption characteristics of monosubstituted benzenes at porous graphitic carbon. J. Electroanal. Chem. 1998, 443, 180–185. [Google Scholar] [CrossRef]
- Harnisch, J.A.; Porter, M.D. Electrochemically modulated liquid chromatography: An electrochemical strategy for manipulating chromatographic retention. Analyst 2001, 126, 1841–1849. [Google Scholar] [CrossRef]
- Abdel-Latif, M.S.; Porter, M.D. Effects of electrolytes on manipulation of the stationary phase in electrochemically modulated liquid chromatography. Anal. Lett. 1998, 31, 1743–1756. [Google Scholar] [CrossRef]
- Ponton, L.M.; Keller, D.W.; Siperko, L.M.; Hayes, M.A.; Porter, M.D. Investigation of adsorption thermodynamics at electrified liquid–solid interfaces by electrochemically modulated liquid chromatography. J. Phys. Chem. C 2019, 123, 28148–28157. [Google Scholar] [CrossRef]
- Soto, R.J.; Hayes, M.A.; Porter, M.D. Electrochemically modulated liquid chromatography in fused silica capillary columns. Anal. Chem. 2019, 91, 13994–14001. [Google Scholar] [CrossRef]
- Nagaoka, T.; Nakao, N.; Tabusa, K.; Yano, J.; Ogura, K. Dynamic elution control in electrochemical ion chromatography using pulse perturbation of stationary phase potential. J. Electroanal. Chem. 1994, 371, 283–286. [Google Scholar] [CrossRef]
- Muna, G.W.; Swope, V.M.; Swain, G.M.; Porter, M.D. Electrochemically modulated liquid chromatography using a boron-doped diamond particle stationary phase. J. Chromatogr. A 2008, 1210, 154–159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Keller, D.W.; Ponton, L.M.; Porter, M.D. Assessment of supporting electrolyte contributions in electrochemically modulated liquid chromatography. J. Chromatogr. A 2005, 1089, 72–81. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Latif, M. Effect of electrolytes on the retention behavior of some benzenesulfonates in electrochemically modulated liquid chromatography. Talanta 1998, 47, 681–687. [Google Scholar] [CrossRef]
- Knizia, M.W.; Vuorilehto, K.; Schrader, J.; Sell, D. Potential-controlled chromatography of short-chain carboxylic acids. Electroanalysis 2003, 15, 49–54. [Google Scholar] [CrossRef]
- Kocak, F.; Vuorilehto, K.; Schrader, J.; Sell, D. Potential-controlled chromatography for the separation of amino acids and peptides. J. Appl. Electrochem. 2005, 35, 1231–1237. [Google Scholar] [CrossRef]
- Brammen, M.; Fraga-Garcia, P.; Berensmeier, S. Carbon nanotubes-A resin for electrochemically modulated liquid chromatography. J. Sep. Sci. 2016, 1–9. [Google Scholar] [CrossRef]
- Speltini, A.; Merli, D.; Profumo, A. Analytical application of carbon nanotubes, fullerenes and nanodiamonds in nanomaterials-based chromatographic stationary phases: A review. Anal. Chim. Acta 2013, 783, 1–16. [Google Scholar] [CrossRef]
- Li, Y.; Chen, Y.; Xiang, R.; Ciuparu, D.; Pfefferle, L.D.; Horváth, C.; Wilkins, J.A. Incorporation of single-wall carbon nanotubes into an organic polymer monolithic stationary phase for mu-HPLC and capillary electrochromatography. Anal. Chem. 2005, 77, 1398–1406. [Google Scholar] [CrossRef]
- Chang, Y.X.; Zhou, L.L.; Li, G.X.; Li, L.; Yuan, L.M. Single-wall carbon nanotubes used as stationary phase in HPLC. J. Liq. Chrom. Relat. Tech. 2007, 30, 2953–2958. [Google Scholar] [CrossRef]
- Zhao, L.; Ai, P.; Duan, A.-H.; Yuan, L.-M. Single-walled carbon nanotubes for improved enantioseparations on a chiral ionic liquid stationary phase in GC. Anal. Bioanal. Chem. 2011, 399, 143–147. [Google Scholar] [CrossRef] [PubMed]
- André, C.; Gharbi, T.; Guillaume, Y.-C. A novel stationary phase based on amino derivatized nanotubes for HPLC separations: Theoretical and practical aspects. J. Sep. Sci. 2009, 32, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Rubianes, M.D.; Rivas, G.A. Carbon nanotubes paste electrode. Electrochem. Commun. 2003, 5, 689–694. [Google Scholar] [CrossRef]
- Nasrabadi, A.T.; Foroutan, M. Ion-separation and water-purification using single-walled carbon nanotube electrodes. Desalination 2011, 277, 236–243. [Google Scholar] [CrossRef]
- Ratajczak, P.; Suss, M.E.; Kaasik, F.; Béguin, F. Carbon electrodes for capacitive technologies. Energy Storage Mater. 2019, 16, 126–145. [Google Scholar] [CrossRef]
- Bulusheva, L.G.; Okotrub, A.V.; Kurenya, A.G.; Zhang, H.; Zhang, H.; Chen, X.; Song, H. Electrochemical properties of nitrogen-doped carbon nanotube anode in Li-ion batteries. Carbon 2011, 49, 4013–4023. [Google Scholar] [CrossRef]
- Saeed, K.; Kahn, I. Carbon nanotubes - properties and applications: A review. Carbon letters 2013, 131–144. [Google Scholar]
- Yang, E.; Thordarson, P.; Gooding, J.; Ringer, S.; Braet, F. Carbon nanotubes for biological and biomedical application. Nanotechnology 2007, 18, 412001. [Google Scholar] [CrossRef]
- Li, C.; Thostenson, E.T.; Chou, T.-W. Sensors and actuators based on carbon nanotubes and their composites: A review. Compos. Sci. Technol. 2008, 68, 1227–1249. [Google Scholar] [CrossRef]
- Iijima, S. Carbon nanotubes: Past, present, and future. Physica B 2002, 323, 1–5. [Google Scholar] [CrossRef]
- Han, C.; Shi, R.; Zhou, D.; Li, H.; Xu, L.; Zhang, T.; Li, J.; Kang, F.; Wang, G.; Li, B. High-energy and high-power nonaqueous lithium-ion capacitors based on polypyrrole/carbon nanotube composites as pseudocapacitive cathodes. ACS Appl. Mater. Interfaces 2019, 11, 15646–15655. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Yuan, X.; Zhang, S.; Wang, J.; Huang, Q.; Yu, N.; Zhu, Y.; Fu, L.; Wang, F.; Chen, Y.; et al. Three-dimensional ordered porous electrode materials for electrochemical energy storage. NPG Asia Mater. 2019, 11, 12. [Google Scholar] [CrossRef]
- Krieg, T.; Sydow, A.; Schröder, U.; Schrader, J.; Holtmann, D. Reactor concepts for bioelectrochemical syntheses and energy conversion. Trends Biotechnol. 2014, 32, 645–655. [Google Scholar] [CrossRef]
- Al-zubaidi, A.; Inoue, T.; Matsushita, T.; Ishii, Y.; Hashimoto, T.; Kawasaki, S. Cyclic voltammogram profile of single-walled carbon nanotube electric double-layer capacitor electrode reveals dumbbell shape. J. Phys. Chem. C 2012, 116, 7681–7686. [Google Scholar] [CrossRef]
- Kaempgen, M. Eigenschaften und Anwendungen von Netzwerken aus Kohlenstoff-Nanoröhren. Ph.D. Thesis, Universität Stuttgart, Stuttgart, Germany, 2006. [Google Scholar]
- Peigney, A.; Laurent, C.; Flahaut, E.; Bacsa, R.R.; Rousset, A. Specific surface area of carbon nanotubes and bundles of carbon nanotubes. Carbon 2001, 39, 507–514. [Google Scholar] [CrossRef]
- Birch, M.E.; Ruda-Eberenz, T.A.; Chai, M.; Andrews, R.; Hatfield, R.L. Properties that influence the specific surface areas of carbon nanotubes and nanofibers. Ann. Occup. Hyg. 2013, 57, 1148–1166. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Frackowiak, E.; Metenier, K.; Bertagna, V.; Beguin, F. Supercapacitor electrodes from multiwalled carbon nanotubes. Appl. Phys. Lett. 2000, 77, 2421–2423. [Google Scholar] [CrossRef]
- Li, S.; Li, H.; Wang, X.; Song, Y.; Liu, Y.; Jiang, L.; Zhu, D. Super-hydrophobicity of large-area honeycomb-like aligned carbon nanotubes. J. Phys. Chem. B 2002, 106, 9274–9276. [Google Scholar] [CrossRef]
- Shanmugam, S.; Gedanken, A. Generation of hydrophilic, bamboo-shaped multiwalled carbon nanotubes by solid-state pyrolysis and its electrochemical studies. J. Phys. Chem. B 2006, 110, 2037–2044. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Naguib, N.; Libera, J.A. In situ chemical experiments in carbon nanotubes. Chem. Phys. Lett. 2002, 365, 354–360. [Google Scholar] [CrossRef]
- Ye, J.-S.; Liu, X.; Cui, H.F.; Zhang, W.-D.; Sheu, F.-S.; Lim, T.M. Electrochemical oxidation of multi-walled carbon nanotubes and its application to electrochemical double layer capacitors. Electrochem. Commun. 2005, 7, 249–255. [Google Scholar] [CrossRef]
- Schwaminger, S.P.; Begovic, B.; Schick, L.; Jumani, N.A.; Brammen, M.W.; Fraga-García, P.; Berensmeier, S. Potential-controlled tensiometry: A tool for understanding wetting and surface properties of conductive powders by electroimbibition. Anal. Chem. 2018, 90, 14131–14136. [Google Scholar] [CrossRef] [PubMed]
- Tomo, Y.; Askounis, A.; Ikuta, T.; Takata, Y.; Sefiane, K.; Takahashi, K. Superstable ultrathin water film confined in a hydrophilized carbon nanotube. Nano Lett. 2018, 18, 1869–1874. [Google Scholar] [CrossRef] [PubMed]
- Ndunda, E.N.; Mizaikoff, B. Multi-walled carbon nanotubes: Innovative sorbents for pre-concentration of polychlorinated biphenyls in aqueous environments. Anal. Methods 2015, 7, 8034–8040. [Google Scholar] [CrossRef]
- Zajíčková, L.; Kučerová, Z.; Buršíková, V.; Eliáš, M.; Houdková, J.; Synek, P.; Maršíková, H.; Jašek, O. Carbon Nanotubes Functionalized in Oxygen and Water Low Pressure Discharges used as Reinforcement of Polyurethane Composites. Plasma Processes Polym. 2009, 6, S864–S869. [Google Scholar] [CrossRef]
- Cheng, X.; Zhong, J.; Meng, J.; Yang, M.; Jia, F.; Xu, Z.; Kong, H.; Xu, H. Characterization of multiwalled carbon nanotubes dispersing in water and association with biological effects. J. Nanomater. 2011, 2011, 1–12. [Google Scholar] [CrossRef]
- Törnkvist, A.; Markides, K.E.; Nyholm, L. Chromatographic behaviour of oxidised porous graphitic carbon columns. Analyst 2003, 128, 844–848. [Google Scholar] [CrossRef]
- Balavoine, F.; Schultz, P.; Richard, C.; Mallouh, V.; Ebbesen, T.W.; Mioskowski, C. Helical crystallization of proteins on carbon nanotubes: A first step towards the development of new biosensors. Angew. Chem. Int. 1999, 38, 1912–1915. [Google Scholar] [CrossRef]
- Barisci, J.N.; Wallace, G.G.; Chattopadhyay, D.; Papadimitrakopoulos, F.; Baughman, R.H. Electrochemical properties of single-wall carbon nanotube electrodes. J. Electrochem. Soc. 2003, 150, E409. [Google Scholar] [CrossRef]
- Hodge, S.A.; Fogden, S.; Howard, C.A.; Skipper, N.T.; Shaffer, M.S.P. Electrochemical processing of discrete single-walled carbon nanotube anions. ACS Nano 2013, 7, 1769–1778. [Google Scholar] [CrossRef]
- Saitoh, K.; Koichi, K.; Yabiku, F.; Noda, Y.; Porter, M.D.; Shibukawa, M. On-column electrochemical redox derivatization for enhancement of separation selectivity of liquid chromatography use of redox reaction as secondary chemical equilibrium. J. Chromatogr. A 2008, 1180, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.M.; Islam, M.A.; Shima, H.; Hasan, M.; Lee, M. Alignment of carbon nanotubes in carbon nanotube fibers through nanoparticles: A route for controlling mechanical and electrical properties. Appl. Mater. Inter. 2017, 9, 5530–5542. [Google Scholar] [CrossRef] [PubMed]
- Minnikanti, S.; Skeath, P.; Peixoto, N. Electrochemical characterization of multi-walled carbon nanotube coated electrodes for biological applications. Carbon 2009, 47, 884–893. [Google Scholar] [CrossRef]
- Pan, L.; Wang, X.; Gao, Y.; Zhang, Y.; Chen, Y.; Sun, Z. Electrosorption of anions with carbon nanotube and nanofibre composite film electrodes. Desalination 2009, 244, 139–143. [Google Scholar] [CrossRef]
- Chen, J.H.; Li, W.Z.; Wang, D.Z.; Yang, S.X.; Wen, J.G.; Ren, Z.F. Electrochemical characterization of carbon nanotubes as electrode in electrochemical double-layer capacitors. Carbon 2002, 40, 1193–1197. [Google Scholar] [CrossRef]
- Gu, W.; Yushin, G. Review of nanostructured carbon materials for electrochemical capacitor applications: Advantages and limitations of activated carbon, carbide-derived carbon, zeolite-templated carbon, carbon aerogels, carbon nanotubes, onion-like carbon, and graphene. WIREs Energy Environ. 2014, 3, 424–473. [Google Scholar] [CrossRef]
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).