Fluctuations in the Homogeneity of Cell Medium Distinguish Benign from Malignant Lymphocytes in a Cellular Model of Acute T Cells Leukemia



{kind=link}
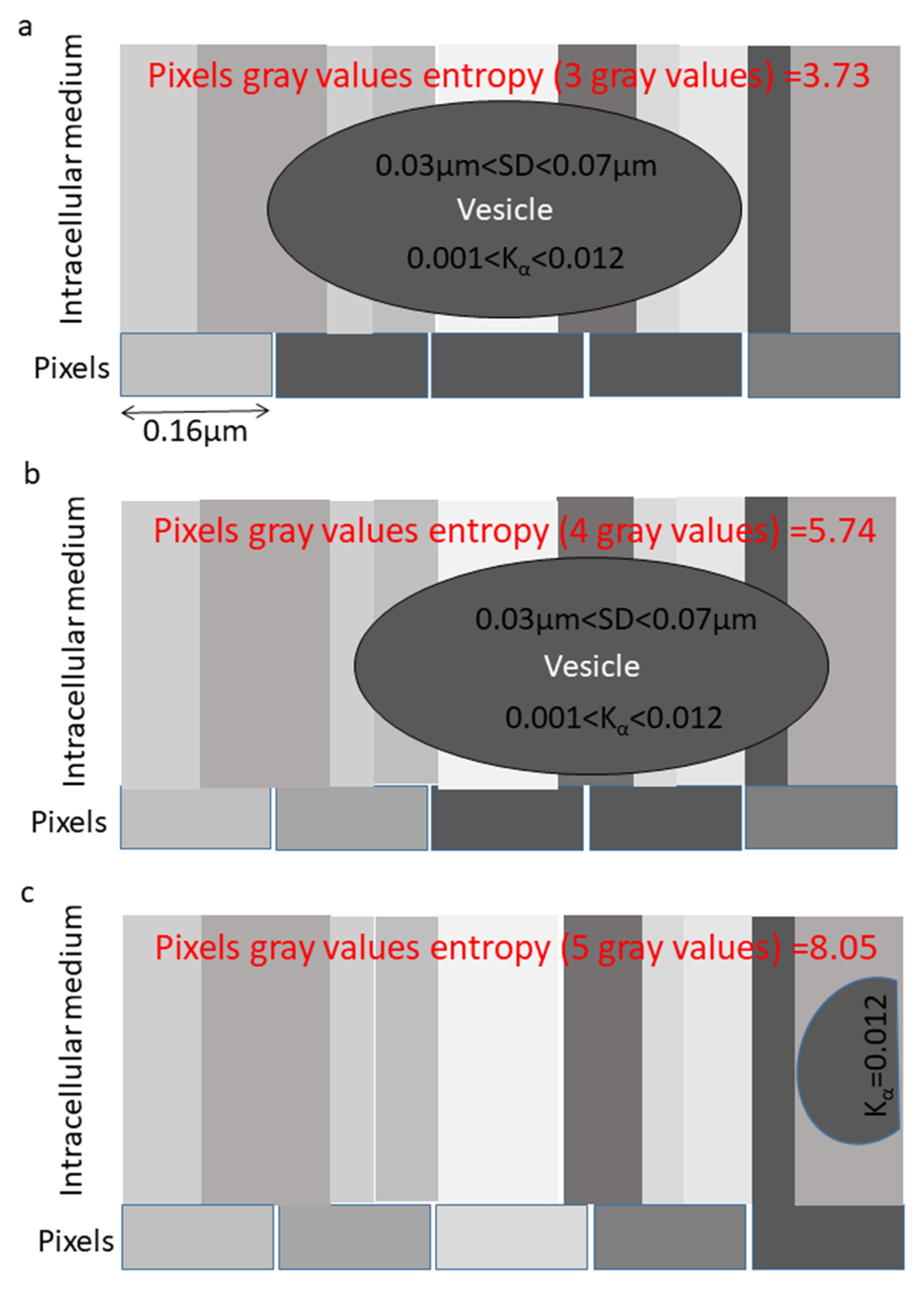{kind=link}
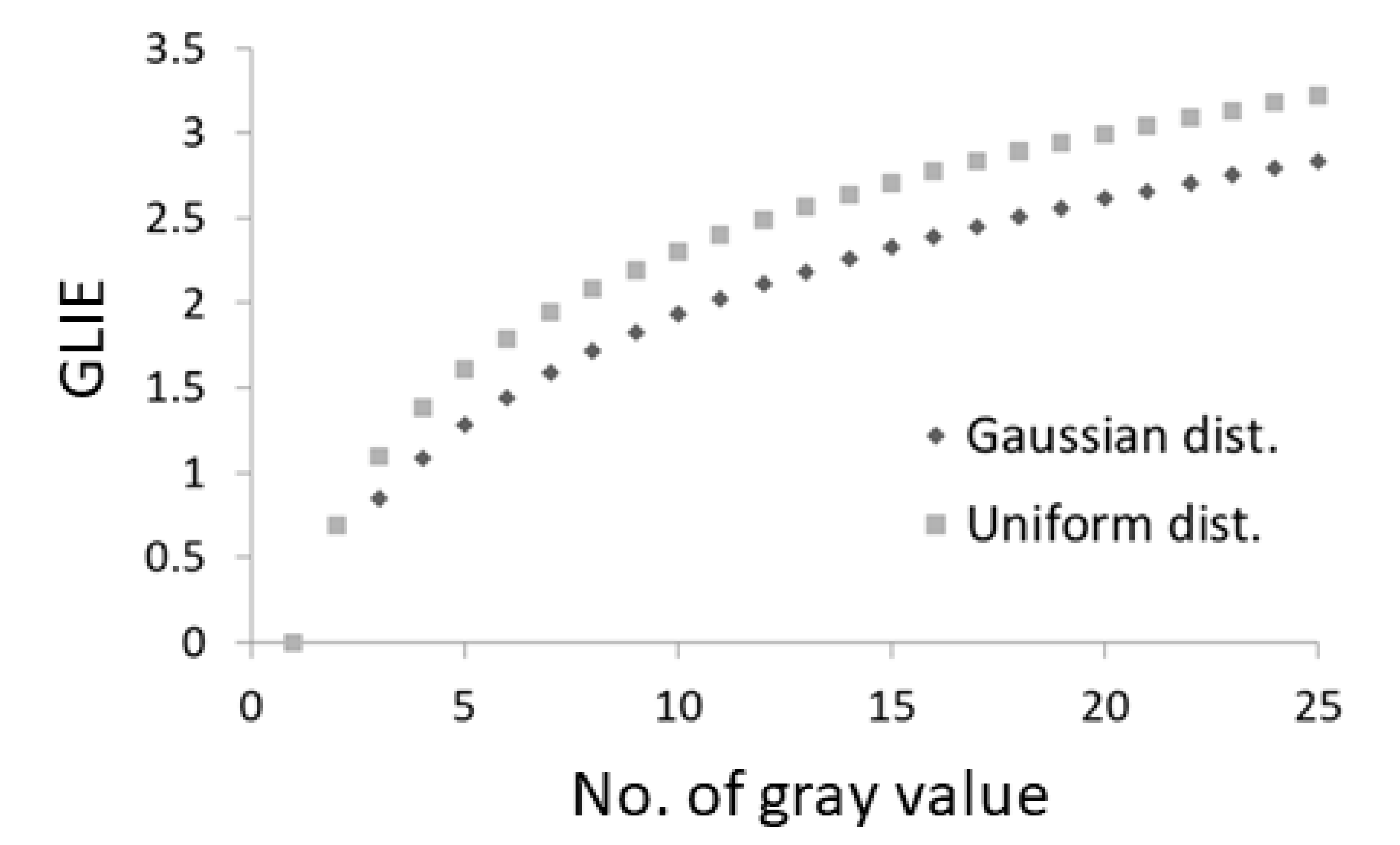{kind=link}
{kind=link}
{kind=link}
{kind=link}
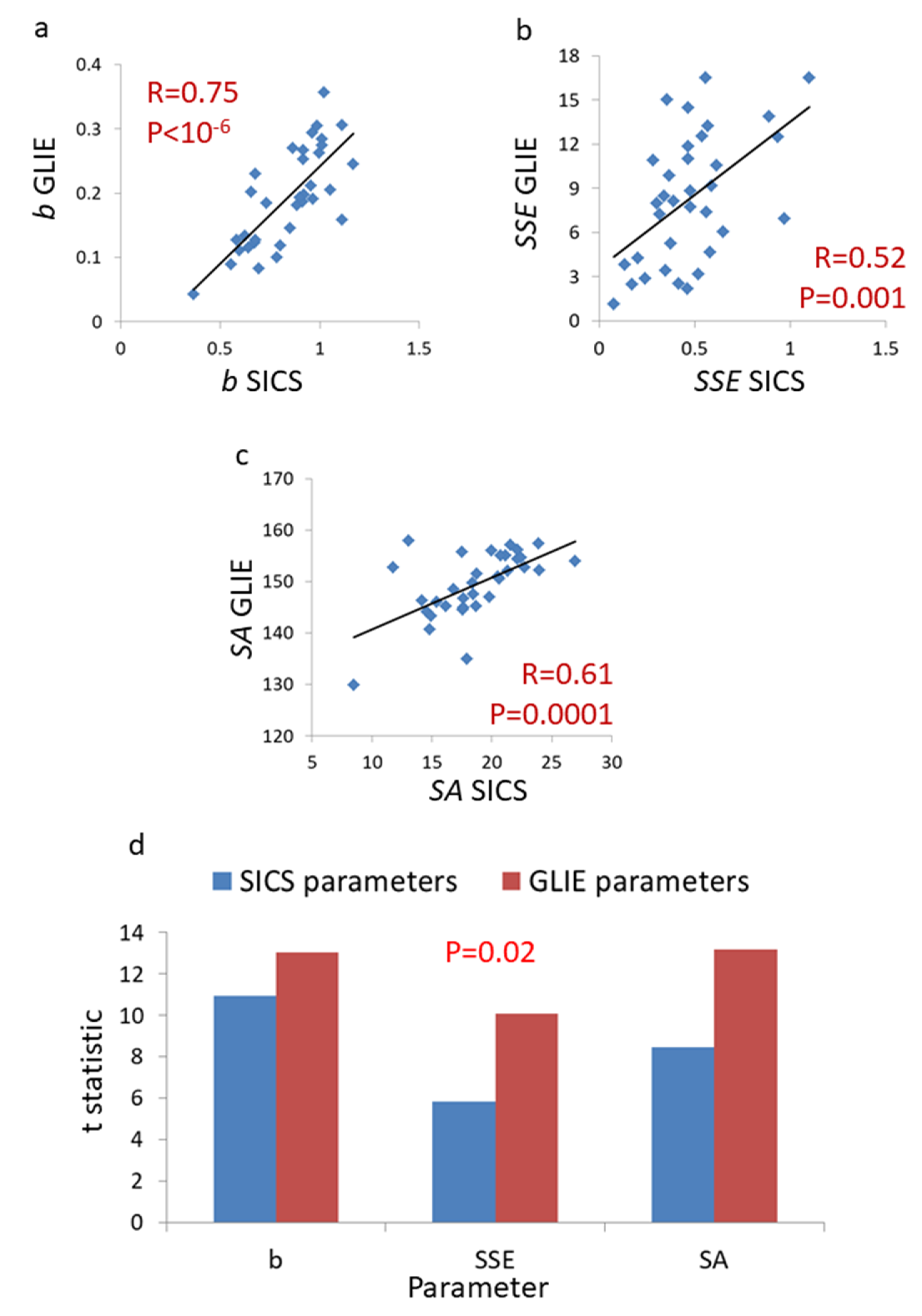{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
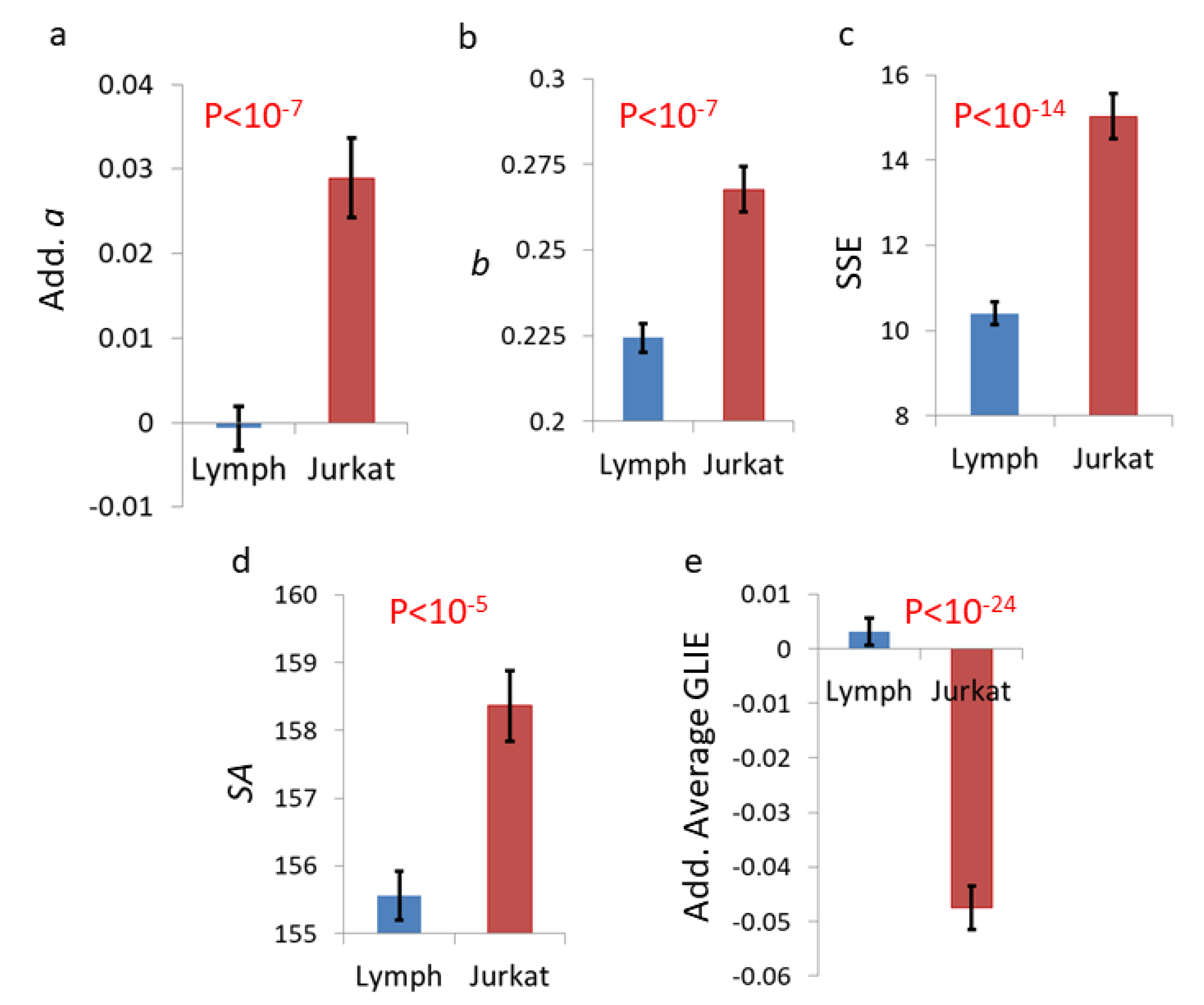{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Line
2.3. Sample Preparation
2.4. Treatment of Jurkat Cells with Rotenone and 2-Deoxy-d-glucose for ATP Depletion
2.5. Treatment of Primary T Cells Anti CD3
2.6. Treatment of Primary T Cells with PMA
2.7. Cells Fixation
2.8. PI Staining
2.9. Microscope
2.10. Image Correlation Spectroscopy (ICS) Calculations
2.11. Gray Level Information Entropy (GLIE) Fluctuation Calculations
2.12. Statistical Analyses
3. Results
3.1. Gray Level Information Entropy (GLIE) Is Sensitive to the Motion of Intracellular Particles
3.2. Comparing Pixel Gray-Levels Information Entropy (GLIE) Fluctuations and Spatial Image Correlation Spectroscopy (SICS) Fluctuations in Live Jurkat Cells before and after ATP Depletion
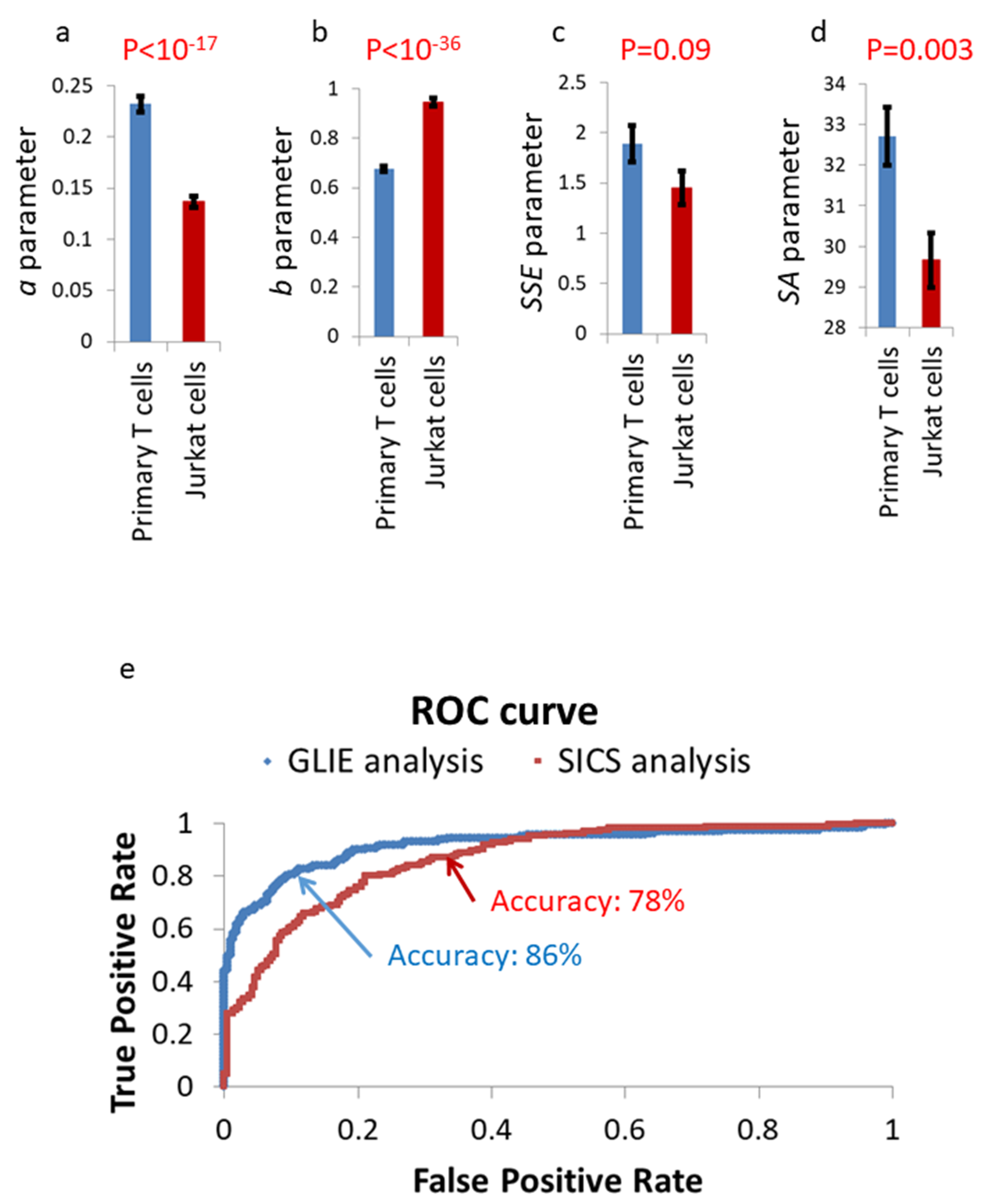
3.3. Comparing GLIE Fluctuations in Live Jurkat and Primary T Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vale, R.D. The Molecular Motor Toolbox for Intracellular Transport. Cell 2003, 112, 467–480. [Google Scholar] [CrossRef]
- Manfred, S.; Woehlke, G. Molecular Motors. Nature 2003, 422, 45–55. [Google Scholar] [CrossRef]
- Brangwynne, C.P.; Koenderink, G.H.; Mackintosh, F.C.; Weitz, D.A. Intracellular transport by active diffusion. Trends Cell Biol. 2009, 19, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.; Ehrlicher, A.J.; Jensen, M.H.; Renz, M.; Moore, J.R.; Goldman, R.D.; Lippincott-Schwartz, J.; Mackintosh, F.C.; Weitz, D.A. Probing the Stochastic, Motor-Driven Properties of the Cytoplasm Using Force Spectrum Microscopy. Cell 2014, 158, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Ishijima, A.; Kojima, H.; Funatsu, T.; Tokunaga, M.; Higuchi, H.; Tanaka, H.; Yanagida, T. Simultaneous Observation of Individual ATPase and Mechanical Events by a Single Myosin Molecule during Interaction with Actin. Cell 1998, 92, 161–171. [Google Scholar] [CrossRef]
- Wohl, I.; Sherman, E. ATP-Dependent Diffusion Entropy and Homogeneity in Living Cells. Entropy 2019, 21, 962. [Google Scholar] [CrossRef]
- Tuvia, S.; Levin, S.; Bitler, A.; Korenstein, R. Mechanical Fluctuations of the Membrane–Skeleton Are Dependent on F-Actin ATPase in Human Erythrocytes. J. Cell Biol. 1998, 141, 1551–1561. [Google Scholar] [CrossRef]
- Wohl, I.; Zurgil, N.; Hakuk, Y.; Deutsch, M.S.A.M. In Situ Evaluation of Physiological Activity and Mitochondrial Dysfunction via Novo Label-Free Measures Based on Fluctuation of Image Gray Values. J. Anal. Bioanal. Tech. 2016, 7, 308. [Google Scholar] [CrossRef]
- Lau, A.W.C.; Hoffman, B.D.; Davies, A.; Crocker, J.C.; Lubensky, T.C. Microrheology, Stress Fluctuations, and Active Behavior of Living Cells. Phys. Rev. Lett. 2003, 91, 198101. [Google Scholar] [CrossRef]
- Li, Y.; Schnekenburger, J.; Duits, M.H.G. Intracellular particle tracking as a tool for tumor cell characterization. J. Biomed. Opt. 2009, 14, 064005. [Google Scholar] [CrossRef]
- Suh, J.; Wirtz, D.; Hanes, J. Real-Time Intracellular Transport of Gene Nanocarriers Studied by Multiple Particle Tracking. Biotechnol. Prog. 2008, 20, 598–602. [Google Scholar] [CrossRef] [PubMed]
- Reverey, J.F.; Jeon, J.-H.; Bao, H.; Leippe, M.; Metzler, R.; Selhuber-Unkel, C. Superdiffusion dominates intracellular particle motion in the supercrowded cytoplasm of pathogenic Acanthamoeba castellanii. Sci. Rep. 2015, 5, srep11690. [Google Scholar] [CrossRef] [PubMed]
- Zam, A.; Kolios, M.C. Measuring intracellular motion in cancer cell using optical coherence tomography. Dyn. Fluct. Biomed. Photonics XIII 2016, 9707, 97070. [Google Scholar] [CrossRef]
- Nicolay, K.; Braun, K.P.J.; De Graaf, R.A.; Dijkhuizen, R.M.; Kruiskamp, M.J. Diffusion NMR spectroscopy. NMR Biomed. 2001, 14, 94–111. [Google Scholar] [CrossRef] [PubMed]
- Wohl, I.; Zurgil, N.; Hakuk, Y.; Sobolev, M.; Bar-On, Z.E. Fluctuation of Information Entropy Measures in Cell Image. Entropy 2017, 19, 565. [Google Scholar] [CrossRef]
- Wohl, I.; Zurgil, N.; Hakuk, Y.; Sobolev, M.; Galmidi, M.; Deutsch, M. In situ label-free static cytometry by monitoring spatiotemporal fluctuations of image gray values. J. Biomed. Opt. 2015, 20, 105013. [Google Scholar] [CrossRef]
- Jeon, J.-H.; Tejedor, V.; Burov, S.; Barkai, E.; Selhuber-Unkel, C.; Berg-Sørensen, K.; Oddershede, L.B.; Metzler, R. In VivoAnomalous Diffusion and Weak Ergodicity Breaking of Lipid Granules. Phys. Rev. Lett. 2011, 106, 048103. [Google Scholar] [CrossRef]
- Regner, B.M.; Vučinić, D.; Domnisoru, C.; Bartol, T.M.; Hetzer, M.W.; Tartakovsky, D.M.; Sejnowski, T.J. Anomalous diffusion of single particles in cytoplasm. Biophys. J. 2013, 104, 1652–1660. [Google Scholar] [CrossRef]
- Krapf, D.; Marinari, E.; Metzler, R.; Oshanin, G.; Xu, X.; Squarcini, A. Power spectral density of a single Brownian trajectory: What one can and cannot learn from it. New J. Phys. 2018, 20, 023029. [Google Scholar] [CrossRef]
- Krapf, D.; Lukat, N.; Marinari, E.; Metzler, R.; Oshanin, G.; Selhuber-Unkel, C.; Squarcini, A.; Stadler, L.; Weiss, M.; Xu, X. Spectral Content of a Single Non-Brownian Trajectory. Phys. Rev. X 2019, 9, 011019. [Google Scholar] [CrossRef]
- Sposini, V.; Metzler, R.; Oshanin, G. Single-trajectory spectral analysis of scaled Brownian motion. New J. Phys. 2019, 21, 073043. [Google Scholar] [CrossRef]
- Pantic, I.; Pantic, S.; Paunovic, J. Aging Increases Nuclear Chromatin Entropy of Erythroid Precursor Cells in Mice Spleen Hematopoietic Tissue. Microsc. Microanal. 2012, 18, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Pantic, I.; Pantic, S.; Basta-Jovanovic, G. Gray Level Co-Occurrence Matrix Texture Analysis of Germinal Center Light Zone Lymphocyte Nuclei: Physiology Viewpoint with Focus on Apoptosis. Microsc. Microanal. 2012, 18, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Pantic, I.; Pantic, S. Germinal Center Texture Entropy as Possible Indicator of Humoral Immune Response: Immunophysiology Viewpoint. Mol. Imaging Biol. 2011, 14, 534–540. [Google Scholar] [CrossRef]
- De Arruda, P.F.F.; Gatti, M.; Junior, F.N.F.; De Arruda, J.G.F.; Moreira, R.D.; Junior, L.O.M.; De Arruda, L.F.; De Godoy, M.F. Quantification of fractal dimension and Shannon’s entropy in histological diagnosis of prostate cancer. BMC Clin. Pathol. 2013, 13, 6. [Google Scholar] [CrossRef]
- Chithra Devi, M.; Audithan, S. Analysis of different types of entropy measures for breast cancer diagnosis using ensemble classification. Biomed. Res. 2017, 28, 3182–3186. [Google Scholar]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial Complex I Inhibitor Rotenone Induces Apoptosis through Enhancing Mitochondrial Reactive Oxygen Species Production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar] [CrossRef]
- Martin, J.A.; Martini, A.; Molinari, A.; Morgan, W.; Ramalingam, W.; Buckwalter, J.A.; McKinley, T.O. Mitochondrial electron transport and glycolysis are coupled in articular cartilage. Osteoarthr. Cartil. 2012, 20, 323–329. [Google Scholar] [CrossRef]
- Kolin, D.L.; Wiseman, P. Advances in Image Correlation Spectroscopy: Measuring Number Densities, Aggregation States, and Dynamics of Fluorescently labeled Macromolecules in Cells. Cell Biophys. 2007, 49, 141–164. [Google Scholar] [CrossRef]
- Tucher, C.; Bode, K.; Schiller, P.; Claßen, L.; Birr, C.; Souto-Carneiro, M.M.; Blank, N.; Lorenz, H.-M.; Schiller, M. Extracellular Vesicle Subtypes Released From Activated or Apoptotic T-Lymphocytes Carry a Specific and Stimulus-Dependent Protein Cargo. Front. Immunol. 2018, 9, 534. [Google Scholar] [CrossRef]
- Hyman, A.A.; Weber, C.A.; Jülicher, F. Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol. 2014, 30, 39–58. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P.; Tompa, P.; Pappu, R.V. Polymer physics of intracellular phase transitions. Nat. Phys. 2015, 11, 899–904. [Google Scholar] [CrossRef]
- Wohl, I.; Yakovian, O.; Razvag, Y.; Reches, M.; Sherman, E. Fast and synchronized fluctuations of cortical actin negatively correlate with nucleoli liquid–liquid phase separation in T cells. Eur. Biophys. J. 2020, 49, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.J.; Jeon, D.I.; Ahn, S.-G.; Yoon, J.-H.; Kim, S.; Lee, B.-H. Full-field optical coherence microscopy for identifying live cancer cells by quantitative measurement of refractive index distribution. Opt. Express 2010, 18, 23285–23295. [Google Scholar] [CrossRef]
- Phillips, K.G.; Velasco, C.R.; Li, J.; Kolatkar, A.; Luttgen, M.; Bethel, K.; Duggan, B.; Kuhn, P.; Mccarty, O.J.T. Optical Quantification of Cellular Mass, Volume, and Density of Circulating Tumor Cells Identified in an Ovarian Cancer Patient. Front. Oncol. 2012, 2, 72. [Google Scholar] [CrossRef]
- Dey, P.; Mohanty, S.K. Fractal dimensions of breast lesions on cytology smears. Diagn. Cytopathol. 2003, 29, 85–86. [Google Scholar] [CrossRef]
- Harkins, K.D.; Galons, J.-P.; Divijak, J.L.; Trouard, T.P. Changes in intracellular water diffusion and energetic metabolism in response to ischemia in perfused C6 rat glioma cells. Magn. Reson. Med. 2011, 66, 859–867. [Google Scholar] [CrossRef][Green Version]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nat. Cell Biol. 2006, 443, 787–795. [Google Scholar] [CrossRef]
- Gibson, G.E.; Starkov, A.; Blass, J.P.; Ratan, R.R.; Beal, M.F. Cause and consequence: Mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2010, 1802, 122–134. [Google Scholar] [CrossRef]
- Wiley, C.D.; Velarde, M.C.; Lecot, P.; Liu, S.; Sarnoski, E.A.; Freund, A.; Shirakawa, K.; Lim, H.W.; Davis, S.S.; Ramanathan, A.; et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab. 2016, 23, 303–314. [Google Scholar] [CrossRef]
- Legido, A.; Jethva, R.; Goldenthal, M.J. Mitochondrial Dysfunction in Autism. Semin. Pediatr. Neurol. 2013, 20, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Myhill, S.; Booth, N.E.; McLaren-Howard, J. Chronic fatigue syndrome and mitochondrial dysfunction. Int. J. Clin. Exp. Med. 2009, 2, 1–16. [Google Scholar] [PubMed]
- Pelicano, H.; Zhang, W.; Liu, J.; Hammoudi, N.; Dai, J.; Xu, R.-H.; Pusztai, L.; Huang, P. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: Role of mTOR pathway and therapeutic potential. Breast Cancer Res. 2014, 16, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Feichtinger, R.G.; Sperl, W.; Bauer, J.W.; Kofler, B. Mitochondrial dysfunction: A neglected component of skin diseases. Exp. Dermatol. 2014, 23, 607–614. [Google Scholar] [CrossRef]
- Kim, J.-A.; Wei, Y.; Sowers, J.R. Role of Mitochondrial Dysfunction in Insulin Resistance. Circ. Res. 2008, 102, 401–414. [Google Scholar] [CrossRef]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wohl, I.; Yakovian, O.; Sherman, E. Fluctuations in the Homogeneity of Cell Medium Distinguish Benign from Malignant Lymphocytes in a Cellular Model of Acute T Cells Leukemia. Appl. Sci. 2020, 10, 8894. https://doi.org/10.3390/app10248894
Wohl I, Yakovian O, Sherman E. Fluctuations in the Homogeneity of Cell Medium Distinguish Benign from Malignant Lymphocytes in a Cellular Model of Acute T Cells Leukemia. Applied Sciences. 2020; 10(24):8894. https://doi.org/10.3390/app10248894
Chicago/Turabian StyleWohl, Ishay, Oren Yakovian, and Eilon Sherman. 2020. "Fluctuations in the Homogeneity of Cell Medium Distinguish Benign from Malignant Lymphocytes in a Cellular Model of Acute T Cells Leukemia" Applied Sciences 10, no. 24: 8894. https://doi.org/10.3390/app10248894
APA StyleWohl, I., Yakovian, O., & Sherman, E. (2020). Fluctuations in the Homogeneity of Cell Medium Distinguish Benign from Malignant Lymphocytes in a Cellular Model of Acute T Cells Leukemia. Applied Sciences, 10(24), 8894. https://doi.org/10.3390/app10248894