Targeted Therapy in Metastatic Bladder Cancer: Present Status and Future Directions

 ,
, 
Abstract
1. Introduction
2. Methods
3. Pharmacological Inhibition of Fibroblast Growth Factor Receptor Signaling
4. Targetting ErbB Receptor Family Signaling
5. MAPK and Pi3K/AKT/mTOR Pathway Alterations
6. Targeting UC Vascularization with VEGF Inhibitors
7. Promising Targeted Treatment Approach: Antibody–Drug Conjugates
8. Sampling and Trial Setup
9. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Antoni, S.; Ferlay, J.; Soerjomataram, I.; Znaor, A.; Jemal, A.; Bray, F. Bladder Cancer Incidence and Mortality: A Global Overview and Recent Trends. Eur. Urol. 2017, 71, 96–108. [Google Scholar] [PubMed]
- Burger, M.; Catto, J.W.F.; Dalbagni, G.; Grossman, H.B.; Herr, H.; Karakiewicz, P.; Kassouf, W.; Kiemeney, L.A.; La Vecchia, C.; Shariat, S.; et al. Epidemiology and Risk Factors of Urothelial Bladder Cancer. Eur. Urol. 2013, 63, 234–241. [Google Scholar] [CrossRef] [PubMed]
- von der Maase, H.; Hansen, S.W.; Roberts, J.T.; Dogliotti, L.; Oliver, T.; Moore, M.J.; Bodrogi, I.; Albers, P.; Knuth, A.; Lippert, C.M.; et al. Gemcitabine and cisplatin versus methotrexate, vinblastine, doxorubicin, and cisplatin in advanced or metastatic bladder cancer: Results of a large, randomized, multinational, multicenter, phase III study. J. Clin. Oncol. 2000, 18, 3068–3077. [Google Scholar] [CrossRef] [PubMed]
- Galsky, M.D.; Chen, G.J.; Oh, W.K.; Bellmunt, J.; Roth, B.J.; Petrioli, R.; Dogliotti, L.; Dreicer, R.; Sonpavde, G. Comparative effectiveness of cisplatin-based and carboplatin-based chemotherapy for treatment of advanced urothelial carcinoma. Ann. Oncol. 2012, 23, 406–410. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; de Wit, R.; Vaughn, D.J.; Fradet, Y.; Lee, J.L.; Fong, L.; Vogelzang, N.J.; Climent, M.A.; Petrylak, D.P.; Choueiri, T.K.; et al. Pembrolizumab as Second-Line Therapy for Advanced Urothelial Carcinoma. N. Engl. J. Med. 2017, 376, 1015–1026. [Google Scholar] [CrossRef]
- Powles, T.; Duran, I.; van der Heijden, M.S.; Loriot, Y.; Vogelzang, N.J.; De Giorgi, U.; Oudard, S.; Retz, M.M.; Castellano, D.; Bamias, A.; et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): A multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018, 391, 748–757. [Google Scholar] [CrossRef]
- Kamoun, A.; de Reynies, A.; Allory, Y.; Sjodahl, G.; Robertson, A.G.; Seiler, R.; Hoadley, K.A.; Groeneveld, C.S.; Al-Ahmadie, H.; Choi, W.; et al. A Consensus Molecular Classification of Muscle-invasive Bladder Cancer. Eur. Urol. 2020, 77, 420–433. [Google Scholar] [CrossRef]
- Robertson, A.G.; Kim, J.; Al-Ahmadie, H.; Bellmunt, J.; Guo, G.; Cherniack, A.D.; Hinoue, T.; Laird, P.W.; Hoadley, K.A.; Akbani, R.; et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017, 171, 540–556.e25. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Kim, J.; Creighton, C.J.; Akbani, R.; Hoadley, K.A.; Kim, W.Y.; Morgan, M.B.; Hinoue, T.; Rosenberg, J.E.; Bajorin, D.F.; et al. Comprehensive molecular profiling of urothelial bladder cancer at the DNA, RNA, and protein levels: A TCGA project. J. Clin. Oncol. 2014, 32, 4509. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Comprehensive molecular characterization of urothelial bladder carcinoma. Nature 2014, 507, 315–322. [Google Scholar] [CrossRef]
- Shaw, R.J.; Cantley, L.C. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature 2006, 441, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Pouessel, D.; Neuzillet, Y.; Mertens, L.S.; van der Heijden, M.S.; de Jong, J.; Sanders, J.; Peters, D.; Leroy, K.; Manceau, A.; Maille, P.; et al. Tumor heterogeneity of fibroblast growth factor receptor 3 (FGFR3) mutations in invasive bladder cancer: Implications for perioperative anti-FGFR3 treatment. Ann. Oncol. 2016, 27, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- van Rhijn, B.W.; van Tilborg, A.A.; Lurkin, I.; Bonaventure, J.; de Vries, A.; Thiery, J.P.; van der Kwast, T.H.; Zwarthoff, E.C.; Radvanyi, F. Novel fibroblast growth factor receptor 3 (FGFR3) mutations in bladder cancer previously identified in non-lethal skeletal disorders. Eur. J. Hum. Genet. 2002, 10, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, D.C.; Baldo, O.; Harnden, P.; Knowles, M.A. FGFR3 protein expression and its relationship to mutation status and prognostic variables in bladder cancer. J. Pathol. 2007, 213, 91–98. [Google Scholar] [CrossRef]
- Guancial, E.A.; Werner, L.; Bellmunt, J.; Bamias, A.; Choueiri, T.K.; Ross, R.; Schutz, F.A.; Park, R.S.; O’Brien, R.J.; Hirsch, M.S.; et al. FGFR3 expression in primary and metastatic urothelial carcinoma of the bladder. Cancer Med. 2014, 3, 835–844. [Google Scholar] [CrossRef]
- van Rhijn, B.W.G.; Mertens, L.S.; Mayr, R.; Bostrom, P.J.; Real, F.X.; Zwarthoff, E.C.; Boormans, J.L.; Abas, C.; van Leenders, G.J.L.H.; Götz, S.; et al. FGFR3 Mutation Status and FGFR3 Expression in a Large Bladder Cancer Cohort Treated by Radical Cystectomy: Implications for Anti-FGFR3 Treatment? Eur. Urol. 2020. [Google Scholar] [CrossRef]
- Helsten, T.; Elkin, S.; Arthur, E.; Tomson, B.N.; Carter, J.; Kurzrock, R. The FGFR Landscape in Cancer: Analysis of 4853 Tumors by Next-Generation Sequencing. Clin. Cancer Res. 2016, 22, 259–267. [Google Scholar]
- Tomlinson, D.C.; Knowles, M.A. Altered splicing of FGFR1 is associated with high tumor grade and stage and leads to increased sensitivity to FGF1 in bladder cancer. Am. J. Pathol. 2010, 177, 2379–2386. [Google Scholar] [CrossRef]
- Tomlinson, D.C.; Baxter, E.W.; Loadman, P.M.; Hull, M.A.; Knowles, M.A. FGFR1-induced epithelial to mesenchymal transition through MAPK/PLCγ/COX-2-mediated mechanisms. PLoS ONE 2012, 7, e38972. [Google Scholar] [CrossRef]
- Cheng, T.; Roth, B.; Choi, W.; Black, P.C.; Dinney, C.; McConkey, D.J. Fibroblast growth factor receptors-1 and -3 play distinct roles in the regulation of bladder cancer growth and metastasis: Implications for therapeutic targeting. PLoS ONE 2013, 8, e57284. [Google Scholar] [CrossRef]
- Pal, S.K.; Rosenberg, J.E.; Hoffman-Censits, J.H.; Berger, R.; Quinn, D.I.; Galsky, M.D.; Wolf, J.; Dittrich, C.; Keam, B.; Delord, J.P.; et al. Efficacy of BGJ398, a Fibroblast Growth Factor Receptor 1-3 Inhibitor, in Patients with Previously Treated Advanced Urothelial Carcinoma with FGFR3 Alterations. Cancer Discov. 2018, 8, 812–821. [Google Scholar] [PubMed]
- Pal, S.K.; Bajorin, D.; Dizman, N.; Hoffman-Censits, J.; Quinn, D.I.; Petrylak, D.P.; Galsky, M.D.; Vaishampayan, U.; De Giorgi, U.; Gupta, S.; et al. Infigratinib in upper tract urothelial carcinoma versus urothelial carcinoma of the bladder and its association with comprehensive genomic profiling and/or cell-free DNA results. Cancer 2020, 126, 2597–2606. [Google Scholar] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [PubMed]
- Schuler, M.; Cho, B.C.; Sayehli, C.M.; Navarro, A.; Soo, R.A.; Richly, H.; Cassier, P.A.; Tai, D.; Penel, N.; Nogova, L.; et al. Rogaratinib in patients with advanced cancers selected by FGFR mRNA expression: A phase 1 dose-escalation and dose-expansion study. Lancet Oncol. 2019, 20, 1454–1466. [Google Scholar] [PubMed]
- Loriot, Y.; Necchi, A.; Park, S.H.; Garcia-Donas, J.; Huddart, R.; Burgess, E.; Fleming, M.; Rezazadeh, A.; Mellado, B.; Varlamov, S.; et al. Erdafitinib in Locally Advanced or Metastatic Urothelial Carcinoma. N. Engl. J. Med. 2019, 381, 338–348. [Google Scholar] [PubMed]
- Casalini, P.; Iorio, M.V.; Galmozzi, E.; Menard, S. Role of HER receptors family in development and differentiation. J. Cell Physiol. 2004, 200, 343–350. [Google Scholar]
- Eriksson, P.; Sjodahl, G.; Chebil, G.; Liedberg, F.; Hoglund, M. HER2 and EGFR amplification and expression in urothelial carcinoma occurs in distinct biological and molecular contexts. Oncotarget 2017, 8, 48905–48914. [Google Scholar]
- Rebouissou, S.; Bernard-Pierrot, I.; de Reynies, A.; Lepage, M.L.; Krucker, C.; Chapeaublanc, E.; Herault, A.; Kamoun, A.; Caillault, A.; Letouze, E.; et al. EGFR as a potential therapeutic target for a subset of muscle-invasive bladder cancers presenting a basal-like phenotype. Sci. Transl. Med. 2014, 6, 244ra291. [Google Scholar]
- Hussain, M.; Daignault, S.; Agarwal, N.; Grivas, P.D.; Siefker-Radtke, A.O.; Puzanov, I.; MacVicar, G.R.; Levine, E.G.; Srinivas, S.; Twardowski, P.; et al. A randomized phase 2 trial of gemcitabine/cisplatin with or without cetuximab in patients with advanced urothelial carcinoma. Cancer 2014, 120, 2684–2693. [Google Scholar] [CrossRef]
- Fleischmann, A.; Rotzer, D.; Seiler, R.; Studer, U.E.; Thalmann, G.N. Her2 amplification is significantly more frequent in lymph node metastases from urothelial bladder cancer than in the primary tumours. Eur. Urol. 2011, 60, 350–357. [Google Scholar]
- Kolla, S.B.; Seth, A.; Singh, M.K.; Gupta, N.P.; Hemal, A.K.; Dogra, P.N.; Kumar, R. Prognostic significance of Her2/neu overexpression in patients with muscle invasive urinary bladder cancer treated with radical cystectomy. Int. Urol. Nephrol. 2008, 40, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Powles, T.; Huddart, R.A.; Elliott, T.; Jones, R.; Hussain, S.A.; Crabb, S.J.; Ackerman, C.; Jagdev, S.; Chester, J.D.; Hilman, S.; et al. A phase II/III, double-blind, randomized trial comparing maInt.enance lapatinib versus placebo after first line chemotherapy in HER1/2 positive metastatic bladder cancer patients. J. Clin. Oncol. 2015, 33, 4505. [Google Scholar] [CrossRef]
- Oudard, S.; Culine, S.; Vano, Y.; Goldwasser, F.; Théodore, C.; Nguyen, T.; Voog, E.; Banu, E.; Vieillefond, A.; Priou, F.; et al. Multicentre randomised phase II trial of gemcitabine+platinum, with or without trastuzumab, in advanced or metastatic urothelial carcinoma overexpressing Her2. Eur. J. Cancer 2015, 51, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.S.; Wang, K.; Khaira, D.; Ali, S.M.; Fisher, H.A.; Mian, B.; Nazeer, T.; Elvin, J.A.; Palma, N.; Yelensky, R.; et al. Comprehensive genomic profiling of 295 cases of clinically advanced urothelial carcinoma of the urinary bladder reveals a high frequency of clinically relevant genomic alterations. Cancer 2016, 122, 702–711. [Google Scholar] [CrossRef]
- Ross, J.S.; Wang, K.; Gay, L.M.; Al-Rohil, R.N.; Nazeer, T.; Sheehan, C.E.; Jennings, T.A.; Otto, G.A.; Donahue, A.; He, J.; et al. A high frequency of activating extracellular domain ERBB2 (HER2) mutation in micropapillary urothelial carcinoma. Clin. Cancer Res. 2014, 20, 68–75. [Google Scholar]
- Kamat, A.M.; Dinney, C.P.; Gee, J.R.; Grossman, H.B.; Siefker-Radtke, A.O.; Tamboli, P.; Detry, M.A.; Robinson, T.L.; Pisters, L.L. Micropapillary bladder cancer: A review of the University of Texas M. D. Anderson Cancer Center experience with 100 consecutive patients. Cancer 2007, 110, 62–67. [Google Scholar] [CrossRef]
- Kiss, B.; Wyatt, A.W.; Douglas, J.; Skuginna, V.; Mo, F.; Anderson, S.; Rotzer, D.; Fleischmann, A.; Genitsch, V.; Hayashi, T.; et al. Her2 alterations in muscle-invasive bladder cancer: Patient selection beyond protein expression for targeted therapy. Sci. Rep. 2017, 7, 42713. [Google Scholar] [CrossRef]
- Braicu, C.; Buse, M.; Busuioc, C.; Drula, R.; Gulei, D.; Raduly, L.; Rusu, A.; Irimie, A.; Atanasov, A.G.; Slaby, O.; et al. A Comprehensive Review on MAPK: A Promising Therapeutic Target in Cancer. Cancers 2019, 11, 1618. [Google Scholar] [CrossRef]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef]
- Felsenstein, K.M.; Theodorescu, D. Precision medicine for urothelial bladder cancer: Update on tumour genomics and immunotherapy. Nat. Rev. Urol. 2018, 15, 92–111. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Pearson, A.; Campbell, J.; Shnyder, S.D.; Knowles, M.A.; Ashworth, A.; Turner, N.C. Parallel RNA Int.erference screens identify EGFR activation as an escape mechanism in FGFR3-mutant cancer. Cancer Discov. 2013, 3, 1058–1071. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Šuštić, T.; Leite de Oliveira, R.; Lieftink, C.; Halonen, P.; van de Ven, M.; Beijersbergen, R.L.; van den Heuvel, M.M.; Bernards, R.; van der Heijden, M.S. A Functional Genetic Screen Identifies the Phosphoinositide 3-kinase Pathway as a Determinant of Resistance to Fibroblast Growth Factor Receptor Inhibitors in FGFR Mutant Urothelial Cell Carcinoma. Eur. Urol. 2017, 71, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Datta, J.; Damodaran, S.; Parks, H.; Ocrainiciuc, C.; Miya, J.; Yu, L.; Gardner, E.P.; Samorodnitsky, E.; Wing, M.R.; Bhatt, D.; et al. Akt Activation Mediates Acquired Resistance to Fibroblast Growth Factor Receptor Inhibitor BGJ398. Mol. Cancer Ther. 2017, 16, 614–624. [Google Scholar] [CrossRef] [PubMed]
- Bellmunt, J.; Lalani, A.K.A.; Jacobus, S.; Wankowicz, S.A.; Polacek, L.; Takeda, D.Y.; Harshman, L.C.; Wagle, N.; Moreno, I.; Lundgren, K.; et al. Everolimus and pazopanib (E/P) benefit genomically selected patients with metastatic urothelial carcinoma. Br. J. Cancer 2018, 119, 707–712. [Google Scholar] [CrossRef]
- Necchi, A.; Lo Vullo, S.; Raggi, D.; Perrone, F.; Giannatempo, P.; Calareso, G.; Togliardi, E.; Nicolai, N.; Piva, L.; Biasoni, D.; et al. Neoadjuvant sorafenib, gemcitabine, and cisplatin administration preceding cystectomy in patients with muscle-invasive urothelial bladder carcinoma: An open-label, single-arm, single-center, phase 2 study. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2018; Volume 36, pp. 8.e1–8.e8. [Google Scholar] [CrossRef]
- Shah, C.H.; Pappot, H.; Agerbæk, M.; Holmsten, K.; Jäderling, F.; Yachnin, J.; Grybäck, P.; von der Maase, H.; Ullén, A. Safety and Activity of Sorafenib in Addition to Vinflunine in Post-Platinum Metastatic Urothelial Carcinoma (Vinsor): Phase I Trial. Oncologist 2019, 24, 745. [Google Scholar] [CrossRef] [PubMed]
- Apte, R.S.; Chen, D.S.; Ferrara, N. VEGF in Signaling and Disease: Beyond Discov.ery and Development. Cell 2019, 176, 1248–1264. [Google Scholar] [CrossRef]
- Ferrara, N.; Adamis, A.P. Ten years of anti-vascular endothelial growth factor therapy. Nat. Rev. Drug Discov. 2016, 15, 385–403. [Google Scholar]
- Hahn, N.M.; Stadler, W.M.; Zon, R.T.; Waterhouse, D.; Picus, J.; Nattam, S.; Johnson, C.S.; Perkins, S.M.; Waddell, M.J.; Sweeney, C.J.; et al. Phase II trial of cisplatin, gemcitabine, and bevacizumab as first-line therapy for metastatic urothelial carcinoma: Hoosier Oncol.ogy Group GU 04-75. J. Clin. Oncol. 2011, 29, 1525–1530. [Google Scholar] [CrossRef]
- Rosenberg, J.E.; Ballman, K.V.; Halabi, S.; Watt, C.; Hahn, O.M.; Steen, P.D.; Dreicer, R.; Flaig, T.W.; Stadler, W.M.; Sweeney, C.; et al. CALGB 90601 (Alliance): Randomized, double-blind, placebo-controlled phase III trial comparing gemcitabine and cisplatin with bevacizumab or placebo in patients with metastatic urothelial carcinoma. J. Clin. Oncol. 2019, 37, 4503. [Google Scholar] [CrossRef]
- Yakes, F.M.; Chen, J.; Tan, J.; Yamaguchi, K.; Shi, Y.; Yu, P.; Qian, F.; Chu, F.; Bentzien, F.; Cancilla, B.; et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 2011, 10, 2298–2308. [Google Scholar] [CrossRef]
- Nadal, R.; Mortazavi, A.; Stein, M.; Pal, S.K.; Davarpanah, N.; Parnes, H.L.; Ning, Y.M.; Cordes, L.M.; Lin, J.; Bagheri, M.; et al. Final results of a phase I study of cabozantinib (cabo) plus nivolumab (nivo) and cabonivo plus ipilimumab (Ipi) in patients (pts) with metastatic urothelial carcinoma (mUC) and oTher. genitourinary (GU) malignancies. Ann. Oncol. 2017, 28, v295. [Google Scholar] [CrossRef]
- Nadal, R.M.; Mortazavi, A.; Stein, M.; Pal, S.K.; Davarpanah, N.N.; Parnes, H.L.; Ning, Y.M.; Cordes, L.M.; Bagheri, M.H.; Lindenberg, L.; et al. Results of phase i plus expansion cohorts of cabozantinib (Cabo) plus nivolumab (Nivo) and CaboNivo plus ipilimumab (Ipi) in patients (PTS) with with metastatic UROTHELIAL CARCINOMA (mUC) and oTher. genitourinary (GU) malignancies. J. Clin. Oncol. 2018, 36, 515. [Google Scholar] [CrossRef]
- Necchi, A.; Giannatempo, P.; Raggi, D.; Anichini, A.; Calareso, G.; Crippa, F.; Mariani, L. Cabozantinib (CABO) plus durvalumab (DURVA) in patients with advanced and chemoTher.apy-treated bladder carcinoma of urothelial and non-urothelial histology: An open-label, single-arm, phase 2 trial. J. Clin. Oncol. 2018, 36, TPS536. [Google Scholar] [CrossRef]
- Raggi, D.; Giannatempo, P.; Anichini, A.; Calareso, G.; Crippa, F.; Mariani, L.; Necchi, A. Cabozantinib (CABO) plus durvalumab (DURVA) in patients with advanced and chemoTher.apy-treated bladder carcinoma, of urothelial and non-urothelial histology: The open-label, single-arm, phase 2 ARCADIA trial. Eur. Urol. Suppl. 2018, 17, e1150. [Google Scholar] [CrossRef]
- Maia, M.C.; Agarwal, N.; McGregor, B.A.; Vaishampayan, U.N.; Choueiri, T.K.; Green, M.C.; Hessel, C.; Scheffold, C.; Schwab, G.; Powles, T.; et al. Phase 1b trial of cabozantinib in combination with atezolizumab in patients with locally advanced or metastatic urothelial carcinoma (UC) or renal cell carcinoma (RCC). J. Clin. Oncol. 2018, 36, TPS42. [Google Scholar] [CrossRef]
- Pal, S.K.; Vaishampayan, U.N.; Castellano, D.E.; Necchi, A.; Van Herpen, C.M.L.; Ramsingh, G.; Loriot, Y.; Agarwal, N. Phase lb (COSMIC-021) trial of cabozantinib (C) in urothelial carcinoma (UC) or C in combination with atezolizumab (A) in patients (pts) with UC, castrate resistant prostate cancer (CRPC) or renal cell carcinoma (RCC). J. Clin. Oncol. 2019, 37, TPS683. [Google Scholar] [CrossRef]
- Powles, T.; Choueiri, T.K.; Agarwal, N.; Necchi, A.; Loriot, Y.; McKay, R.R.; Chang, P.Y.; Kondo, A.; Wang, E.; McGregor, B.; et al. Phase 1b Trial of Cabozantinib in Combination with Atezolizumab in Patients with Locally Advanced or Metastatic Renal Cell Carcinoma and Urothelial Carcinoma. Eur. Urol. Suppl. 2017, 16, e2789. [Google Scholar] [CrossRef]
- Agarwal, N.; Loriot, Y.; McGregor, B.A.; Dreicer, R.; Dorff, T.B.; Maughan, B.L.; Kelly, W.K.; Pagliaro, L.C.; Srinivas, S.; Squillante, C.M.; et al. Cabozantinib (C) in combination with atezolizumab (A) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC): Results of Cohort 6 of the COSMIC-021 Study. J. Clin. Oncol. 2020, 38, 139. [Google Scholar] [CrossRef]
- O‘Mahony, D.; Bishop, M.R. Monoclonal antibody Therapy. Front. BioSci. 2006, 11, 1620–1635. [Google Scholar] [CrossRef]
- Lucas, A.T.; Price, L.S.L.; Schorzman, A.N.; Storrie, M.; PiScitelli, J.A.; Razo, J.; Zamboni, W.C. Factors Affecting the Pharmacology of Antibody-Drug Conjugates. Antibodies 2018, 7, 10. [Google Scholar] [CrossRef]
- Khongorzul, P.; Ling, C.J.; Khan, F.U.; Ihsan, A.U.; Zhang, J. Antibody–Drug Conjugates: A Comprehensive Review. Mol. Cancer Res. 2020, 18, 3–19. [Google Scholar] [CrossRef]
- Tsuchikama, K.; An, Z. Antibody-drug conjugates: Recent advances in conjugation and linker chemistries. Protein Cell 2018, 9, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Petrylak, D.P.; Balar, A.V.; O’Donnell, P.H.; McGregor, B.A.; Heath, E.I.; Yu, E.Y.; Galsky, M.D.; Hahn, N.M.; Gartner, E.M.; Pinelli, J.; et al. EV-201: Results of enfortumab vedotin monotherapy for locally advanced or metastatic urothelial cancer previously treated with platinum and immune checkpoint inhibitors. J. Clin. Oncol. 2019, 37, 4505. [Google Scholar] [CrossRef]
- Hanna, K.S. Clinical Overview of Enfortumab Vedotin in the Management of Locally Advanced or Metastatic Urothelial Carcinoma. Drugs 2020, 80, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Shen, L.; Wang, W.; Fang, J. Safety, pharmacokinetics and efficacy of RC48-ADC in a phase I study in patients with HER2-overexpression advanced solid cancer. J. Clin. Oncol. 2018, 36, e16059. [Google Scholar] [CrossRef]
- Sheng, X.; Zhou, A.-P.; Yao, X.; Shi, Y.; Luo, H.; Shi, B.; Liu, J.; Yu, G.; He, Z.; Hu, C.; et al. A phase II study of RC48-ADC in HER2-positive patients with locally advanced or metastatic urothelial carcinoma. J. Clin. Oncol. 2019, 37, 4509. [Google Scholar] [CrossRef]
- Li, B.T.; Makker, V.; Buonocore, D.J.; Offin, M.D.; Olah, Z.T.; Panora, E.; Shen, R.; Ho, A.L.; Yaeger, R.; Iyer, G.; et al. A multi-histology basket trial of ado-trastuzumab emtansine in patients with HER2 amplified cancers. J. Clin. Oncol. 2018, 36, 2502. [Google Scholar] [CrossRef]
- Strzebonska, K.; Waligora, M. Umbrella and basket trials in Oncol.ogy: Ethical challenges. BMC Med. Ethics 2019, 20, 58. [Google Scholar] [CrossRef]
- Bahleda, R.; Italiano, A.; Hierro, C.; Mita, A.; Cervantes, A.; Chan, N.; Awad, M.; Calvo, E.; Moreno, V.; Govindan, R.; et al. Multicenter Phase I Study of Erdafitinib (JNJ-42756493), Oral Pan-Fibroblast Growth Factor Receptor Inhibitor, in Patients with Advanced or Refractory Solid Tumors. Clin. Cancer Res. 2019, 25, 4888–4897. [Google Scholar] [CrossRef]
- Faltas, B.M.; Prandi, D.; Tagawa, S.T.; Molina, A.M.; Nanus, D.M.; Sternberg, C.; Rosenberg, J.; Mosquera, J.M.; Robinson, B.; Elemento, O.; et al. Clonal evolution of chemoTher.apy-resistant urothelial carcinoma. Nat. Genet. 2016, 48, 1490–1499. [Google Scholar] [CrossRef]
- Nordentoft, I.; Lamy, P.; Birkenkamp-Demtröder, K.; Shumansky, K.; Vang, S.; Hornshøj, H.; Juul, M.; Villesen, P.; Hedegaard, J.; Roth, A.; et al. Mutational context and diverse clonal development in early and late bladder cancer. Cell Rep. 2014, 7, 1649–1663. [Google Scholar] [CrossRef] [PubMed]
- Lamy, P.; Nordentoft, I.; Birkenkamp-Demtröder, K.; Thomsen, M.B.; Villesen, P.; Vang, S.; Hedegaard, J.; Borre, M.; Jensen, J.B.; Høyer, S.; et al. Paired Exome Analysis Reveals Clonal Evolution and Potential Ther.apeutic Targets in Urothelial Carcinoma. Cancer Res. 2016, 76, 5894–5906. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, M.B.; Nordentoft, I.; Lamy, P.; Høyer, S.; Vang, S.; Hedegaard, J.; Borre, M.; Jensen, J.B.; Ørntoft, T.F.; Dyrskjøt, L. Spatial and temporal clonal evolution during development of metastatic urothelial carcinoma. Mol. Oncol. 2016, 10, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Birkenkamp-Demtröder, K.; Christensen, E.; Nordentoft, I.; Knudsen, M.; Taber, A.; Høyer, S.; Lamy, P.; Agerbæk, M.; Jensen, J.B.; Dyrskjøt, L. Monitoring Treatment Response and Metastatic Relapse in Advanced Bladder Cancer by Liquid Biopsy Analysis. Eur. Urol. 2018, 73, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.; Birkenkamp-Demtröder, K.; Sethi, H.; Shchegrova, S.; Salari, R.; Nordentoft, I.; Wu, H.-T.; Knudsen, M.; Lamy, P.; Lindskrog, S.V.; et al. Early Detection of Metastatic Relapse and Monitoring of Ther.apeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients With Urothelial Bladder Carcinoma. J. Clin. Oncol. 2019, 37, 1547–1557. [Google Scholar] [CrossRef] [PubMed]
- Christensen, E.; Birkenkamp-Demtröder, K.; Nordentoft, I.; Høyer, S.; van der Keur, K.; van Kessel, K.; Zwarthoff, E.; Agerbæk, M.; Ørntoft, T.F.; Jensen, J.B.; et al. Liquid Biopsy Analysis of FGFR3 and PIK3CA Hotspot Mutations for Disease Surveillance in Bladder Cancer. Eur. Urol. 2017, 71, 961–969. [Google Scholar] [CrossRef] [PubMed]
- Vandekerkhove, G.; Todenhöfer, T.; Annala, M.; Struss, W.J.; Wong, A.; Beja, K.; Ritch, E.; Brahmbhatt, S.; Volik, S.V.; Hennenlotter, J.; et al. Circulating Tumor DNA Reveals Clinically Actionable Somatic Genome of Metastatic Bladder Cancer. Clin. Cancer Res. 2017, 23, 6487–6497. [Google Scholar] [CrossRef] [PubMed]


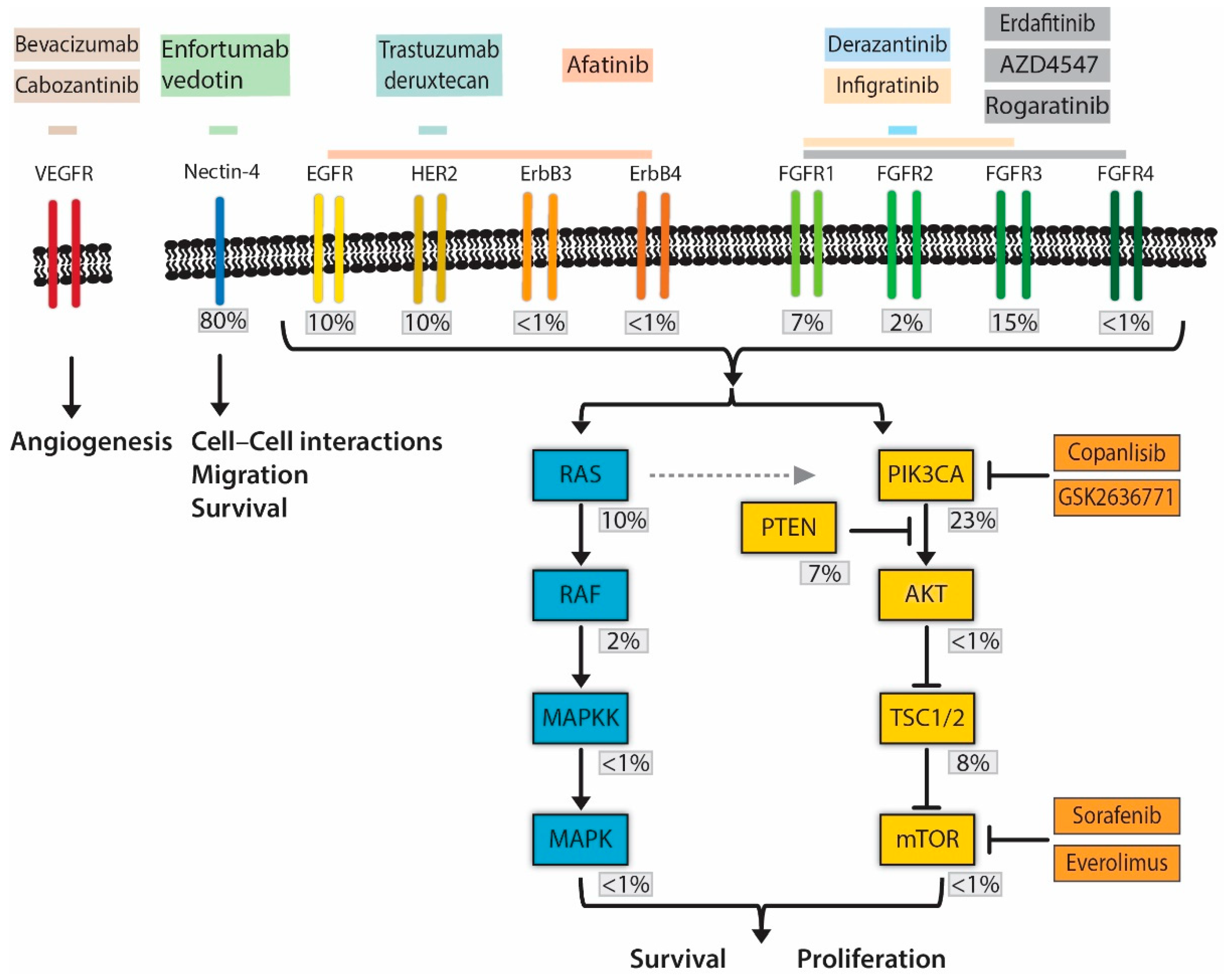{kind=link}
| Drug | Target | Clinical Trials |
|---|---|---|
| erdafitinib | FGFR1–4 | NCT03473743, NCT02465060 (MATCH) |
| infigratinib (BGJ-398) | FGFR1–3 | NCT04197986 (PROOF 302), |
| AZD4547 | FGFR1–4, Kit, CSF1R | NCT02546661 (BISCAY), NCT02465060 (MATCH) |
| rogaratinib (BAY 1163877) | FGFR1–4 | NCT03410693 (FORT-1) |
| derazantinib (ARQ 087) | FGFR2 | NCT04045613 (FIDES-02) |
| afatinib (BIBW2992) | ErbB1–4 | NCT02780687 (LUX Bladder-1) |
| trastuzumab deruxtecan (DS-8201a) | Erbb2 | NCT03523572 |
| cabozantinib (XL184) | VEGFR, c-MET, AXL, RET | NCT01688999, NCT03866382, NCT03824691 (ARCADIA), NCT03170960 (COSMIC-021) |
| copanlisib | mTOR | NCT02465060 (MATCH) |
| GSK2636771 | mTOR | NCT02465060 (MATCH) |
| valemetostat (DS3201) | EZH2 | NCT04388852 |
| tazemetostat (EPZ-6438) | EZH2 | NCT03854474 |
| rucaparib | PARP1 | NCT03397394 (ATLAS) |
| olaparib | PARP1 | NCT03375307, NCT02546661 (BISCAY) |
| entinostat | HDAC I | NCT03978624 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scholtes, M.; Akbarzadeh, M.; Zwarthoff, E.; Boormans, J.; Mahmoudi, T.; Zuiverloon, T. Targeted Therapy in Metastatic Bladder Cancer: Present Status and Future Directions. Appl. Sci. 2020, 10, 7102. https://doi.org/10.3390/app10207102
Scholtes M, Akbarzadeh M, Zwarthoff E, Boormans J, Mahmoudi T, Zuiverloon T. Targeted Therapy in Metastatic Bladder Cancer: Present Status and Future Directions. Applied Sciences. 2020; 10(20):7102. https://doi.org/10.3390/app10207102
Chicago/Turabian StyleScholtes, Mathijs, Maryam Akbarzadeh, Ellen Zwarthoff, Joost Boormans, Tokameh Mahmoudi, and Tahlita Zuiverloon. 2020. "Targeted Therapy in Metastatic Bladder Cancer: Present Status and Future Directions" Applied Sciences 10, no. 20: 7102. https://doi.org/10.3390/app10207102
APA StyleScholtes, M., Akbarzadeh, M., Zwarthoff, E., Boormans, J., Mahmoudi, T., & Zuiverloon, T. (2020). Targeted Therapy in Metastatic Bladder Cancer: Present Status and Future Directions. Applied Sciences, 10(20), 7102. https://doi.org/10.3390/app10207102