Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3


 , ,
, , 

Abstract
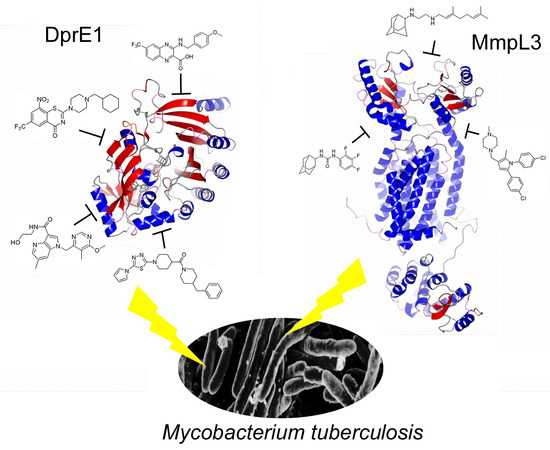
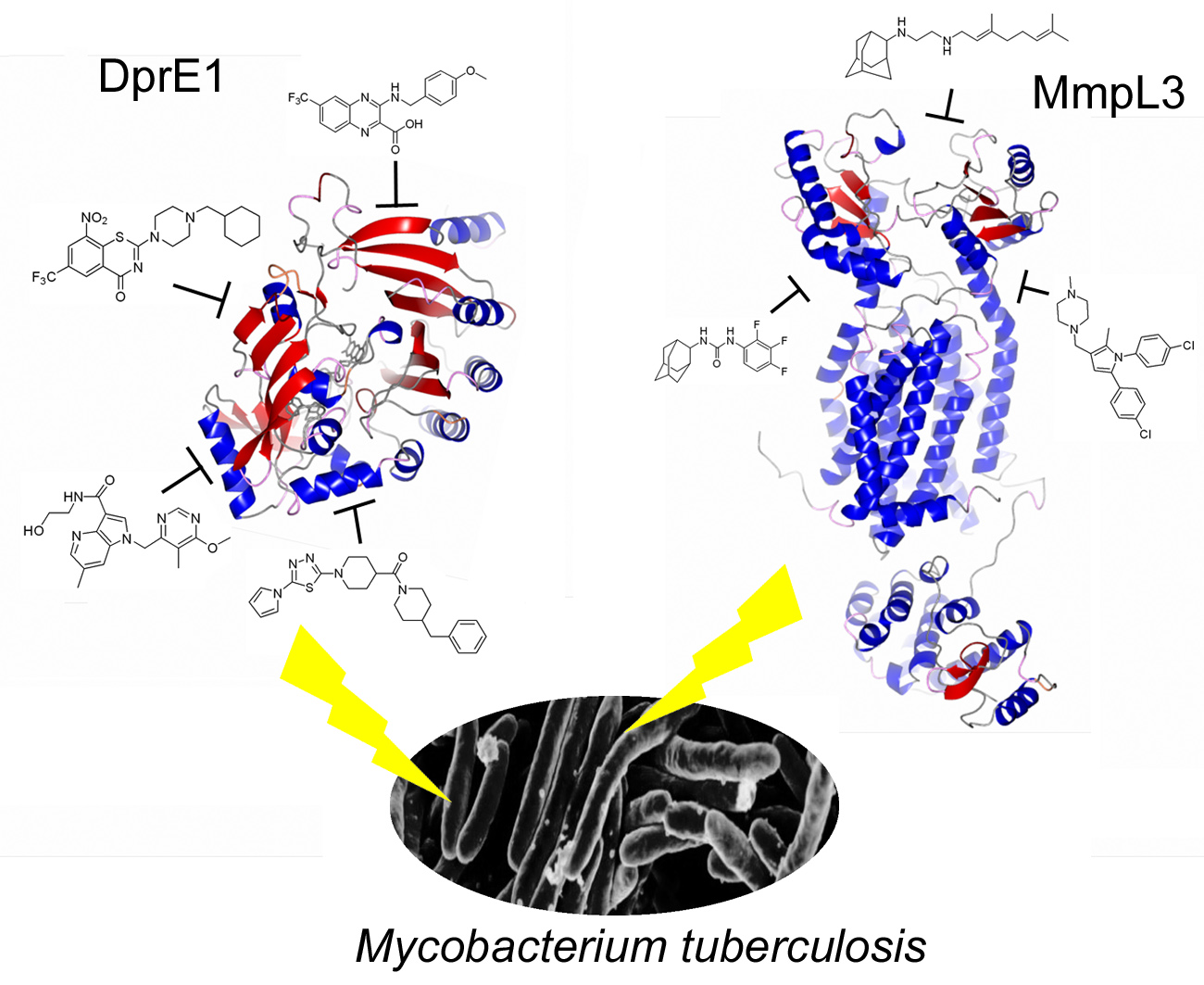
1. The Added Value of Promiscuous Targets for Antitubercular Drug Discovery
2. The First Discovery of DprE1 and MmpL3 as Drug Target
3. DprE1: The Hot TB Target of the Moment
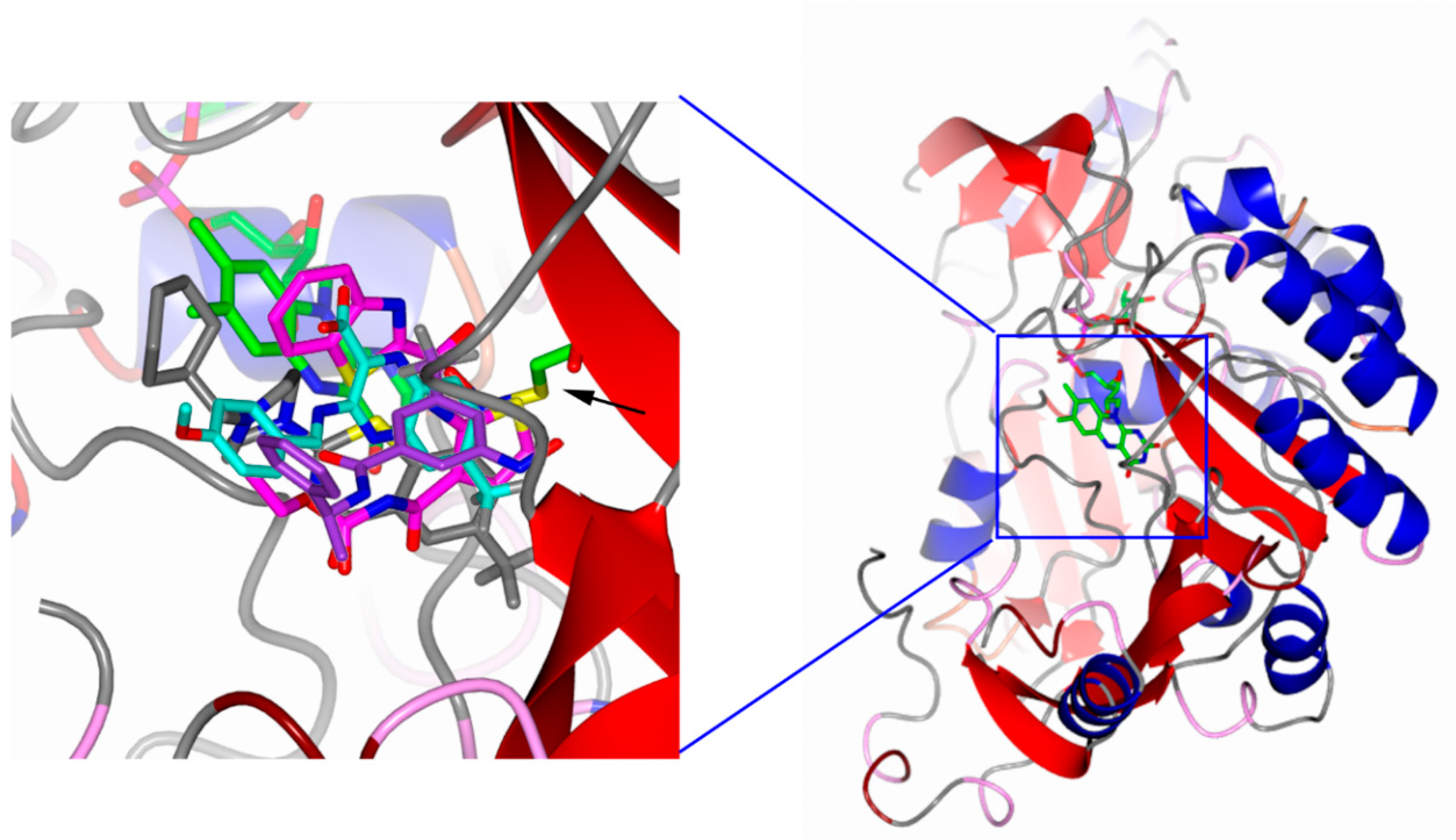
3.1. Why DprE1 Is a Promiscuous Target
3.2. DprE1 Inhibitors, Which Are the Current Status and Future Perspectives
3.3. DprE2: A Promising Target for Future Drug Discovery
4. Mmpl3 Transporter: The Other Hot TB Target of the Moment
4.1. MmpL3: A Mycolic Acid Transporter
4.2. MmpL3: The Achilles Heel of Mycobacterium Tuberculosis
5. Future Perspectives for DprE1 and MmpL3 Inhibitors in Clinical Therapy
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report 2019. Available online: https://www.who.int/tb/publications/global_report/en/ (accessed on 7 January 2020).
- Lechartier, B.; Rybniker, J.; Zumla, A.; Cole, S.T. Tuberculosis drug discovery in the post-post-genomic era. EMBO Mol. Med. 2014, 6, 158–168. [Google Scholar] [CrossRef]
- Chiarelli, L.R.; Mori, G.; Esposito, M.; Orena, B.S.; Pasca, M.R. New and old hot drug targets in tuberculosis. Curr. Med. Chem. 2016, 23, 3813–3846. [Google Scholar] [CrossRef]
- Meneghetti, F.; Villa, S.; Gelain, A.; Barlocco, D.; Chiarelli, L.R.; Pasca, M.R.; Costantino, L. Iron acquisition pathways as targets for antitubercular drugs. Curr. Med. Chem. 2016, 23, 4009–4026. [Google Scholar] [CrossRef]
- Mori, M.; Sammartino, J.C.; Costantino, L.; Gelain, A.; Meneghetti, F.; Villa, S.; Chiarelli, L.R. An Overview on the Potential Antimycobacterial Agents Targeting Serine/Threonine Protein Kinases from Mycobacterium tuberculosis. Curr. Top. Med. Chem. 2019, 19, 646–661. [Google Scholar] [CrossRef] [PubMed]
- Andries, K.; Verhasselt, P.; Guillemont, J.; Göhlmann, H.W.; Neefs, J.M.; Winkler, H.; Van Gestel, J.; Timmerman, P.; Zhu, M.; Lee, E.; et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 2005, 307, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, M.; Hashizume, H.; Tomishige, T.; Kawasaki, M.; Tsubouchi, H.; Sasaki, H.; Shimokawa, Y.; Komatsu, M. OPC-67683, a nitro-dihydro-imidazooxazole derivative with promising action against tuberculosis in vitro and in mice. PLoS Med. 2006, 3, e466. [Google Scholar] [CrossRef] [PubMed]
- Stover, C.K.; Warrener, P.; VanDevanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Ryan, N.J.; Lo, J.H. Delamanid: First global approval. Drugs 2014, 74, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.J. Pretomanid: First Approval. Drugs 2019, 79, 1797–1803. [Google Scholar] [CrossRef]
- Cole, S.T. Inhibiting Mycobacterium tuberculosis within and without. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371, 20150506. [Google Scholar] [CrossRef]
- Lee, B.S.; Pethe, K. Therapeutic potential of promiscuous targets in Mycobacterium tuberculosis. Curr. Opin. Pharmacol. 2018, 42, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Campaniço, A.; Moreira, R.; Lopes, F. Drug discovery in tuberculosis. New drug targets and antimycobacterial agents. Eur. J. Med. Chem. 2018, 150, 525–545. [Google Scholar] [CrossRef] [PubMed]
- Mikusová, K.; Huang, H.; Yagi, T.; Holsters, M.; Vereecke, D.; D’Haeze, W.; Scherman, M.S.; Brennan, P.J.; McNeil, M.R.; Crick, D.C. Decaprenylphosphoryl arabinofuranose, the donor of the D-arabinofuranosyl residues of mycobacterial arabinan, is formed via a two-step epimerization of decaprenylphosphoryl ribose. J. Bacteriol. 2005, 187, 8020–8025. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Meshcheryakov, V.A.; Poce, G.; Chng, S.S. MmpL3 is the flippase for mycolic acids in mycobacteria. Proc. Natl. Acad. Sci. USA 2017, 114, 7993–7998. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Upadhyay, A.; Fontes, F.L.; North, E.J.; Wang, Y.; Crans, D.C.; Grzegorzewicz, A.E.; Jones, V.; Franzblau, S.G.; Lee, R.E.; et al. Novel insights into the mechanism of inhibition of MmpL3, a target of multiple pharmacophores in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 6413–6423. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Cox, J.A.; Spivey, V.L.; Loman, N.J.; Pallen, M.J.; Constantinidou, C.; Fernández, R.; Alemparte, C.; Remuiñán, M.J.; Barros, D.; et al. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE 2012, 7, e52951. [Google Scholar] [CrossRef]
- Pethe, K.; Bifani, P.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Wilson, R.; Kumar, P.; Parashar, V.; Vilchèze, C.; Veyron-Churlet, R.; Freundlich, J.S.; Barnes, S.W.; Walker, J.R.; Szymonifka, M.J.; Marchiano, E.; et al. Antituberculosis thiophenes define a requirement for Pks13 in mycolic acid biosynthesis. Nat. Chem. Biol. 2013, 9, 499–506. [Google Scholar] [CrossRef]
- Aggarwal, A.; Parai, M.K.; Shetty, N.; Wallis, D.; Woolhiser, L.; Hastings, C.; Dutta, N.K.; Galaviz, S.; Dhakal, R.C.; Shrestha, R.; et al. Development of a Novel Lead that Targets M. tuberculosis Polyketide Synthase 13. Cell 2017, 170, 249–259. [Google Scholar] [CrossRef]
- Grant, S.S.; Kawate, T.; Nag, P.P.; Silvis, M.R.; Gordon, K.; Stanley, S.A.; Kazyanskaya, E.; Nietupski, R.; Golas, A.; Fitzgerald, M.; et al. Identification of novel inhibitors of nonreplicating Mycobacterium tuberculosis using a carbon starvation model. ACS Chem. Biol. 2013, 8, 2224–2234. [Google Scholar] [CrossRef]
- Bonnett, S.A.; Dennison, D.; Files, M.; Bajpai, A.; Parish, T. A class of hydrazones are active against non-replicating Mycobacterium tuberculosis. PLoS ONE 2018, 13, e0198059. [Google Scholar] [CrossRef] [PubMed]
- Boldrin, F.; Degiacomi, G.; Serafini, A.; Kolly, G.S.; Ventura, M.; Sala, C.; Provvedi, R.; Palù, G.; Cole, S.T.; Manganelli, R. Promoter mutagenesis for fine-tuning expression of essential genes in Mycobacterium tuberculosis. Microb. Biotechnol. 2018, 11, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Lupien, A.; Vocat, A.; Foo, C.S.; Blattes, E.; Gillon, J.Y.; Makarov, V.; Cole, S.T. Optimized background regimen for treatment of active tuberculosis with the next-generation benzothiazinone Macozinone (PBTZ169). Antimicrob. Agents Chemother. 2018, 62, e00840-18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Li, J.; Yang, X.; Wu, L.; Zhang, J.; Yang, Y.; Zhao, Y.; Zhang, L.; Cheng, X.; Liu, Z.; et al. Crystal structures of membrane transporter MmpL3, an anti-TB drug target. Cell 2019, 176, 636–648. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.newtbdrugs.org/pipeline/compound/sq109 (accessed on 4 December 2019).
- Available online: https://www.newtbdrugs.org/pipeline/compound/tba-7371 (accessed on 4 December 2019).
- Available online: https://www.newtbdrugs.org/pipeline/trials/phase-2-telacebec-q203-eba (accessed on 4 December 2019).
- Available online: https://www.newtbdrugs.org/pipeline/compound/macozinone-mcz-pbtz-169 (accessed on 4 December 2019).
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Wolucka, B.A. Biosynthesis of D-arabinose in mycobacteria—A novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 2008, 275, 2691–2711. [Google Scholar] [CrossRef]
- Christophe, T.; Jackson, M.; Jeon, H.K.; Fenistein, D.; Contreras-Dominguez, M.; Kim, J.; Genovesio, A.; Carralot, J.P.; Ewann, F.; Kim, E.H.; et al. High content screening identifies decaprenyl-phosphoribose 2′ epimerase as a target for intracellular antimycobacterial inhibitors. PLoS Pathog. 2009, 5, e1000645. [Google Scholar] [CrossRef]
- Magnet, S.; Hartkoorn, R.C.; Székely, R.; Pató, J.; Triccas, J.A.; Schneider, P.; Szántai-Kis, C.; Orfi, L.; Chambon, M.; Banfi, D.; et al. Leads for antitubercular compounds from kinase inhibitor library screens. Tuberculosis 2010, 90, 354–360. [Google Scholar] [CrossRef]
- Matsoso, L.G.; Kana, B.D.; Crellin, P.K.; Lea-Smith, D.J.; Pelosi, A.; Powell, D.; Dawes, S.S.; Rubin, H.; Coppel, R.L.; Mizrahi, V. Function of the cytochrome bc1-aa3 branch of the respiratory network in mycobacteria and network adaptation occurring in response to its disruption. J. Bacteriol. 2005, 187, 6300–6308. [Google Scholar] [CrossRef]
- Kolly, G.S.; Boldrin, F.; Sala, C.; Dhar, N.; Hartkoorn, R.C.; Ventura, M.; Serafini, A.; McKinney, J.D.; Manganelli, R.; Cole, S.T. Assessing the essentiality of the decaprenyl-phospho-d-arabinofuranose pathway in Mycobacterium tuberculosis using conditional mutants. Mol. Microbiol. 2014, 92, 194–211. [Google Scholar] [CrossRef]
- La Rosa, V.; Poce, G.; Canseco, J.O.; Buroni, S.; Pasca, M.R.; Biava, M.; Raju, R.M.; Porretta, G.C.; Alfonso, S.; Battilocchio, C.; et al. MmpL3 is the cellular target of the antitubercular pyrrole derivative BM212. Antimicrob. Agents Chemother. 2012, 56, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Tahlan, K.; Wilson, R.; Kastrinsky, D.B.; Arora, K.; Nair, V.; Fischer, E.; Barnes, S.W.; Walker, J.R.; Alland, D.; Barry, C.E.; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar] [CrossRef] [PubMed]
- Grzegorzewicz, A.E.; Pham, H.; Gundi, V.A.; Scherman, M.S.; North, E.J.; Hess, T.; Jones, V.; Gruppo, V.; Born, S.E.; Korduláková, J.; et al. Inhibition of mycolic acid transport across the Mycobacterium tuberculosis plasma membrane. Nat. Chem. Biol. 2012, 8, 334–341. [Google Scholar] [CrossRef]
- Degiacomi, G.; Benjak, A.; Madacki, J.; Boldrin, F.; Provvedi, R.; Palù, G.; Kordulakova, J.; Cole, S.T.; Manganelli, R. Essentiality of mmpL3 and impact of its silencing on Mycobacterium tuberculosis gene expression. Sci. Rep. 2017, 7, 43495. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef]
- Neres, J.; Pojer, F.; Molteni, E.; Chiarelli, L.R.; Dhar, N.; Boy-Rottger, S.; Buroni, S.; Fullam, E.; Degiacomi, G.; Lucarelli, A.P.; et al. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci. Trans. Med. 2012, 4, 150ra121. [Google Scholar] [CrossRef]
- Batt, S.M.; Cacho Izquierdo, M.; Castro Pichel, J.; Stubbs, C.J.; Del Peral, L.V.-G.; Pérez-Herrán, E.; Dhar, N.; Mouzon, B.; Rees, M.; Hutchinson, J.P.; et al. Whole cell target engagement identifies novel inhibitors of Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose oxidase. ACS Infect. Dis 2015, 1, 615–626. [Google Scholar] [CrossRef]
- Batt, S.M.; Jabeen, T.; Bhowruth, V.; Quill, L.; Lund, P.A.; Eggeling, L.; Alderwick, L.J.; Fütterer, K.; Besra, G.S. Structural basis of inhibition of Mycobacterium tuberculosis DprE1 by benzothiazinone inhibitors. Proc. Natl. Acad. Sci. USA 2012, 109, 11354–11359. [Google Scholar] [CrossRef]
- Landge, S.; Mullick, A.B.; Nagalapur, K.; Neres, J.; Subbulakshmi, V.; Murugan, K.; Ghosh, A.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; et al. Discovery of benzothiazoles as antimycobacterial agents: Synthesis, structure-activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-β-d-ribose 2′-oxidase. Bioorg. Med. Chem. 2015, 23, 7694–7710. [Google Scholar] [CrossRef]
- Piton, J.; Foo, C.S.; Cole, S.T. Structural studies of Mycobacterium tuberculosis DprE1 interacting with its inhibitors. Drug Discov. Today 2017, 22, 526–533. [Google Scholar] [CrossRef]
- Chikhale, R.V.; Barmade, M.A.; Murumkar, P.R.; Yadav, M.R. Overview of the development of dpre1 inhibitors for combating the menace of tuberculosis. J. Med. Chem. 2018, 61, 8563–8593. [Google Scholar] [CrossRef] [PubMed]
- Mori, G.; Chiarelli, L.R.; Riccardi, G.; Pasca, M.R. New prodrugs against tuberculosis. Drug Discov. Today 2017, 22, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, C.; Škovierová, H.; Buroni, S.; Bobovská, A.; Nenci, S.; Molteni, E.; Pojer, F.; Pasca, M.R.; Makarov, V.; Cole, S.T.; et al. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-β-d-ribofuranose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; van der Sar, A.M.; Raadsen, S.A.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef]
- Richter, A.; Rudolph, I.; Möllmann, U.; Voigt, K.; Chung, C.W.; Singh, O.M.P.; Rees, M.; Mendoza-Losana, A.; Bates, R.; Ballell, L.; et al. Novel insight into the reaction of nitro, nitroso and hydroxylamino benzothiazinones and of benzoxacinones with Mycobacterium tuberculosis DprE1. Sci. Rep. 2018, 8, 13473. [Google Scholar] [CrossRef]
- Stanley, S.A.; Grant, S.S.; Kawate, T.; Iwase, N.; Shimizu, M.; Wivagg, C.; Silvis, M.; Kazyanskaya, E.; Aquadro, J.; Golas, A.; et al. Identification of novel inhibitors of M. tuberculosis growth using whole cell based high-throughput screening. ACS Chem. Biol. 2012, 7, 1377–1384. [Google Scholar] [CrossRef]
- De Jesus Lopes Ribeiro, A.L.; Degiacomi, G.; Ewann, F.; Buroni, S.; Incandela, M.L.; Chiarelli, L.R.; Mori, G.; Kim, J.; Contreras-Dominguez, M.; Park, Y.-S.; et al. Analogous mechanisms of resistance to benzothiazinones and dinitrobenzamides in Mycobacterium smegmatis. PLoS ONE 2011, 6, e26675. [Google Scholar] [CrossRef]
- Li, H.; Jogl, G. Crystal structure of decaprenylphosphoryl-β-d-ribose 2′-epimerase from Mycobacterium smegmatis. Proteins 2013, 81, 538–543. [Google Scholar] [CrossRef]
- Landge, S.; Ramachandran, V.; Kumar, A.; Neres, J.; Murugan, K.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; Vachaspati, P.; Raichurkar, A.; et al. Nitroarenes as antitubercular agents: Stereoelectronic modulation to mitigate mutagenicity. ChemMedChem 2016, 11, 331–339. [Google Scholar] [CrossRef]
- Wang, F.; Sambandan, D.; Halder, R.; Wang, J.; Batt, S.M.; Weinrick, B.; Ahmad, I.; Yang, P.; Zhang, Y.; Kim, J.; et al. Identification of a small molecule with activity against drug-resistant and persistent tuberculosis. Proc. Natl. Acad. Sci. USA 2013, 110, E2510–E2517. [Google Scholar] [CrossRef]
- Liu, R.; Lyu, X.; Batt, S.M.; Hsu, M.H.; Harbut, M.B.; Vilchèze, C.; Cheng, B.; Ajayi, K.; Yang, B.; Yang, Y.; et al. Determinants of the inhibition of DprE1 and CYP2C9 by antitubercular thiophenes. Angew. Chem. Int. Ed. Engl. 2017, 56, 13011–13015. [Google Scholar] [CrossRef] [PubMed]
- Piton, J.; Vocat, A.; Lupien, A.; Foo, C.S.; Riabova, O.; Makarov, V.; Cole, S.T. Structure-based drug design and characterization of sulfonyl-piperazine benzothiazinone inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00681-18. [Google Scholar] [CrossRef] [PubMed]
- Neres, J.; Hartkoorn, R.C.; Chiarelli, L.R.; Gadupudi, R.; Pasca, M.R.; Mori, G.; Venturelli, A.; Savina, S.; Makarov, V.; Kolly, G.S.; et al. 2-carboxyquinoxalines kill Mycobacterium tuberculosis through noncovalent inhibition of DprE1. ACS Chem. Biol. 2015, 10, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, K.A.; Besra, G.S. Mycobacterial cell wall biosynthesis: A multifaceted antibiotic target. Parasitology 2018, 145, 116–133. [Google Scholar] [CrossRef] [PubMed]
- Brecik, M.; Centárová, I.; Mukherjee, R.; Kolly, G.S.; Huszár, S.; Bobovská, A.; Kilacsková, E.; Mokošová, V.; Svetlíková, Z.; Šarkan, M.; et al. DprE1 is a vulnerable tuberculosis drug target due to its cell wall localization. ACS Chem. Biol. 2015, 10, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Chikhale, R.; Menghani, S.; Babu, R.; Bansode, R.; Bhargavi, G.; Karodia, N.; Rajasekharan, M.V.; Paradkar, A.; Khedekar, P. Development of selective DprE1 inhibitors: Design, synthesis, crystal structure and antitubercular activity of benzothiazolylpyrimidine-5-carboxamides. Eur. J. Med. Chem. 2015, 96, 30–46. [Google Scholar] [CrossRef]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Jena, L.K.; Koushik, K.; et al. 4-Aminoquinolone piperidine amides: Noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar] [CrossRef]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [CrossRef]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Prabhakar, K.R.; et al. 1,4-azaindole, a potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef]
- Tiwari, R.; Moraski, G.C.; Krchňák, V.; Miller, P.A.; Colon-Martinez, M.; Herrero, E.; Oliver, A.G.; Miller, M.J. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J. Am. Chem. Soc. 2013, 135, 3539–3549. [Google Scholar] [CrossRef]
- Manina, G.; Bellinzoni, M.; Pasca, M.R.; Neres, J.; Milano, A.; De Jesus Lopes Ribeiro, A.L.; Buroni, S.; Skovierová, H.; Dianišková, P.; Mikušová, K.; et al. Biological and structural characterization of the Mycobacterium smegmatis nitroreductase NfnB, and its role in benzothiazinone resistance. Mol. Microbiol. 2010, 77, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Neres, J.; Hartkoorn, R.C.; Ryabova, O.B.; Kazakova, E.; Šarkan, M.; Huszár, S.; Piton, J.; Kolly, G.S.; Vocat, A.; et al. The 8-Pyrrole-Benzothiazinones Are Noncovalent Inhibitors of DprE1 from Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4446–4452. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, R.; Miller, P.A.; Chiarelli, L.R.; Mori, G.; Šarkan, M.; Centárová, I.; Cho, S.; Mikusová, K.; Franzblau, S.G.; Oliver, A.G.; et al. Design, syntheses, and anti-tb activity of 1,3-benzothiazinone azide and click chemistry products inspired by BTZ043. ACS Med. Chem. Lett. 2016, 7, 266–270. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 inhibitors from scaffold morphing efforts, kill Mycobacterium tuberculosis and are efficacious in vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef]
- Shirude, P.S.; Shandil, R.K.; Manjunatha, M.R.; Sadler, C.; Panda, M.; Panduga, V.; Reddy, J.; Saralaya, R.; Nanduri, R.; Ambady, A.; et al. Lead optimization of 1,4-azaindoles as antimycobacterial agents. J. Med. Chem. 2014, 57, 5728–5737. [Google Scholar] [CrossRef]
- Jankute, M.; Byng, C.V.; Alderwick, L.J.; Besra, G.S. Elucidation of a protein-protein interaction network involved in Corynebacterium glutamicum cell wall biosynthesis as determined by bacterial two-hybrid analysis. Glycoconj. J. 2014, 31, 475–483. [Google Scholar] [CrossRef]
- Bhutani, I.; Loharch, S.; Gupta, P.; Madathil, R.; Parkesh, R. Structure, dynamics, and interaction of Mycobacterium tuberculosis (Mtb) DprE1 and DprE2 examined by molecular modeling, simulation, and electrostatic studies. PLoS ONE 2015, 10, e0119771. [Google Scholar] [CrossRef]
- Gawad, J.; Bonde, C. Decaprenyl-phosphoryl-ribose 2′-epimerase (DprE1): Challenging target for antitubercular drug discovery. Chem. Cent. J. 2018, 12, 72. [Google Scholar] [CrossRef]
- Camacho, L.R.; Constant, P.; Raynaud, C.; Laneelle, M.A.; Triccas, J.A.; Gicquel, B.; Daffe, M.; Guilhot, C. Analysis of the phthiocerol dimycocerosate locus of Mycobacterium tuberculosis. Evidence that this lipid is involved in the cell wall permeability barrier. J. Biol. Chem. 2001, 276, 19845–19854. [Google Scholar] [CrossRef]
- Converse, S.E.; Mougous, J.D.; Leavell, M.D.; Leary, J.A.; Bertozzi, C.R.; Cox, J.S. MmpL8 is required for sulfolipid-1 biosynthesis and Mycobacterium tuberculosis virulence. Proc. Natl. Acad. Sci. USA 2003, 100, 6121–6126. [Google Scholar] [CrossRef]
- Pacheco, S.A.; Hsu, F.F.; Powers, K.M.; Purdy, G.E. MmpL11 protein transports mycolic acid-containing lipids to the mycobacterial cell wall and contributes to biofilm formation in Mycobacterium smegmatis. J. Biol. Chem. 2013, 288, 24213–24222. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.M.; Jones, C.M.; Xi, Z.; Speer, A.; Danilchanka, O.; Doornbos, K.S.; Sun, P.; Wu, F.; Tian, C.; Niederweis, M. Discovery of a siderophore export system essential for virulence of Mycobacterium tuberculosis. PLoS Pathog. 2013, 9, e1003120. [Google Scholar] [CrossRef] [PubMed]
- Belardinelli, J.M.; Larrouy-Maumus, G.; Jones, V.; de Carvalho, L.P.S.; McNeil, M.R.; Jackson, M. Biosynthesis and translocation of unsulfated acyltrehaloses in Mycobacterium tuberculosis. J. Biol. Chem. 2014, 289, 27952–27965. [Google Scholar] [CrossRef] [PubMed]
- Domenech, P.; Reed, M.B.; Barry, C.E. Contribution of the Mycobacterium tuberculosis MmpL protein family to virulence and drug resistance. Infect. Immun. 2005, 73, 3492–3501. [Google Scholar] [CrossRef]
- Li, W.; Obregón-Henao, A.; Wallach, J.B.; North, E.J.; Lee, R.E.; Gonzalez-Juarrero, M.; Schnappinger, D.; Jackson, M. Therapeutic potential of the Mycobacterium tuberculosis mycolic acid transporter, MmpL3. Antimicrob. Agents Chemother. 2016, 60, 5198–5207. [Google Scholar] [CrossRef]
- Belardinelli, J.M.; Yazidi, A.; Yang, L.; Fabre, L.; Li, W.; Jacques, B.; Angala, S.K.; Rouiller, I.; Zgurskaya, H.I.; Sygusch, J.; et al. Structure-function profile of MmpL3, the essential mycolic acid transporter from Mycobacterium tuberculosis. ACS Infect. Dis. 2016, 2, 702–713. [Google Scholar] [CrossRef]
- Carel, C.; Nukdee, K.; Cantaloube, S.; Bonne, M.; Diagne, C.T.; Laval, F.; Daffé, M.; Zerbib, D. Mycobacterium tuberculosis proteins involved in mycolic acid synthesis and transport localize dynamically to the old growing pole and septum. PLoS ONE 2014, 9, e97148. [Google Scholar] [CrossRef]
- Belardinelli, J.M.; Stevens, C.M.; Li, W.; Tan, Y.Z.; Jones, V.; Mancia, F.; Zgurskaya, H.I.; Jackson, M. The MmpL3 interactome reveals a complex crosstalk between cell envelope biosynthesis and cell elongation and division in mycobacteria. Sci. Rep. 2019, 9, 10728. [Google Scholar] [CrossRef]
- Vilchèze, C.; Jacobs, W.R. The mechanism of isoniazid killing: Clarity through the scope of genetics. Annu. Rev. Microbiol. 2007, 61, 35–50. [Google Scholar] [CrossRef]
- Medjahed, H.; Reyrat, J.M. Construction of Mycobacterium abscessus defined glycopeptidolipid mutants: Comparison of genetic tools. Appl. Environ. Microbiol. 2009, 75, 1331–1338. [Google Scholar] [CrossRef]
- Seeliger, J.C.; Holsclaw, C.M.; Schelle, M.W.; Botyanszki, Z.; Gilmore, S.A.; Tully, S.E.; Niederweis, M.; Cravatt, B.F.; Leary, J.A.; Bertozzi, C.R. Elucidation and chemical modulation of sulfolipid-1 biosynthesis in Mycobacterium tuberculosis. J. Biol. Chem. 2012, 287, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Su, C.C.; Klenotic, P.A.; Bolla, J.R.; Purdy, G.E.; Robinson, C.V.; Yu, E.W. MmpL3 is a lipid transporter that binds trehalose monomycolate and phosphatidylethanolamine. Proc. Natl. Acad. Sci. USA 2019, 116, 11241–11246. [Google Scholar] [CrossRef] [PubMed]
- Murakami, S.; Nakashima, R.; Yamashita, E.; Yamaguchi, A. Crystal structure of bacterial multidrug efflux transporter AcrB. Nature 2002, 419, 587–593. [Google Scholar] [CrossRef]
- Sennhauser, G.; Bukowska, M.A.; Briand, C.; Grütter, M.G. Crystal structure of the multidrug exporter MexB from Pseudomonas aeruginosa. J. Mol. Biol. 2009, 389, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Bolla, J.R.; Su, C.C.; Do, S.V.; Radhakrishnan, A.; Kumar, N.; Long, F.; Chou, T.H.; Delmar, J.A.; Lei, H.T.; Rajashankar, K.R.; et al. Crystal structure of the Neisseria gonorrhoeae MtrD inner membrane multidrug efflux pump. PLoS ONE 2014, 9, e97903. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Su, C.C.; Chou, T.H.; Radhakrishnan, A.; Delmar, J.A.; Rajashankar, K.R.; Yu, E.W. Crystal structures of the Burkholderia multivorans hopanoid transporter HpnN. Proc. Natl. Acad. Sci. USA 2017, 114, 6557–6562. [Google Scholar] [CrossRef]
- Gong, X.; Qian, H.; Zhou, X.; Wu, J.; Wan, T.; Cao, P.; Huang, W.; Zhao, X.; Wang, X.; Wang, P.; et al. Structural insights into the Niemann-Pick C1 (NPC1)-mediated cholesterol transfer and ebola infection. Cell 2016, 165, 1467–1478. [Google Scholar] [CrossRef]
- Rao, S.P.; Lakshminarayana, S.B.; Kondreddi, R.R.; Herve, M.; Camacho, L.R.; Bifani, P.; Kalapala, S.K.; Jiricek, J.; Ma, N.L.; Tan, B.H.; et al. Indolcarboxamide is a preclinical candidate for treating multidrug-resistant tuberculosis. Sci. Transl. Med. 2013, 5, 214ra168. [Google Scholar] [CrossRef]
- Remuiñán, M.J.; Pérez-Herrán, E.; Rullás, J.; Alemparte, C.; Martínez-Hoyos, M.; Dow, D.J.; Afari, J.; Mehta, N.; Esquivias, J.; Jiménez, E.; et al. Tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide and N-benzyl-6′,7′-dihydrospiro[piperidine-4,4′-thieno[3,2-c]pyran] analogues with bactericidal efficacy against Mycobacterium tuberculosis targeting MmpL3. PLoS ONE 2013, 8, e60933. [Google Scholar] [CrossRef]
- Dupont, C.; Viljoen, A.; Dubar, F.; Blaise, M.; Bernut, A.; Pawlik, A.; Bouchier, C.; Brosch, R.; Guérardel, Y.; Lelièvre, J.; et al. A new piperidinol derivative targeting mycolic acid transport in Mycobacterium abscessus. Mol. Microbiol. 2016, 101, 515–529. [Google Scholar] [CrossRef]
- Shetty, A.; Xu, Z.; Lakshmanan, U.; Hill, J.; Choong, M.L.; Chng, S.S.; Yamada, Y.; Poulsen, A.; Dick, T.; Gengenbacher, M. Novel acetamide indirectly targets mycobacterial transporter MmpL3 by proton motive force disruption. Front. Microbiol. 2018, 9, 2960. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Williams, J.T.; Coulson, G.B.; Haiderer, E.R.; Abramovitch, R.B. HC2091 Kills Mycobacterium tuberculosis by targeting the MmpL3 mycolic acid transporter. Antimicrob. Agents Chemother. 2018, 62, e02459-17. [Google Scholar] [CrossRef] [PubMed]
- Protopopova, M.; Hanrahan, C.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Einck, L.; Nacy, C.A. Identification of a new antitubercular drug candidate, SQ109, from a combinatorial library of 1,2-ethylenediamines. J. Antimicrob. Chemother. 2005, 56, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Gearhart, J.; Protopopova, M.; Einck, L.; Nacy, C.A. Synergistic interactions of SQ109, a new ethylene diamine, with front-line antitubercular drugs in vitro. J. Antimicrob. Chemother. 2006, 58, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.M.; Einck, L.; Andries, K.; Nacy, C.A. In vitro interactions between new antitubercular drug candidates SQ109 and TMC207. Antimicrob. Agents Chemother. 2010, 54, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Nikonenko, B.V.; Protopopova, M.; Samala, R.; Einck, L.; Nacy, C.A. Drug therapy of experimental tuberculosis (TB): Improved outcome by combining SQ109, a new diamine antibiotic, with existing TB drugs. Antimicrob. Agents Chemother. 2007, 51, 1563–1565. [Google Scholar] [CrossRef]
- Heinrich, N.; Dawson, R.; du Bois, J.; Narunsky, K.; Horwith, G.; Phipps, A.J.; Nacy, C.A.; Aarnoutse, R.E.; Boeree, M.J.; Gillespie, S.H.; et al. Early phase evaluation of SQ109 alone and in combination with rifampicin in pulmonary TB patients. J. Antimicrob. Chemother. 2015, 70, 1558–1566. [Google Scholar] [CrossRef]
- Veiga-Santos, P.; Li, K.; Lameira, L.; de Carvalho, T.M.; Huang, G.; Galizzi, M.; Shang, N.; Li, Q.; Gonzalez-Pacanowska, D.; Hernandez-Rodriguez, V.; et al. SQ109, a new drug lead for Chagas disease. Antimicrob. Agents Chemother. 2015, 59, 1950–1961. [Google Scholar] [CrossRef]
- García-García, V.; Oldfield, E.; Benaim, G. Inhibition of Leishmania mexicana growth by the tuberculosis drug SQ109. Antimicrob. Agents Chemother. 2016, 60, 6386–6389. [Google Scholar] [CrossRef]
- Li, W.; Stevens, C.M.; Pandya, A.N.; Darzynkiewicz, Z.; Bhattarai, P.; Tong, W.; Gonzalez-Juarrero, M.; North, E.J.; Zgurskaya, H.I.; Jackson, M. Direct inhibition of MmpL3 by novel antitubercular compounds. ACS Infect. Dis. 2019, 5, 1001–1012. [Google Scholar] [CrossRef]
- Seeger, M.A.; Diederichs, K.; Eicher, T.; Brandstätter, L.; Schiefner, A.; Verrey, F.; Pos, K.M. The AcrB efflux pump: Conformational cycling and peristalsis lead to multidrug resistance. Curr. Drug Targets 2008, 9, 729–749. [Google Scholar] [CrossRef]
- Tsukazaki, T.; Mori, H.; Echizen, Y.; Ishitani, R.; Fukai, S.; Tanaka, T.; Perederina, A.; Vassylyev, D.G.; Kohno, T.; Maturana, A.D.; et al. Structure and function of a membrane component SecDF that enhances protein export. Nature 2011, 474, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, G.; Tyagi, S.; Bishai, W.R. Designer arrays for defined mutant analysis to detect genes essential for survival of Mycobacterium tuberculosis in mouse lungs. Infect. Immun. 2005, 73, 2533–2540. [Google Scholar] [CrossRef] [PubMed]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar] [CrossRef]
- Oh, S.; Park, Y.; Engelhart, C.A.; Wallach, J.B.; Schnappinger, D.; Arora, K.; Manikkam, M.; Gac, B.; Wang, H.; Murgolo, N.; et al. Discovery and structure-activity-relationship study of N-Alkyl-5-hydroxypyrimidinone carboxamides as novel antitubercular agents targeting decaprenylphosphoryl-β-d-ribose 2′-oxidase. J. Med. Chem. 2018, 61, 9952–9965. [Google Scholar] [CrossRef] [PubMed]
- Borisov, S.E.; Bogorodskaya, E.M.; Volchenkov, G.V.; Kulchavenya, E.V.; Maryandyshev, A.O.; Skornyakov, S.N.; Talibov, O.B.; Tikhonov, A.M.; Vasilyeva, I.A. Efficiency and safety of chemotherapy regimen with SQ109 in those suffering from multiple drug resistant tuberculosis. Tuberc. Lung Dis. 2018, 96, 6–18. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Chemical Class | Compound | Structure | M. tuberculosis MIC | Ref. |
|---|---|---|---|---|
| Covalent inhibitors | ||||
| Benzothiazinones | BTZ043 |  | 0.0023 µM | [30] |
| PBTZ169 |  | 0.001 µM | [49] | |
| Dinitrobenzamides | DNB1 |  | 0.69 µM | [32] |
| Nitro-quinoxalines | VI-9376 |  | 3.1 µM | [33] |
| Triazoles | 377790 |  | 0.48 µM | [51] |
| Noncovalent inhibitors | ||||
| Benzothiazoles | TCA1 |  | 0.51 µM | [55] |
| 7a |  | 0.08 µM | [61] | |
| Thiadiazoles | GSK-710 |  | 4 µM | [42] |
| Carboxy-quinoxalines | Ty38c |  | 3.1 µM | [58] |
| Aminoquinolones | 3 |  | 0.8 µM | [62] |
| Pyrazolopyridones | 19 |  | 0.1 µM | [63] |
| Azaindoles | TBA-7371 |  | 0.01 µM | [64] |
| Chemical Class | Compound | Structure | M. tuberculosis MIC | Ref. |
|---|---|---|---|---|
| Diarylpyrroles | BM212 |  | 3.6 µM | [36] |
| 1,2-diamine derivatives | DA5 |  | 9.97 µM | [37] |
| SQ109 |  | 0.78 µM | [37] | |
| Adamantyl urea | AU1235 |  | 0.3 µM | [38] |
| Benzimidazoles | C215 |  | 16.0 µM | [51] |
| Indolcarboxamides | NIDT349 |  | 0.023 µM | [93] |
| Tetrahydropyrazolo pyrimidine | THP P |  | 0.3 µM | [94] |
| Dihydrospiro[piperidine-4,4′-thieno[3,2-c]pyrans] | Spiro |  | 0.3 µM | [94] |
| Piperidinols | PIPD1 |  | 1.28 µM | [95] |
| Acetamides | E11 |  | 8.0 µM | [96] |
| Carboxamides | HC2091 |  | 19.3 µM | [97] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Degiacomi, G.; Belardinelli, J.M.; Pasca, M.R.; De Rossi, E.; Riccardi, G.; Chiarelli, L.R. Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Appl. Sci. 2020, 10, 623. https://doi.org/10.3390/app10020623
Degiacomi G, Belardinelli JM, Pasca MR, De Rossi E, Riccardi G, Chiarelli LR. Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Applied Sciences. 2020; 10(2):623. https://doi.org/10.3390/app10020623
Chicago/Turabian StyleDegiacomi, Giulia, Juan Manuel Belardinelli, Maria Rosalia Pasca, Edda De Rossi, Giovanna Riccardi, and Laurent Roberto Chiarelli. 2020. "Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3" Applied Sciences 10, no. 2: 623. https://doi.org/10.3390/app10020623
APA StyleDegiacomi, G., Belardinelli, J. M., Pasca, M. R., De Rossi, E., Riccardi, G., & Chiarelli, L. R. (2020). Promiscuous Targets for Antitubercular Drug Discovery: The Paradigm of DprE1 and MmpL3. Applied Sciences, 10(2), 623. https://doi.org/10.3390/app10020623