Development of Fast Sampling and High Recovery Extraction Method for Stable Isotope Measurement of Gaseous Mercury


Abstract
1. Introduction
2. Materials and Methods
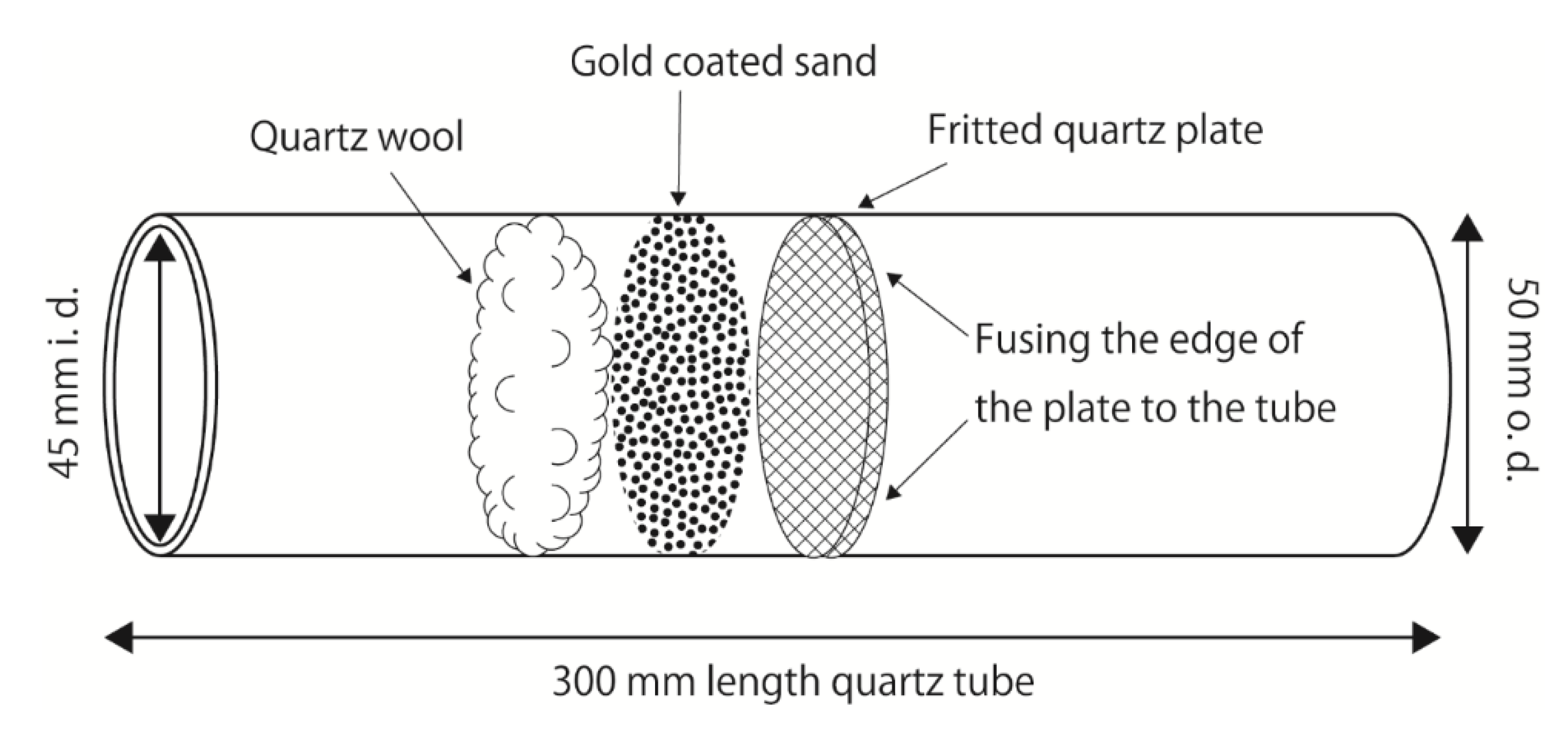
2.1. Sampling and Preconcentration of TGM
2.2. Plastic Bag Extraction of TGM
2.3. CV–MC–ICP–MS Measurements
2.4. Methodology Test
3. Results and Discussion
3.1. The BAuT Collection Efficiency
3.2. Plastic Bag Extraction
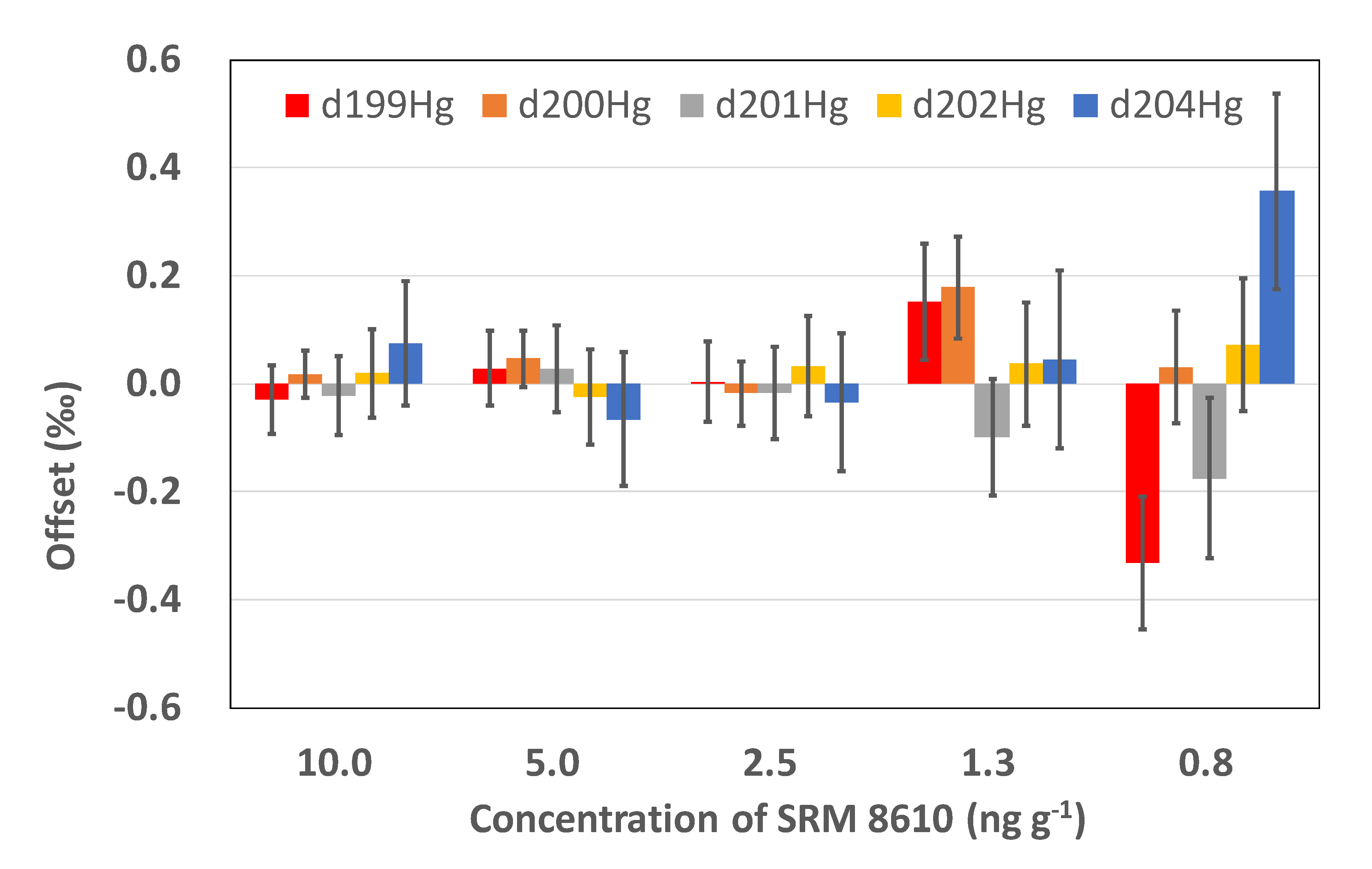
3.3. Methodology Tests
4. Conclusions
Supplementary Materials
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Concise International Chemical Assessment Document 50, Elemental Mercury and Inorganic Mercury Compounds: Human Health Aspects; World Health Organization: Geneva, Switzerland, 2003. [Google Scholar]
- Kwon, S.Y.; Blum, J.D.; Yin, R.; Tsui, M.T.-K.; Yang, Y.H.; Choi, J.W. Mercury stable isotopes for monitoring the effectiveness of the Minamata Convention on Mercury. Earth Sci. Rev. 2020, 203, 103111. [Google Scholar] [CrossRef]
- Zambardi, T.; Sonke, J.E.; Toutain, J.; Sortino, F.; Shinohara, H. Mercury emissions and stable isotopic compositions at Vulcano Island (Italy). Earth Planet. Sci. Lett. 2009, 277, 236–243. [Google Scholar] [CrossRef]
- Gill, G.A.; Fitzgerald, W.F. Mercury in surface waters of the open ocean. Glob. Biogiochemical Cycles 1987, 1, 199–212. [Google Scholar] [CrossRef]
- Demers, J.D.; Blum, J.D.; Zak, D.R. Mercury isotopes in a forested ecosystem: Implications for air-surface exchange dynamics and the global mercury cycle. Glob. Biogeochemical Cycles 2013, 27, 222–238. [Google Scholar] [CrossRef]
- Schuster, P.F.; Schaefer, K.; Aiken, G.R.; Antweiler, R.C.; DeWild, J.F.; Gryziec, J.D.; Gusmeroli, A.; Hugelius, G.; Jafarov, E.E.; Krabbenhoft, D.P.; et al. Permafrost stores a globally significant amount of mercury. Geophys. Res. Lett. 2018, 45, 1463–1471. [Google Scholar] [CrossRef]
- Blum, J.; Bergquist, B.A. Reporting of variations in the natural isotopic composition of mercury. Anal. Bioanal. Chem. 2007, 388, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Gratz, L.E.; Keeler, G.J.; Blum, J.D.; Sherman, L.S. Isotopic composition and fractionation of mercury in Great Lakes precipitation and ambient air. Environ. Sci. Technol. 2010, 44, 7764–7770. [Google Scholar] [CrossRef]
- Yamakawa, A.; Takami, A.; Takeda, Y.; Kato, S.; Kajii, Y. Emerging investigator series: Investigation of mercury emission sources using Hg isotopic compositions of atmospheric mercury at the Cape Hedo Atmosphere and Aerosol Monitoring Station (CHAAMS), Japan. Environ. Sci. Process. Impacts 2019, 21, 809–818. [Google Scholar] [CrossRef]
- Fu, X.; Marusczak, N.; Wang, X.; Gheusi, F.; E Sonke, J. Isotopic composition of gaseous elemental mercury in the free troposphere of the Pic du Midi observatory, France. Environ. Sci. Technol. 2016, 50, 5641–5650. [Google Scholar] [CrossRef]
- Szponar, N.; McLagan, D.S.; Kaplan, R.J.; Mitchell, C.P.; Wania, F.; Steffen, A.; Stupple, G.W.; Monaci, F.; Bergquist, B.A. Isotopic characterization of atmospheric gaseous elemental mercury by passive air sampling. Environ. Sci. Technol. 2020, 54, 10533–10543. [Google Scholar] [CrossRef]
- Jiskra, M.; Marusczak, N.; Leung, K.-H.; Hawkins, L.; Prestbo, E.M.; E Sonke, J. Automated stable isotope sampling of gaseous elemental mercury (ISO-GEM): Insights into GEM emissions from building surfaces. Environ. Sci. Technol. 2019, 53, 4346–4354. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Enrico, M.; Heimbürger, L.-E.; Scott, C.; Sonke, J.E. A double-stage tube furnace-acid trapping protocol for the pre-concentration of mercury from solid samples for isotopic analysis. Anal. Bionalytical Chem. 2013, 405, 6771–6781. [Google Scholar] [CrossRef]
- Yamakawa, A.; Moriya, K.; Yoshinaga, J. Determination of isotopic composition of atmospheric mercury in urban-industrial and coastal regions of Chiba, Japan, using cold vapor multicollector inductively coupled plasma mass spectrometry. Chem. Geol. 2017, 448, 84–92. [Google Scholar] [CrossRef]
- Janssen, S.E.; Lepak, R.; Tate, M.; Ogorek, J.; DeWild, J.; Babiarz, C.; Hurley, J.; Krabbenhoft, D. Rapid pre-concentration of mercury in solids and water for isotopic analysis. Anal. Chim. Acta 2019, 1054, 95–103. [Google Scholar] [CrossRef]
- Fu, X.; Heimbürger, L.-E.; Sonke, J.E. Collection of atmospheric gaseous mercury for stable isotope analysis using iodine- and chlorine-impregnated activated carbon traps. J. Anal. At. Spectrom. 2014, 29, 841. [Google Scholar] [CrossRef]
- Irei, S.; Huang, L.; Collin, F.; Zhang, W.; Hastie, D.; Rudolph, J. Flow Reactor Studies of Stable Carbon Isotopic Composition of Secondary Particulate Organic Matter Generated by OH-Radical-Induced Reactions of Toluene. Atmos. Environ. 2006, 40, 5858–5867. [Google Scholar] [CrossRef]
- Irei, S.; Rudolph, J.; Huang, L.; Auld, J.; Hastie, D. Stable carbon isotope ratio of secondary particulate organic matter formed by photooxidation of toluene in indoor smog chamber. Atmos. Environ. 2011, 45, 856–862. [Google Scholar] [CrossRef]
- Landis, M.S.; Stevens, R.K.; Schaldlich, F.; Prestbo, E.M. Development and characterization of an annular denuder methodology for the measurement of divalent inorganic reactive gaseous mercury in ambient air. Environ. Sci. Technol. 2002, 36, 3000–3009. [Google Scholar] [CrossRef]
- Ministry of Environment. Results of Background Monitoring Survey for Atmospheric Mercury and Other Metal Element Concentrations in Aerosols. 2017. Available online: https://www.env.go.jp/press/files/jp/110018.pdf (accessed on 9 September 2020). (In Japanese)
- Irei, S.; Takami, A.; Hara, K.; Hayashi, M. Evaluation of Transboundary Secondary Organic Aerosol in the Urban Air of Western Japan: Direct Comparison of Two Site Observations. ACS Earth Space Chem. 2018, 2, 1231–1239. [Google Scholar] [CrossRef]
- Chen, J.; Hintelmann, H.; Feng, X.; Dimock, B. Unusual fractionation of both odd and even mercury isotopes in precipitation from Peterborough, ON, Canada. Geochim. Cosmochim. Acta 2012, 90, 33–46. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Sampling Duration (h) | Flow Rate (L min−1) | Total Volume (m3) | Hg in Backup BAuT (pg) | Collection Efficiency † (%) |
|---|---|---|---|---|---|
| Laboratory air | 6 | 100 | 36 | 7 | 99.99 |
| Clean room air | 24 | 100 | 144 | 115 | 99.86 |
| Reactor air | 1.3 | 20 | 1.5 | 74.8 | 99.93 |
| Sample | n | Spiked Hg (ng) | Extraction Duration (h) | Recovery Yield (%) | δ199Hg § (‰) | δ200Hg § (‰) | δ201Hg § (‰) | δ202Hg § (‰) | δ204Hg § (‰) |
|---|---|---|---|---|---|---|---|---|---|
| Referenc δxHg for SRM 8610 † | N.A. ‡ | N.A. ‡ | N.A. ‡ | −0.17 ± 0.01 | −0.27 ± 0.01 | −0.46 ± 0.02 | −0.56 ± 0.03 | −0.82 ± 0.07 | |
| 40% inversed aqua regia | 6 | 103 ± 2 | 24 | 102 ± 5 | −0.19 ± 0.05 | −0.30 ± 0.06 | −0.51 ± 0.06 | −0.62 ± 0.09 | −0.91 ± 0.07 |
| 30% inversed aqua regia | 4 | 103 ± 2 | 24 | 100 ± 2 | −0.20 ± 0.01 | −0.30 ± 0.03 | −0.52 ± 0.02 | −0.63 ± 0.04 | −0.92 ± 0.05 |
| 20% inversed aqua regia | 3 | 103 ± 2 | 24 | 103 ± 3 | −0.10 ± 0.02 | −0.18 ± 0.03 | −0.40 ± 0.03 | −0.45 ± 0.06 | −0.67 ± 0.06 |
| 40% inversed aqua regia | 3 | 103 ± 2 | 12 | 97 ± 2 | −0.19 ± 0.05 | −0.29 ± 0.05 | −0.48 ± 0.09 | −0.59 ± 0.08 | −0.94 ± 0.15 |
| 30% inversed aqua regia | 3 | 103 ± 2 | 12 | 99.1 ± 0.5 | −0.20 ± 0.01 | −0.31 ± 0.03 | −0.52 ± 0.03 | −0.62 ± 0.05 | −0.92 ± 0.06 |
| 40% inversed aqua regia | 3 | 103 ± 2 | 6 | 97 ± 2 | −0.11 ± 0.03 | −0.18 ± 0.02 | −0.36 ± 0.06 | −0.44 ± 0.03 | −0.67 ± 0.03 |
| 30% inversed aqua regia | 3 | 103 ± 2 | 6 | 99 ± 2 | −0.19 ± 0.05 | −0.27 ± 0.05 | −0.44 ± 0.08 | −0.55 ± 0.06 | −0.87 ± 0.08 |
| 20% inversed aqua regia | 2 | 103 ± 2 | 6 | 92 ± 6 | −0.15 ± 0.01 | −0.19 ± 0.05 | −0.28 ± 0.09 | −0.33 ± 0.14 | −0.49 ± 0.34 |
| 40% inversed aqua regia | 3 | 103 ± 2 | 3 | 87 ± 11 | −0.15 ± 0.01 | −0.21 ± 0.03 | −0.38 ± 0.04 | −0.46 ± 0.08 | −0.69 ± 0.21 |
| 30% inversed aqua regia | 2 | 103 ± 2 | 3 | 96 ± 5 | −0.15 ± 0.05 | −0.24 ± 0.09 | −0.44 ± 0.17 | −0.50 ± 0.20 | −0.74 ± 0.30 |
| This Method | Yamakawa et al. [14] | Sun et al. [13] | Demers et al. [5] | Janssen et al. [15] | |
|---|---|---|---|---|---|
| Tested Hg quantity (ng) | 103 | unknown | 0.5–2.7 | 30–80 | 4.7–200 |
| Type of acid used for the extraction | 40% inversed aqua regia | 1% KMnO4 in 5% H2SO4 | inversed aqua regia | 1% KMnO4 | 0.03–1% KMnO4 or 40% HNO3:BrCl |
| Extraction time (h) | 24 | 2 | unknown | unknown | 0.7 |
| Extraction method | plastic bag | purging under the programmed heating | purging under the programmed heating | purging under the programmed heating | purging under the programmed heating |
| Recovery yield (%) | 100–103 | 99.1–99.6 | 75–102 | 89–94 | 96–101 |
| Sample | Flow Rate (L min–1) | Sampling Duration (h | Air Volume (m3 | Conc. of Extract † (ng g–1 | Conc. in the Air ‡ (ng m–3 | δ199Hg (‰) | δ200Hg (‰) | δ201Hg (‰) | δ202Hg (‰) | δ204Hg (‰) |
|---|---|---|---|---|---|---|---|---|---|---|
| Noyaki, 24 March | 75 | 0.9 | 4.2 | 3.1 | 3.7 | –0.58 | –0.58 | –1.01 | –0.93 | –1.40 |
| Noyaki, 6 April | 78 | 0.8 | 3.5 | 1.0 | 1.4 | –1.14 | –0.69 | –1.25 | –1.39 | 1.19 |
| Background air, 31 March | 54 | 2.1 | 6.7 | 1.1 | 0.8 | –0.05 | 0.44 | 0.35 | 1.06 | 1.30 |
| Background air, 23 May | 80 | 4.2 | 20.1 | 3.0 | 0.7 | –0.02 | 0.13 | –0.06 | 0.16 | 0.49 |
| The average of the background air | N.A. § | N.A. § | N.A. § | N.A. § | 0.8 | –0.04 | 0.28 | 0.14 | 0.61 | 0.89 |
| Outdoor air | 75 | 22.3 | 100.2 | 17.6 | 0.9 | –0.12 | 0.08 | 0.01 | 0.29 | 0.48 |
| Laboratory air | 100 | 6.0 | 36.0 | 23.2 | 3.2 | –0.12 | –0.11 | –0.22 | –0.12 | –0.16 |
| Clean room air | 100 | 24.0 | 144.0 | 17.0 | 0.6 | –0.23 | –0.20 | –0.45 | –0.40 | –0.59 |
| Sample ID | δ199Hg (‰) | δ200Hg (‰) | δ201Hg (‰) | δ202Hg (‰) | δ204Hg (‰) | Δ199Hg (‰) | Δ200Hg (‰) | Δ201Hg (‰) | Δ204Hg (‰) |
|---|---|---|---|---|---|---|---|---|---|
| Noyaki, 24 March | −0.73 | −0.82 | −1.33 | −1.35 | −2.03 | −0.39 | −0.14 | −0.31 | −0.01 |
| Noyaki, 6 April | −2.53 | −0.72 | −0.84 | −1.18 | 1.10 | −2.24 | −0.13 | 0.05 | 2.87 |
| Sample | Δ199Hg (‰) | Δ200Hg (‰) | Δ201Hg (‰) | Δ204Hg (‰) |
|---|---|---|---|---|
| Outdoor air | 0.11 | 0.55 | 0.71 | 1.87 |
| Laboratory air | 0.12 | 0.36 | 0.48 | 1.23 |
| Clean room air | 0.00 | 0.27 | 0.25 | 0.80 |
| Reactor GEM | 8.96 | –1.43 | 5.44 | 10.52 |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Irei, S. Development of Fast Sampling and High Recovery Extraction Method for Stable Isotope Measurement of Gaseous Mercury. Appl. Sci. 2020, 10, 6691. https://doi.org/10.3390/app10196691
Irei S. Development of Fast Sampling and High Recovery Extraction Method for Stable Isotope Measurement of Gaseous Mercury. Applied Sciences. 2020; 10(19):6691. https://doi.org/10.3390/app10196691
Chicago/Turabian StyleIrei, Satoshi. 2020. "Development of Fast Sampling and High Recovery Extraction Method for Stable Isotope Measurement of Gaseous Mercury" Applied Sciences 10, no. 19: 6691. https://doi.org/10.3390/app10196691
APA StyleIrei, S. (2020). Development of Fast Sampling and High Recovery Extraction Method for Stable Isotope Measurement of Gaseous Mercury. Applied Sciences, 10(19), 6691. https://doi.org/10.3390/app10196691