Two Decades of TB Drug Discovery Efforts—What Have We Learned?


Abstract
- Serendipitous drug discovery—early chemotherapy;
- Modification of drug scaffolds;
- Revisiting targets that have clinically validated drugs against them (referred to as established targets);
- Target-based screening;
- Phenotypic screening.
1. Serendipitous Drug Discovery: Early Chemotherapy
- In vitro MIC (Minimum Inhibitory Concentration) was insufficient to predict efficacy in humans. Requirement for a combination therapy of drugs.
- The best regimen required six months of treatment to achieve cure.
- Each of the drugs in the combination had a unique role to play in leading to the cure.
2. Modification of Drug Scaffolds: Analogs of Known Drugs or the Literature Compounds
2.1. Nitroimidazole as a Starting Point
2.2. Rifampicin Analogs
2.3. Ethambutol Derivatives
2.4. Isoniazid Analogs
3. Revisiting Established Targets: Revisiting Targets Proven as Druggable by Using Broad-Spectrum Compounds
3.1. Protein-Synthesis Inhibitors
3.2. Beta Lactams as Antimycobacterials
- -
- Priming of resistance against the antibiotic among normal gut bacteria due to the long-term treatment required for TB. This priming could lead to the selection of resistant mutants and subsequently spread of resistance to other pathogens in the gut.
- -
- Effect of the antibiotic on ‘latent MTB bacteria’: Latent bacteria could also be primed, leading to probability of a drug-resistant infection on reactivation.
- -
- Drugs with several new targets, like the ones discussed above, can be introduced into novel combinations, thus enabling the treatment of drug resistant (DR) MTB patients.
- -
- The effectiveness of drugs like moxifloxacin or linezolid establish the vulnerability of the target, thus promoting the search for new compounds that can inhibit the same target. This approach of revisiting ‘established/vulnerable targets’ continues to be explored by using several newer assets, like new libraries, which are novel screening formats, including those enabled by the availability of the molecular structures. Two novel compounds that have entered clinical development, GSK070 shown in Figure 2a [37] and SPR20 shown in Figure 2b [38], are examples of this approach. GSK 3036656 (GSK-070) belongs to the oxaborole class of compounds and has been shown to be a Leucine tRNA synthase inhibitor. The compound is currently in Phase 2 clinical trials [37]. SPR 720 is a GyrB ATPase inhibitor that belongs to the benzimidazole class. The molecule is also in Phase 2 clinical trials [38,39]. A very recent report shows that SPR 720 obtained an orphan disease status from FDA to treat non-tuberculosis mycobacteria (NTM) [40].
3.3. Gyrase Inhibitors
3.4. The AstraZeneca India (AZI) Effort
- -
- A variety of novel chemical structures could be identified as potent starting points (Table 1).
- -
- Several of these enzyme inhibitors showed an IC50-MIC correlation.
- -
- The inhibitors worked through different mechanisms; hence, they have a potential to avoid cross-resistance.
- -
- These inhibitors were shown to have a higher potency against the MTB enzyme target, as compared to other bacterial gyrases, that translated into selectivity in their antimicrobial activity.
4. Target-Based Screening
- The availability of the MTB genome sequence in 1998 [51] promised a new era both for studying the ‘biology’ of the pathogen and investigating novel pathways suitable for drug development. Several publications appeared, proving the essentiality of biochemical targets based on gene knockout studies in vitro, as well as investigations on the survival of the gene knockouts of MTB in the mouse model, confirming the essentiality of a variety of metabolic targets in vivo [52].
- Chris Lipinski et al. published the ‘Lipinski rule of 5′ for oral drugs [53]. The poor physiochemical properties of lead compounds vis a vis the ‘Lipinski rule of 5′ was shown to be the leading reason for the failure of potent compounds in the clinical trials, which was the direct result of poor pharmacokinetics. This led to the understanding of the concept of ‘lead-like compounds’ [54]. These rules served as guidelines for the selection/prioritization of ‘hits’ with a higher probability of being converted to drugs with favorable pharmacokinetics.
- This period also saw an enhanced efficiency in solving crystal structures; the macromolecular structures of several mycobacterial enzymes were solved and became key tools for the rationale design of novel inhibitors. A consortium of structural genomics groups was formed for facilitating the determination and analysis of structures of proteins from MTB [55].
- In addition to these facilitators of drug development, several public–private partnership consortia were also formed, which propelled interactions between the pharmaceutical industry and academic laboratories to engage in the TB drug discovery process. Examples include ‘The Action TB program’ (1996); the ‘Global Alliance for TB’ (GATB), which was launched in 2000; and the EU FW6/7 program entitled ‘New Medicines for Tuberculosis’ (NM4TB), which was launched in 2005, followed by the ‘More Medicines for Tuberculosis’ (MM4TB) program. Several pharmaceutical companies turned to target-based screening to find novel ‘lead compounds’ with a potential to inhibit the growth of MTB cells [56].
- Examples of efforts at AZI toward finding potent inhibitors by using target-based screening are summarized in Table 2. Though there are several publications on these efforts, it is interesting to view them in the context of the learning and the impact. A few of these programs were also in collaboration between AZI and partners of the NM4TB and MM4TB, the EU FW6, and seven programs, as well as with the Global Alliance for TB.
- The patterns obtained throw important light on both the characteristics of the target and the characteristics of the inhibitor.
- The question of target vulnerability: Gene knockout studies for PanK demonstrated the essentiality of the gene product. However, elegant studies with the PanK target showed that modulation of enzyme levels (inferred based on regulating gene expression) altered the sensitivity of the MTB cell to inhibitors of the enzyme [62]. This change in vulnerability of the target was the reason attributed to the failure to obtaining bactericidal effects with the potent inhibitors.
- Type of inhibition: Most of the inhibitors described in Table 2 were ‘competitive’ in nature, competing with the ATP site for binding. High-throughput screening mostly identifies ‘competitive inhibitors’ because of the design of the assay. The inhibition brought about by this type of inhibition is influenced by the ‘substrate concentration’, resulting in the modulation of target vulnerability [65].
- Promiscuous targets: These types of targets are discussed later, in the lead generation approaches.
- Established target: As described under Section 3.4, several inhibitors that have progressed to anti-TB drugs establish the ‘vulnerability’ of a target; ATP synthase (ATPS) is one such target. Bedaquiline, a non-competitive inhibitor of ATPS, is a front-line treatment for MDR TB [67,68].
5. Phenotypic Screening
- a.
- Bedaquiline: Approved in 2012 for the treatment of MDR TB, Bedaquiline (TMC-207, R207910, shown in Figure 3) has the reputation of being the first FDA approved TB drug in more than four decades. It was discovered by Johnson & Johnson by screening more than 70,000 compounds against Mycobacterium. smegmatis (M.sm). The compound was first described in 2004, at the Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). The target of TMC-207 was later conclusively proven to be the MTB ATP synthase [68]. Subsequently, in 2012, Bedaquiline became an essential part of the MDR regimen [71,72]. Several combinations of Bedaquiline are also being investigated for their potential to reduce the duration of therapy [73]. Discovery of Bedaquiline and the target link has made MTB ATP synthase an attractive validated target.
- b.
- Benzothiazinone (BTZ) and Macozinone (PBTZ): BTZ was discovered at the Russian academy of science and developed with NM4TB, and it is presently being clinically evaluated under the Innovative Medicines for Tuberculosis (iM4TB). The target for BTZ has been shown as decaprenylphosphoryl-β-d-ribose-2′-oxidase (DprE1) by genetic and biochemical studies [74,75]. Based on SAR explorations, a piperazine derivative, PBTZ-169 (Macozinone), which has superior pharmacokinetics (PK), as compared to that of BTZ [76], was discovered and is currently in Phase 2 clinical trials, in addition to BTZ 043. Structures of BTZ and PBTZ are shown in Figure 4.
- c.
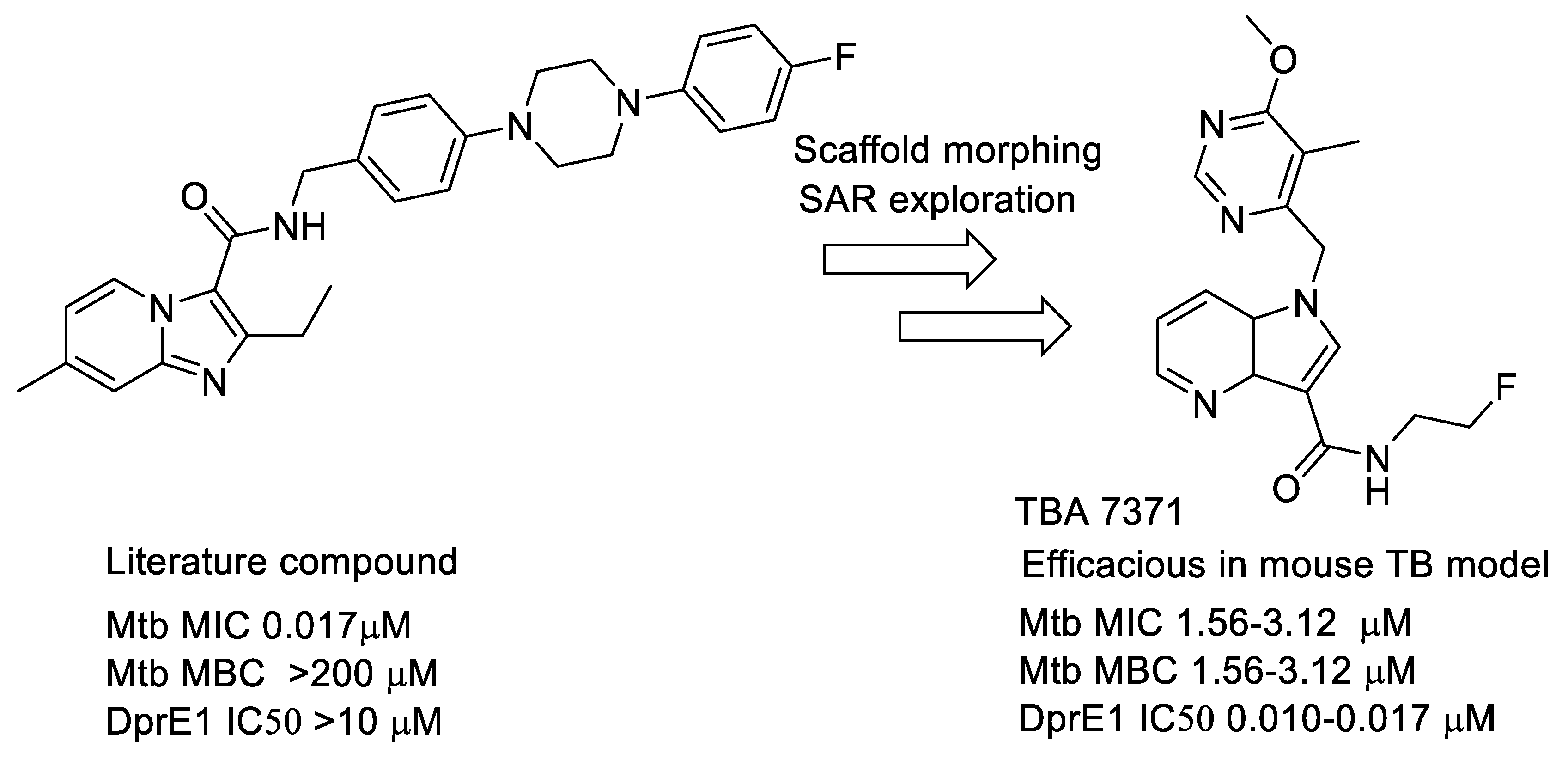
- Azaindoles: These compounds emerged from a literature search for compounds similar to Q-203 [77,78]. The compound had an MIC but was not bactericidal. Scaffold morphing efforts on this compound at AZ Bangalore in a collaboration with TB alliance led to the identification of TBA 7371 [79,80,81], a potent DprE1 inhibitor with non-covalent binding. TBA 7371, jointly owned by Global Alliance for TB and the Foundation for Neglected Disease Research (FNDR), was developed by GATB through Phase 1 safety trials in humans. Recently, GATB has licensed TBA 7371 to Gates Medical Research Institute (GMRI); the news was disclosed in Union World conference of lung health at Hyderabad, India, in November 2019. GMRI is currently testing TBA7371 in Phase 2A clinical trials in TB patients [82]. The progression is depicted in Figure 5.
- d.
- Q203: Telacebec (Q203) is a compound that was discovered by the researchers at Institute Pasteur, Korea, and is structurally very similar to the one discussed above [77,78]. This compound inhibits the cytochrome bc1 complex of MTB that is critical for the electron transport chain. The compound is in Phase 2 clinical trials [83]. Structure of Telacebec is shown in Figure 6.
- e.
- OPC-167832: First discussed in 2016 at the 47th Union World Conference on Lung Health in Liverpool, UK, this is a carbostyril derivative that inhibits DPRE1. The structure shown in Figure 7 represents a balance of hydrophobic and hydrophilic residues. The compound is currently in Phase 2 clinical trials [84]. Structure of OPC-167832 is shown in Figure 7.
Phenotypic-Screening Efforts at AstraZeneca India
6. The Anti-TB Pipeline—The Learning and the Gaps
6.1. The ‘Evolution’ of the Anti-TB Drug Pipeline
6.2. Lead Generation Approaches
6.3. Has the Pipeline Evolved Based on the Significant Increase in the Knowledge of the ‘Biology’ of MTB?
- What is the value of having novel drugs that are active predominantly on one of the physiological states like the hypoxic or the intracellular state? Metronidazole is a known antiparasitic drug that is also active on MTB growing under oxygen-limiting conditions [110]. The addition of metronidazole to a cocktail of drugs for the treatment of MDR TB did not show clinical benefit [111]. On the other hand, several inhibitors of the intracellular survival of the pathogen in the macrophage have been identified, have been shown to be active in animal models of infection [34,67,77,83,112], and are undergoing clinical evaluation. These inhibitors potentially will facilitate the ‘killing’ of the pathogen within the macrophage, which in turn may reflect in ‘faster’ cure.
- How do we develop molecules with activity against only specific subpopulations? The accepted path through the regulatory system is by showing superiority of the new combination over the existing standard of care (SOC) combination—a steep bar to cross. Until the beginning of this decade, very few efficacious drugs were available for the treatment of MDR TB patients. This provided the opportunity for newer efficacious drugs to show non-inferiority against the poorly active regimen, a bar, which was reasonable to go across. However, the current combination regimen for MDR TB treatment is efficacious, and newer compounds will have to demonstrate a clear advantage over the current regimen. Identification of novel biomarkers that can be evaluated during Phase 2a (Early Bactericidal Activity trials) could help in positioning these compounds that are active on specific populations to the best advantage.
6.4. Combination Therapy: Finding Novel Combinations
7. Going Forward
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- TB Facts.org, Information about Tuberculosis. Available online: https://tbfacts.org/history-of-tb-drugs/ (accessed on 13 April 2020).
- Lehmann, J. Para-aminosalicylic acid in the treatment of Tuberculosis. Lancet 1946, 247, 15–16. [Google Scholar] [CrossRef]
- Lehmann, J. Twenty years afterwards, Historical notes on the Discovery of the Antituberculosis Effect of Para-Aminosalicylic Acid (PAS) and the First Clinical Trials. Am. Rev. Respir. Dis. 1964, 90, 953–956. [Google Scholar]
- Lehmann, E. Nicotinic acid in therapy of pulmonary tuberculosis; preliminary therapeutic report. Dtsch. Med. Wochenschr. 1952, 77, 1480–1481. [Google Scholar] [CrossRef]
- Murray, F.M. Nicotinamide: An Oral Antimicrobial Agent with Activity against Both Mycobacterium tuberculosis and Human Immunodeficiency Virus. Clin. Infect. Dis. 2003, 36, 453–460. [Google Scholar] [CrossRef]
- Thayer, J.D.; Seligman, R.B. The anti-tuberculous activity of some derivatives of p-aminosalicylic acid, nicotinic acid, and isonicotinic acid. Antibiot. Chemother. (Northfield) 1955, 5, 129–131. [Google Scholar]
- Mitchison, D.A. Treatment of tuberculosis. The Mitchell lecture 1979. J. R. Coll. Physicians Lond. 1980, 14, 91. [Google Scholar]
- Aquinas, M. Short-course therapy for tuberculosis. Drugs 1982, 24, 118–132. [Google Scholar] [CrossRef]
- Mohammad, A. Rifampin and Their Analogs: A Development of Antitubercular Drugs. World J. Org. Chem. 2013, 1, 14–19. [Google Scholar]
- Ramani, A.V.; Monika, A.; Indira, V.L.; Karyavardhi, G.; Venkatesh, J.; Jeankumar, V.U.; Manjashetty, T.H.; Yogeeswari, P.; Sriram, D. Synthesis of highly potent novel anti-tubercular isoniazid analogues with preliminary pharmacokinetic evaluation. Bioorg. Med. Chem. Lett. 2012, 22, 2764–2767. [Google Scholar] [CrossRef]
- Yamamoto, S.; Toida, I.; Watanabe, N.; Ura, T. In vitro Antimycobacterial Activities of Pyrazinamide Analogs. Antimicrob. Agents Chemother. 1995, 39, 2088–2091. [Google Scholar] [CrossRef][Green Version]
- Häusler, H.; Kawakami, R.P.; Mlaker, E.; Severn, W.B.; Stütz, A.E. Ethambutol Analogues as Potential Antimycobacterial Agents. Bioorg. Med. Chem. Lett. 2001, 11, 1679–1681. [Google Scholar] [CrossRef]
- Goldman, P. The development of 5-nitroimidazole for the treatment and prophylaxis of anaerobic bacterial infections. J. Antimicrob. Chemother. 1982, 10, 23–33. [Google Scholar] [CrossRef]
- Ashtekar, D.R.; Costa-Perira, R.; Nagrajan, K.; Vishvanathan, N.; Bhatt, A.D.; Rittel, W. In vitro and in vivo activities of the nitroimidazole CGI 17341 against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1993, 37, 183–186. [Google Scholar] [CrossRef]
- Stover, C.K.; Warrener, P.; Van Devanter, D.R.; Sherman, D.R.; Arain, T.M.; Langhorne, M.H.; Anderson, S.W.; Towell, J.A.; Yuan, Y.; McMurray, D.N.; et al. A small-molecule nitroimidazopyran drug candidate for the treatment of tuberculosis. Nature 2000, 405, 962–966. [Google Scholar] [CrossRef]
- Liu, Y.; Matsumoto, M.; Ishida, H.; Ohguro, K.; Yoshitake, M.; Gupta, R.; Geiter, L.; Hafkin, J. Delamanid: From discovery to its use for pulmonary multidrug-resistant tuberculosis (MDR-TB). Tuberculosis 2018, 111, 20–30. [Google Scholar] [CrossRef]
- Sensi, P. History of the development of rifampin. Rev. Infect. Dis. 1983, 5, S402–S466. [Google Scholar] [CrossRef]
- Janin, Y.L. Antituberculosis drugs: Ten years of research. Bioorg. Med. Chem. 2007, 15, 2479–2513. [Google Scholar] [CrossRef]
- Bogatcheva, E.; Hanrahan, C.F.; Nikonenko, B.; Samala, R.; Chen, P.; Gearhart, J.; Barbosa, F.; Einck, L.; Nacy, A.C.A.; Protopopova, M. Identification of new diamine scaffolds with activity against Mycobacterium tuberculosis. J. Med. Chem. 2006, 49, 3045–3048. [Google Scholar] [CrossRef]
- Yendapally, R.; Lee, R.E. Design, synthesis, and evaluation of novel ethambutol analogues. Bioorg. Med. Chem. Lett. 2008, 18, 1607–1611. [Google Scholar] [CrossRef][Green Version]
- Kapil, T.; Regina, W.; David, B.K.; Kriti, A.; Vinod, N.; Elizabeth, F.; Barnes, S.W.; John, R.W.; David, A.; Clifton, E.B., III; et al. SQ109 targets MmpL3, a membrane transporter of trehalose monomycolate involved in mycolic acid donation to the cell wall core of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1797–1809. [Google Scholar]
- Sinha, N.; Jain, S.; Tilekar, A.; Upadhayaya, R.S.; Kishore, N.; Jana, G.H.; Arora, S.K. Synthesis of isonicotinic acid N’-arylidene-N-2-oxo-2-(4-aryl-piperazin-1-yl) ethyl-hydrazides as antituberculosis agents. Bioorg. Med. Chem. Lett. 2005, 15, 1573–1576. [Google Scholar] [CrossRef]
- Sinha, R.K.; Arora, S.K.; Sinha, N.; Modak, V.M. In vivo activity of LL4858 against Mycobacterium tuberculosis. In Proceedings of the 44th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC-2004), Washington, DC, USA, 30 October–2 November 2004. [Google Scholar]
- World Health Organization. Tuberculosis—A Global Emergency Case Notification Update: February 1996; WHO Reference Number: WHO/TB/96.197; WHO: Geneva, Switzerland, 1996. [Google Scholar]
- Hinshaw, H.; Pyle, M.M.; Feldman, W.H. Streptomycin in tuberculosis. Am. J. Med. 1947, 2, 429–435. [Google Scholar] [CrossRef]
- Schecter, G.F.; Scott, C.; True, L.; Raftery, A.; Flood, J.; Mase, S. Linezolid in the Treatment of Multidrug-Resistant Tuberculosis. Clin. Infect. Dis. 2010, 50, 49–55. [Google Scholar] [CrossRef]
- Maartens, G.; Benson, C.A. Linezolid for Treating Tuberculosis: A Delicate Balancing Act. BioMedicine 2015, 2, 1568–1569. [Google Scholar] [CrossRef]
- Lee, M.; Song, T.; Kim, Y.; Jeong, I.; Cho, S.N.; Barry, C.E., III. Linezolid for XDR-TB—Final Study Outcomes. N. Engl. J. Med. 2015, 373, 290–291. [Google Scholar] [CrossRef]
- Balasubramanian, V.; Solapure, S.; Shandil, R.; Gaonkar, S.; Mahesh, K.N.; Reddy, J.; Deshpande, A.; Bharath, S.; Kumar, N.; Wright, L.; et al. Pharmacokinetic and pharmacodynamic evaluation of AZD5847 in a mouse model of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 4185–4190. [Google Scholar] [CrossRef][Green Version]
- Balasubramanian, V.; Solapure, S.; Iyer, H.; Ghosh, A.; Sharma, S.; Kaur, P.; Deepthi, R.; Subbulakshmi, V.; Ramya, V.; Ramachandran, V.; et al. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob. Agents Chemother. 2013, 58, 495–502. [Google Scholar] [CrossRef]
- Werngren, J.; Wijkander, M.; Perskvist, N.; Balasubramanian, V.; Sambandamurthy, V.K.; Rodrigues, C.; Hoffner, S. In vitro activity of AZD5847 against geographically diverse clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 4222–4223. [Google Scholar] [CrossRef][Green Version]
- Williams, K.N.; Brickner, S.J.; Stover, C.K.; Zhu, T.; Ogden, A.; Tasneen, R.; Tyagi, S.; Grosset, J.H.; Nuermberger, E.L. Addition of PNU-100480 to First-Line Drugs Shortens the Time Needed to Cure Murine Tuberculosis. Am. J. Respir. Crit. Care Med. 2009, 180, 371–376. [Google Scholar] [CrossRef]
- Lanoix, J.P.; Nuermberger, E. Sutezolid: Oxazolidinone antibacterial treatment of tuberculosis. Drugs Future 2013, 38, 387–394. [Google Scholar] [CrossRef]
- Choi, Y.; Lee, S.W.; Kim, A.; Jang, K.; Nam, H.; Cho, Y.L.; Yu, K.-S.; Chung, J.-Y. Safety, tolerability and pharmacokinetics of 21 day multiple oral administration of a new oxazolidinone antibiotic, LCB01-0371, in healthy male subjects. Antimicrob. Chemother. 2018, 73, 183–190. [Google Scholar] [CrossRef]
- Shoen, C.; DeStefano, M.; Hafkin, B.; Cynamon, M. In vitro and in vivo activity of contezolid (MRX-I) against M. tuberculosis. Antimicrob. Agents Chemother. 2018, 62, e00493-18. [Google Scholar] [CrossRef]
- ClinicalTrails.gov. Phase 2 Trial to Evaluate the Early Bactericidal Activity, Safety and Tolerability of Meropenem Plus Amoxycillin/CA and Faropenem Plus Amoxycillin/CA in Adult Patients with Newly Diagnosed Pulmonary Tuberculosis. Available online: https://clinicaltrials.gov/ct2/show/NCT02349841 (accessed on 15 March 2020).
- Palencia, A.; Li, X.; Bu, W.; Choi, W.; Ding, C.Z.; Easom, E.E.; Feng, L.; Hernandez, V.; Houston, P.; Liu, L.; et al. Discovery of Novel Oral Protein Synthesis Inhibitors of Mycobacterium tuberculosis That Target Leucyl-tRNA Synthetase. Antimicrob. Agents Chemother. 2016, 60, 6271–6280. [Google Scholar] [CrossRef]
- Shoen, C.; DeStefano, M.; Pucci, M.; Cynamon, M. Evaluating the Sterilizing Activity of SPR720 in Combination Therapy against Mycobacterium Tuberculosis Infection in Mice, ASM Microbe 2019. Session P439 Poster AAR-749. Available online: https://www.newtbdrugs.org/pipeline/compound/spr720 (accessed on 19 March 2020).
- Shoen, C.; Pucci, M.; DeStefano, M.; Cynamon, M. Efficacy of SPR720 and SPR750 Gyrase Inhibitors in a Mouse Mycobacterium tuberculosis Infection Model, ASM Microbe 2017. Session 336. Poster 43. Available online: https://www.abstractsonline.com/pp8/#!/4358/presentation/6167 (accessed on 25 March 2020).
- Spero Therapeutics Receives FDA Orphan Drug Designation for SPR720 for the Treatment of on tuberculous Mycobacterial (NTM) Infection. Available online: https://www.globenewswire.com/news-release/2020/03/11/1998722 (accessed on 6 April 2020).
- Narayanan, P. Shortening short course chemotherapy: A randomised clinical trial for treatment of smear positive pulmonary tuberculosis with regimens using ofloxacin in the intensive phase. Indian J. Tuberc. 2002, 49, 27–38. [Google Scholar]
- Stephen, H.G. The role of moxifloxacin in tuberculosis therapy. Eur. Respir. Rev. 2016, 25, 19–28. [Google Scholar]
- Nagaraja, V.; Godbole, A.A.; Henderson, S.R.; Maxwell, A. DNA topoisomerase I and DNA gyrase as targets for TB therapy. Drug Discov. Today 2017, 22, 510–518. [Google Scholar] [CrossRef]
- Shirude, P.S.; Hameed, S. Nonfluoroquinolone-Based Inhibitors of Mycobacterial Type II Topoisomerase as Potential Therapeutic Agents for TB. Annu. Rep. Med. Chem. 2012, 47, 319–330. [Google Scholar]
- Solapure, S.; Mukherjee, K.; Nandi, V.; Waterson, D.; Shandil, R.; Balganesh, M.; Sambandamurthy, V.K.; Raichurkar, A.K.; Deshpande, A.; Ghosh, A.; et al. Optimization of Pyrrolamides as Mycobacterial GyrB ATPase Inhibitors: Structure-Activity Relationship and In Vivo Efficacy in a Mouse Model of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 61–70. [Google Scholar]
- Kale, M.G.; Raichurkar, A.; Waterson, D.; McKinney, D.; Manjunatha, M.R.; Kranthi, U.; Koushik, K.; Jena, L.K.; Shinde, V.; Rudrapatna, S.; et al. Thiazolopyridine Ureas as Novel Antitubercular Agents Acting through Inhibition of DNA Gyrase B. J. Med. Chem. 2013, 56, 8834–8848. [Google Scholar]
- Kale, R.R.; Kale, M.G.; Waterson, D.; Raichurkar, A.; Hameed, S.P.; Manjunatha, M.R.; Reddy, B.K.; Malolanarasimhan, K.; Shinde, V.; Koushik, K.; et al. Thiazolopyridoneureas as DNA gyrase B inhibitors: Optimization of antitubercular activity and efficacy. Bioorg. Med. Chem. Lett. 2014, 24, 870–879. [Google Scholar] [CrossRef]
- Shirude, P.S.; Madhavapeddi, P.; Tucker, J.A.; Murugan, K.; Patil, V.; Basavarajappa, H.D.; Raichurkar, A.V.; Humnabadkar, V.; Hussein, S.; Sharma, S.; et al. Aminopyrazinamides: Novel and Specific GyrB Inhibitors that Kill Replicating and Nonreplicating Mycobacterium tuberculosis. ACS Chem. Biol. 2013, 8, 519–523. [Google Scholar] [CrossRef]
- Hameed, P.S.; Patil, V.; Solapure, S.; Sharma, U.; Madhavapeddi, P.; Raichurkar, A.; Chinnapattu, M.; Manjrekar, P.; Shanbhag, G.; Puttur, J.; et al. Novel N-Linked Aminopiperidine-Based Gyrase Inhibitors with Improved hERG and in Vivo Efficacy against Mycobacterium tuberculosis. J. Med. Chem. 2014, 57, 4889–4905. [Google Scholar]
- Hameed, P.S.; Raichurkar, A.; Madhavapeddi, P.; Menasinakai, S.; Sharma, S.; Kaur, P.; Nandishaiah, R.; Panduga, V.; Reddy, J.; Sambandamurthy, V.K.; et al. Benzimidazoles: Novel Mycobacterial Gyrase Inhibitors from Scaffold Morphing. ACS Med. Chem. Lett. 2014, 5, 820–825. [Google Scholar] [CrossRef]
- Cole, S.T.; Brosch, R.; Parkhill, J.; Garnier, T.; Churcher, C.; Harris, D.; Gordon, S.; Eiglmeier, K.; Gas, S.; Barry, C.E.; et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [Google Scholar] [CrossRef]
- Vashisht, R.; Bhat, A.G.; Kushwaha, S.; Bhardwaj, A.; OSDD Consortium; Brahmachari, S.K. Systems level mapping of metabolic complexity in Mycobacterium tuberculosis to identify high-value drug target. J. Transl. Med. 2014, 12, 263. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The Design of Lead like Combinatorial Libraries. Angew. Chem. Int. Ed. Eng. 1999, 28, 3743–3748. [Google Scholar] [CrossRef]
- Terwilliger, T.C.; Park, M.; Waldo, G.; Berendzen, J.; Hung, L.-W.; Kim, C.-Y.; Smith, C.; Sacchettini, J.; Bellinzoni, M.; Bossi, R.; et al. The TB structural genomics consortium: A resource for Mycobacterium tuberculosis biology. Tuberculosis 2003, 83, 223–249. [Google Scholar] [CrossRef]
- Yuan, T.; Sampson, N.S. Hit Generation in TB Drug Discovery: From Genome to Granuloma. Chem. Rev. 2018, 118, 1887–1916. [Google Scholar] [CrossRef]
- Patil, V.; Kale, M.; Raichurkar, A.; Bhaskar, B.; Prahlad, D.; Balganesh, M.; Nandan, S.; Shahul Hameed, P. Design and synthesis of triazolopyrimidineacylsulfonamides as novel anti-mycobacterial leads acting through inhibition of acetohydroxyacid synthase. Bioorg. Med. Chem. Lett. 2014, 24, 2222–2225. [Google Scholar] [CrossRef]
- Balganesh, M.; Nandan, S. Combination Chemotherapy for Tuberculosis by Synergistic Action of Rifampicin as the RNA Polymerase Inhibitors with Acetolactate Synthase Inhibitors. PCT International Application WO 2007132189 A1 20071122, 22 November 2007. [Google Scholar]
- Bandodkar, B.S.; Naik, M.; Ghorpade, S.; Kale, M.; Shanbhag, G.; Patil, V.; Solapure, S.; Balganesh, M.; Shandil, R.K.; Balasubramanian, B.; et al. Lead Generation via Virtual Screening: Discovery of Pyrazolones as Potent Antimycobacterial Leads through structure based virtual screening of shikimate kinase. In Proceedings of the ICAAC 2009, San Francisco, CA, USA, 12–15 September 2009. [Google Scholar]
- Bandodkar, B.S.; Schmitt, S. Pyrazolone Derivatives for the Treatment of Tuberculosis. PCT International Application No. WO/2007/020426 A1, 22 February 2007. [Google Scholar]
- Bandodkar, B.S. Oral presentation, “A decade of learning”. In Proceedings of the CSIR-NM4TB Symposium, Bangalore, India, 14 December 2009. [Google Scholar]
- Venkatraman, J.; Bhat, J.; Solapure, S.M.; Sandesh, J.; Sarkar, D.; Aishwarya, S.; Mukherjee, K.; Datta, S.; Malolanarasimhan, K.; Bandodkar, B.; et al. Screening, Identification, and Characterization of Mechanistically Diverse Inhibitors of the Mycobacterium Tuberculosis Enzyme, Pantothenate Kinase (CoaA). J. Biomol. Screen. 2012, 17, 293. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.K.K.; Landge, S.; Ravishankar, S.; Patil, V.; Shinde, V.; Tantry, S.; Kale, M.; Raichurkar, A.; Menasinakai, S.; Mudugal, N.V.; et al. Assessment of Mycobacterium tuberculosis Pantothenate Kinase Vulnerability through Target Knockdown and Mechanistically Diverse Inhibitors. Antimicrob. Agents Chemother. 2014, 8, 3312–3326. [Google Scholar] [CrossRef]
- Björkelid, C.; Bergfors, T.; Raichurkar, A.K.V.; Mukherjee, K.; Malolanarasimhan, K.; Bandodkar, B.; Jones, T.A. Structural and Biochemical Characterization of Compounds Inhibiting Mycobacterium tuberculosis Pantothenate Kinase. J. Biol. Chem. 2013, 288, 18260–18270. [Google Scholar] [CrossRef]
- Naik, M.; Raichurkar, A.; Bandodkar, B.S.; Varun, B.V.; Bhat, S.; Kalkhambkar, R.; Murugan, K.; Menon, R.; Bhat, J.; Paul, B.; et al. Structure Guided Lead Generation for M. tuberculosis Thymidylate Kinase (Mtb TMK): Discovery of 3-Cyanopyridone and 1,6-Naphthyridin-2-one as Potent Inhibitors. J. Med. Chem. 2015, 58, 755–766. [Google Scholar]
- Shirude, P.S.; Paul, B.; Choudhury, N.R.; Kedari, C.; Bandodkar, B.; Ugarkar, B.G. Quinolinyl Pyrimidines: Potent Inhibitors of NDH-2 as a Novel Class of Anti-TB Agents. ACS Med. Chem. Lett. 2012, 3, 736–740. [Google Scholar] [CrossRef]
- Andries, K. A diarylquinoline drug active on the ATP Synthase of Mycobacterium tuberculosis. Science 2006, 307, 223–227. [Google Scholar] [CrossRef]
- Koul, A.; Dendouga, N.; Vergauwen, K.; Molenberghs, B.; Vranckx, L.; Willebrords, R.; Ristic, Z.; Lill, H.; Dorange, I.; Guillemont, J.; et al. Diarylquinolines target subunit c of mycobacterial ATP synthase. Nat. Chem. Biol. 2007, 3, 323–324. [Google Scholar] [CrossRef]
- Grzelak, E.M.; Choules, M.P.; Gao, W.; Cai, G.; Wan, B.; Wang, Y.; McAlpine, J.B.; Cheng, J.; Jin, Y.; Lee, H.; et al. Strategies in anti-Mycobacterium tuberculosis drug discovery based on phenotypic screening. J. Antibiot. 2019, 72, 719–728. [Google Scholar] [CrossRef]
- Manjunatha, U.H.; Smith, P.W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorg. Med. Chem. 2015, 23, 5087–5097. [Google Scholar] [CrossRef]
- Matteelli, A.; Carvalho, A.C.; E Dooley, K.; Kritski, A. TMC207: The first compound of a new class of potent anti-tuberculosis drugs. Future Microbiol. 2010, 5, 849–858. [Google Scholar] [CrossRef]
- Deoghare, S. Bedaquiline: A new drug approved for treatment of multidrug-resistant tuberculosis. Indian J. Pharmacol. 2013, 45, 536–537. [Google Scholar] [CrossRef] [PubMed]
- Conradie, F.; Diacon, A.H.; Ngubane, N.; Howell, P.; Everitt, D.; Crook, A.M.; Mendel, C.M.; Egizi, E.; Moreira, J.; Timm, J.; et al. Bedaquiline, pretomanid and linezolid for treatment of extensively drug resistant, intolerant or non-responsive multidrug resistant pulmonary tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones Kill Mycobacterium tuberculosis by Blocking Arabinan Synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Trefzer, C.; Rengifo-Gonzalez, M.; Hinner, M.J.; Schneider, P.; Makarov, V.; Cole, S.T.; Johnsson, K. Benzothiazinones: Prodrugs That Covalently Modify the Decaprenylphosphoryl-D-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Lechartier, B.; Zhang, M.; Neres, J.; Sar, A.M.; A Raadsen, S.; Hartkoorn, R.C.; Ryabova, O.B.; Vocat, A.; Decosterd, L.A.; et al. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 6, 372–383. [Google Scholar] [CrossRef] [PubMed]
- No, Z.; Kim, J.; Brodin, P.B.; Seo, M.J.; Kim, Y.M.; Cechetto, J.; Jeon, H.; Genovesio, A.; Lee, S.; Kang, S.; et al. Anti-Infective Compounds. PCT Publication No. WO 2011/113606 A1, 22 September 2011. [Google Scholar]
- No, Z.; Kim, J.; Brodin, P.; Seo, M.J.; Park, E.; Cechetto, J.; Jeon, H.; Genovesio, A.; Lee, S.; Kang, S.; et al. Anti-Infective pyrido (1,2-a) pyrimidines. PCT Publication No. WO 2011/085990 A1, 21 July 2011. [Google Scholar]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hameed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious In Vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [PubMed]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Kr, P.; et al. 1, 4-Azaindole, a Potential Drug Candidate for Treatment of Tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, M.R.; Shandil, R.K.; Panda, M.; Sadler, C.; Ambady, A.; Panduga, V.; Kumar, N.; Mahadevaswamy, J.; Sreenivasaiah, M.; Narayan, A.; et al. Scaffold Morphing to Identify Novel DprE1 Inhibitors with Antimycobacterial Activity. ACS Med. Chem. Lett. 2019, 10, 1480–1485. [Google Scholar]
- Early Bactericidal Activity of TBA-7371 in Pulmonary Tuberculosis, Identifier: NCT04176250. Available online: https://clinicaltrials.gov (accessed on 2 April 2020).
- Pethe, K.; Bifani, P.J.; Jang, J.; Kang, S.; Park, S.; Ahn, S.; Jiricek, J.; Jung, J.; Jeon, H.K.; Cechetto, J.; et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013, 19, 1157–1160. [Google Scholar] [CrossRef]
- Otsuka Awarded Grant to Advance Development of Novel Anti-Tuberculosis Compound OPC-167832 with Delamanid. Available online: https://www.businesswire.com/news/home/20180129005073/en/ (accessed on 11 April 2020).
- Landge, S.; Mullick, A.B.; Nagalapur, K.; Neres, J.; Subbulakshmi, V.; Murugan, K.; Ghosh, A.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; et al. Discovery of benzothiazoles as antimycobacterial agents: Synthesis, structure–activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-b-D-ribose 20-oxidase. Bioorg. Med. Chem. 2015, 23, 7694–7710. [Google Scholar] [CrossRef]
- Landge, S.; Ramachandran, V.; Kumar, A.; Neres, J.; Murugan, K.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; Vachaspati, P.; Raichurkar, A.; et al. Nitroarenes as Antitubercular Agents: Stereoelectronic Modulation to Mitigate Mutagenicity. Chemmedchem 2016, 11, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Jena, L.K.; Koushik, K.; et al. 4-Aminoquinolone Piperidine Amides: Noncovalent Inhibitors of DprE1 with Long Residence Time and Potent Antimycobacterial Activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of Pyrazolopyridones as a Novel Class of Noncovalent DprE1 Inhibitor with Potent Anti-Mycobacterial Activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [PubMed]
- Tantry, S.J.; Markad, S.D.; Shinde, V.; Bhat, J.; Balakrishnan, G.; Gupta, A.K.; Ambady, A.; Raichurkar, A.; Kedari, C.; Sharma, S.; et al. Discovery of Imidazo[1,2-a]pyridine Ethers and Squaramides as Selective and Potent Inhibitors of Mycobacterial Adenosine Triphosphate (ATP) Synthesis. J. Med. Chem. 2017, 60, 1379–1399. [Google Scholar] [PubMed]
- Tantry, S.J.; Shinde, V.; Balakrishnan, G.; Markad, S.D.; Gupta, A.K.; Bhat, J.; Narayan, A.; Raichurkar, A.; Jena, L.K.; Sharma, S.; et al. Scaffold morphing leading to evolution of 2,4-diaminoquinolines and aminopyrazolopyrimidines as inhibitors of the ATP synthesis pathway. MedChemComm 2016, 7, 1022–1032. [Google Scholar] [CrossRef]
- Gandhi, N.R.; Nunn, P.; Dheda, K.; Schaaf, H.S.; Zignol, M.; Van Soolingen, D.; Jensen, P.; Bayona, J. Multidrug-resistant and extensively drug-resistant tuberculosis: A threat to global control of tuberculosis. Lancet 2010, 375, 1830–1843. [Google Scholar] [CrossRef]
- Nuermberger, E.; Yoshimatsu, T.; Tyagi, S.; O’Brien, R.J.; Vernon, A.N.; Chaisson, R.E.; Bishai, W.R.; Grosset, J. 2004. Moxifloxacin-containing Regimen Greatly Reduces Time to Culture Conversion in Murine Tuberculosis. Am. J. Respir. Crit. Care Med. 2003, 169, 421–426. [Google Scholar] [CrossRef]
- Gillespie, S.H.; Crook, A.M.; McHugh, T.D.; Mendel, C.M.; Meredith, S.K.; Murray, S.R.; Pappas, F.; Phillips, P.P.; Nunn, A.J. Four-Month Moxifloxacin-Based Regimens for Drug-Sensitive Tuberculosis. N. Eng. J. Med. 2014, 351, 1577–1587. [Google Scholar] [CrossRef]
- Mabhula, A.; Singh, V. Drug-resistance in Mycobacterium tuberculosis: Where we stand. MedChemComm 2019, 10, 1342–1360. [Google Scholar] [CrossRef]
- Muñoz-Elías, E.J.; Timm, J.; Botha, T.; Chan, W.T.; Gomez, J.E.; McKinney, J.D. Replication Dynamics of Mycobacterium tuberculosis in Chronically Infected Mice. Infect. Immun. 2005, 73, 546–551. [Google Scholar] [CrossRef]
- Mitchison, D.A. Role of individual drugs in the chemotherapy of tuberculosis. Int. J. Tuberc. Lung Dis. 2004, 4, 796–806. [Google Scholar]
- Rayasam, G.V.; Balganesh, T.S. Exploring the potential of adjunct therapy in tuberculosis. Trends Pharmacol. Sci. 2015, 36, 506–513. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chakaya, J.; Hoelscher, M.; Ntoumi, F.; Rustomjee, R.; Vilaplana, C.; Yeboah-Manu, D.; Rasolofo, V.; Munderi, P.; Singh, N.; et al. Towards host-directed therapies for tuberculosis. Nat. Rev. Drug Discov. 2015, 14, 511–512. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Rao, M.; Wallis, R.S.; Kaufmann, S.H.; Rustomjee, R.; Mwaba, P.; Vilaplana, C.; Yeboah-Manu, D.; Chakaya, J.; Ippolito, G.; et al. Host-directed therapies for infectious diseases: Current status, recent progress and future prospects. Lancet Infect. Dis. 2016, 16, e47–e63. [Google Scholar] [CrossRef]
- Kumar, D.; Nath, L.; Kamal, A.; Varshney, A.; Jain, A.; Singh, S.; Rao, K.V.; Singh, S. Genome-wide analysis of the host intracellular network that regulates survival of Mycobacterium tuberculosis. Cell 2010, 140, 731–743. [Google Scholar] [CrossRef]
- Singhal, A.; Jie, L.; Kumar, P.; Hong, G.S.; Leow, M.K.-S.; Paleja, B.; Tsenova, L.; Kurepina, N.; Chen, J.; Zolezzi, F.; et al. Metformin as adjunct antituberculosis therapy. Sci. Transl. Med. 2014, 263, 263ra159. [Google Scholar] [CrossRef]
- Mishra, R.; Kohli, S.; Malhotra, N.; Bandyopadhyay, P.; Mehta, M.; Munshi, M.; Adiga, V.; Ahuja, V.K.; Shandil, R.K.; Rajmani, R.S.; et al. Targeting redox heterogeneity to counteract drug tolerance in replicating Mycobacterium tuberculosis. Sci. Transl. Med. 2019, 11, eaaw6635. [Google Scholar] [CrossRef]
- Padmapriyadarsini, C.; Bhavani, P.K.; Natrajan, M.; Ponnuraja, C.; Kumar, H.; Gomathy, S.N.; Guleria, R.; Jawahar, S.M.; Singh, M.; Balganesh, T.; et al. Evaluation of metformin in combination with rifampicin containing antituberculosis therapy in patients with new, smear-positive pulmonary tuberculosis (METRIF): Study protocol for a randomised clinical trial. BMJ Open 2019, 9, e024363. [Google Scholar] [CrossRef]
- Mdluli, K.; Kaneko, T.; Upton, A. The Tuberculosis Drug Discovery andDevelopment Pipeline and Emerging Drug Targets. Cold Spring Harb. Perspect. Med. 2015, 5, a021154. [Google Scholar] [CrossRef]
- Roy, K.K.; Wani, M.A. Emerging opportunities of exploiting electron transport chain pathway for drug resistant tuberculosis drug discovery. Expert Opin. Drug Discov. 2020, 15, 231–241. [Google Scholar] [CrossRef]
- Mitchison, D.A. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 1985, 66, 219–225. [Google Scholar] [CrossRef]
- Jindani, A.; Doré, C.J.; Mitchison, D.A. The bactericidal and sterilising activities of antituberculosis drugs during the first 14 days. Am. J. Respir. Crit. Care Med. 2003, 167, 1348–1354. [Google Scholar] [CrossRef]
- Hernandez-Pando, R. Persistence of DNA from Mycobacterium tuberculosis in superficially normal lung tissue during latent infection. Lancet 2004, 356, 2133–2137. [Google Scholar] [CrossRef]
- Mitchison, D.A. The search for new sterilizing anti-tuberculosis drugs. Front. Biosci. 2004, 9, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- Waynel, L.G.; Hilda, A. Metronidazole is bactericidal to dormant cells of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1994, 38, 2054–2058. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.W. Efficacy and safety of metronidazole for pulmonary multidrug resistant tuberculosis. Antimicrob. Agents Chemother. 2013, 57, 3903–3909. [Google Scholar] [CrossRef]
- Tyagi, S.; Nuermberger, E.; Yoshimatsu, T.; Williams, K.; Rosenthal, I.; Lounis, N.; Bishai, W.; Grosset, J. Bactericidal activity of the nitroimidazopyranPA-824 in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2005, 49, 2289–2293. [Google Scholar] [CrossRef]
- Dooley, K.E.; Hanna, D.; Mave, V.; Eisenach, K.; Savic, R.M. Advancing the development of new tuberculosis treatment regimens: The essential role of translational and clinical pharmacology and microbiology. PLoS Med. 2019, 16, e1002842. [Google Scholar] [CrossRef]
- Nimmo, C.; Naidoo, K.; O’Donnell, M.; Bolhuis, M.S.; Van Der Werf, T.S.; Akkerman, O.W.; Conradie, F.; Everitt, D.; Crook, A.M. Treatment of Highly Drug-Resistant Pulmonary Tuberculosis. N. Engl. J. Med. 2020, 382, 893–902. [Google Scholar]
- Diacon, A.H.; Dawson, R.; Von Groote-Bidlingmaier, F.; Symons, G.; Venter, A.; Donald, P.R.; Van Niekerk, C.; Everitt, D.; Winter, H.; Becker, P.; et al. 14-Day bactericidal activity of PA-824, bedaquiline, pyrazinamide, and moxifloxacin combinations: A randomised trial. Lancet 2012, 380, 986–993. [Google Scholar] [CrossRef]
- McHugh, T.D.; Honeyborne, I.; Lipman, M.; Zumla, A. Revolutionary new regimens for multidrug resistant tuberculosis. Lancet 2018, 19, 233–234. [Google Scholar] [CrossRef]
- Working Group on New TB Drugs. Available online: https://www.newtbdrugs.org/ (accessed on 13 April 2020).
- TB Facts.org, New-Tb-Drugs. Available online: https://tbfacts.org/new-tb-drugs/ (accessed on 13 April 2020).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Approach | Target | Inhibitor Series | Mechanism of Inhibition | Reference |
|---|---|---|---|---|
| Following known series | GyrB | Pyrrolamides | ATPase inhibitor | [45] |
| Pharmacophore based library | GyrB | Thiazolopyridine ureas | ATPase inhibitor | [46] |
| Pharmacophore based library, scaffold morphing | GyrB | Thiazolopyridone ureas | ATPase inhibitor | [47] |
| High throughput screening | GyrB | Aminopyrazinamides | ATPase, MTB gyrase specific. Novel binding mode. | [48] |
| Focused library screening | GyrB | Aminopiperidine | Non-ATP site binders, different from fluoroquinolones | [49] |
| Scaffold hopping | GyrB | Benzimidazoles | Non-ATP site binders, different from FQs | [50] |
| Approach | Target | Pathway | Inhibitor Class | Status Reached | Remarks | Reference |
|---|---|---|---|---|---|---|
| Known inhibitors | Acetolactate synthase | Branched chain amino acid biosynthesis | Triazolopyrimidine | IC50~30 nM MIC0.5 μg/mL | Possible auxotrophy | [57,58] |
| Virtual screening | MtSK | Aromatic amino acid biosynthesis | Pyrazolones | IC50~0. 6μM MIC 0.5 μg/mL | Possible promiscuity | [59,60] |
| Targeted Library | Pkn B | Cell division | Bis anilinopyrimidines (BAP) | IC50~40 pM MIC 8 μg/mL | Possible redundancy | [61] |
| HTS | PanK | Coenzyme A (CoA) biosynthesis pathway | Triazoles (ATP competitive) | Nanomolar IC50, not translating into microbial inhibition. | Possible Target vulnerability | [62,63,64] |
| HTS | PanK | Coenzyme A (CoA) biosynthesis pathway | Quinolones (ATP competitive) | Nanomolar IC50, not translating into microbial inhibition. | Possible Target vulnerability | [62,63,64] |
| HTS | PanK | Coenzyme A (CoA) biosynthesis pathway | Biarylacetic acids (ATP non-competitive) | Nanomolar IC50, not translating into microbial inhibition. | Possible Target vulnerability | [62,63,64] |
| HTS | TMK | DNA synthesis | 3-Cyanopyridone | Nanomolar IC50, not translating into microbial inhibition. | Possible redundancy | [65] |
| Fragment screening | TMK | DNA synthesis | 1,6-Naphthyridinone | Nanomolar IC50, not translating into microbial inhibition. | Possible redundancy | [65] |
| HTS | NdH2 | Energy Metabolism | Quinolyl pyrimidines | Ndh2 IC50: 96 nM MIC: <0.5 μg/mL | In progress | [66] |
| Screening Mode | Target | Compound | Comments | Reference |
|---|---|---|---|---|
| HTS | DprE1 | Benzothiazole | Promiscuous target | [85,86] |
| HTS | DprE1 | 4-Aminoquinoline piperidine amides | Promiscuous target | [87] |
| HTS | DprE1 | Pyrazolopryridones | Promiscuous target | [88] |
| HTS | DprE1 | Azaindoles | Promiscuous target | [79,80] |
| Scaffold hopping | DprE1 | Benzimidazole | Promiscuous target | [81] |
| HTS | ATPS | Squaramide | Established/Validated Target | [89] |
| HTS | ATPS | Imidazopyridine | Established/Validated Target | [89] |
| Scaffold morphing | ATPS | Diaminoquinazoline | Established/Validated Target | [90] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bandodkar, B.; Shandil, R.K.; Bhat, J.; Balganesh, T.S. Two Decades of TB Drug Discovery Efforts—What Have We Learned? Appl. Sci. 2020, 10, 5704. https://doi.org/10.3390/app10165704
Bandodkar B, Shandil RK, Bhat J, Balganesh TS. Two Decades of TB Drug Discovery Efforts—What Have We Learned? Applied Sciences. 2020; 10(16):5704. https://doi.org/10.3390/app10165704
Chicago/Turabian StyleBandodkar, Balachandra, Radha Krishan Shandil, Jagadeesh Bhat, and Tanjore S. Balganesh. 2020. "Two Decades of TB Drug Discovery Efforts—What Have We Learned?" Applied Sciences 10, no. 16: 5704. https://doi.org/10.3390/app10165704
APA StyleBandodkar, B., Shandil, R. K., Bhat, J., & Balganesh, T. S. (2020). Two Decades of TB Drug Discovery Efforts—What Have We Learned? Applied Sciences, 10(16), 5704. https://doi.org/10.3390/app10165704