Electrochemical Ion Pumping Device for Blue Energy Recovery: Mixing Entropy Battery



Abstract
1. Introduction
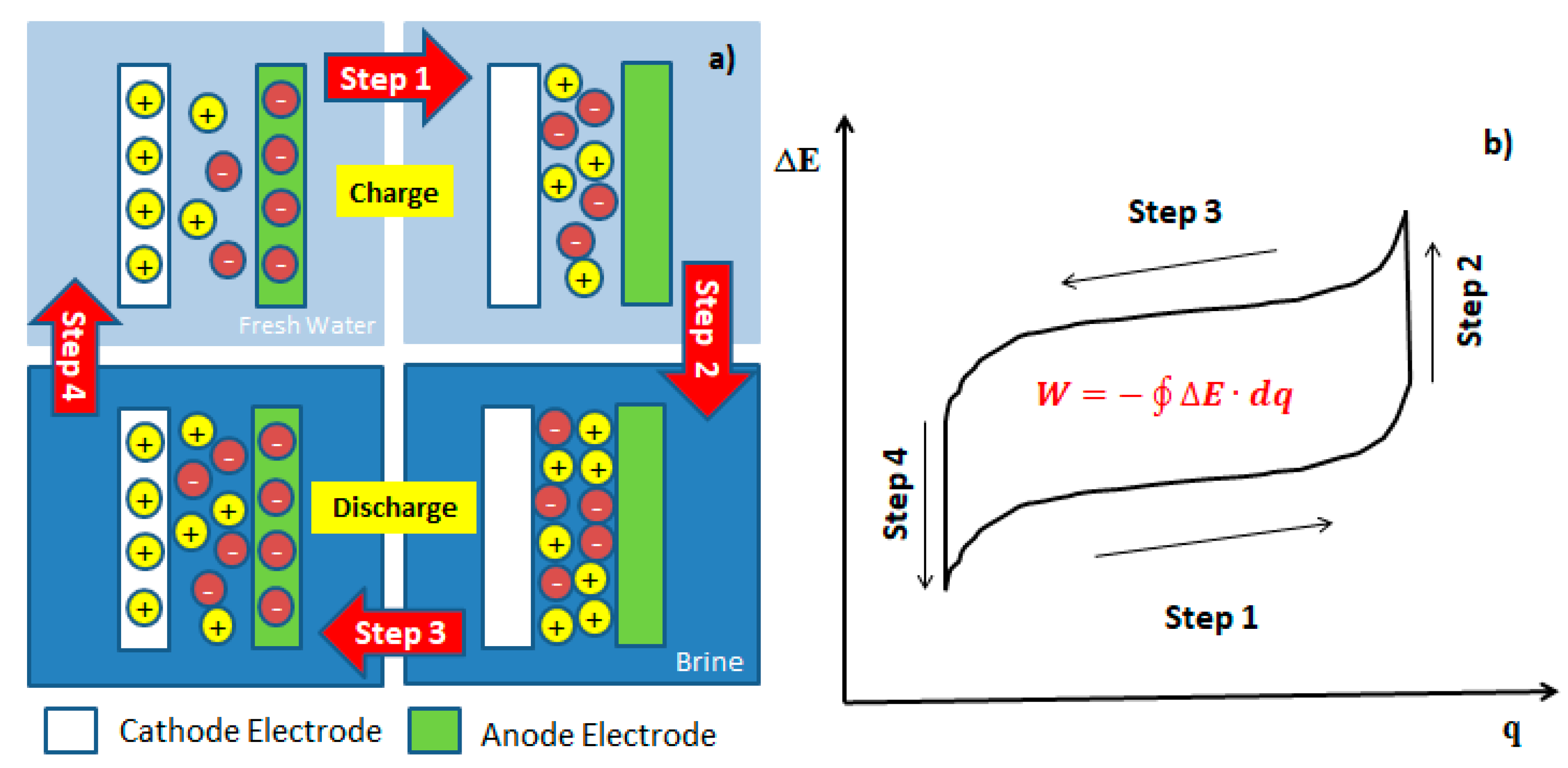
2. Theoretical Background for Mixing Process
3. Energy Consumption in the Process of Mixing Entropy Battery (MEB)
3.1. Thermodynamic Energy Consumption
3.1.1. Salt Capturing Method
3.1.2. Selective Exchange Method
3.2. Kinetic Energy Consumption
3.2.1. Salt Capturing Method
3.2.2. Selective Exchange Method
3.2.3. Salt Capturing Method
3.2.4. Selective Exchange Method
4. Natural Solutions as Electrolyte for MEB
5. Intercalation Materials as Cathode Electrode in MEB
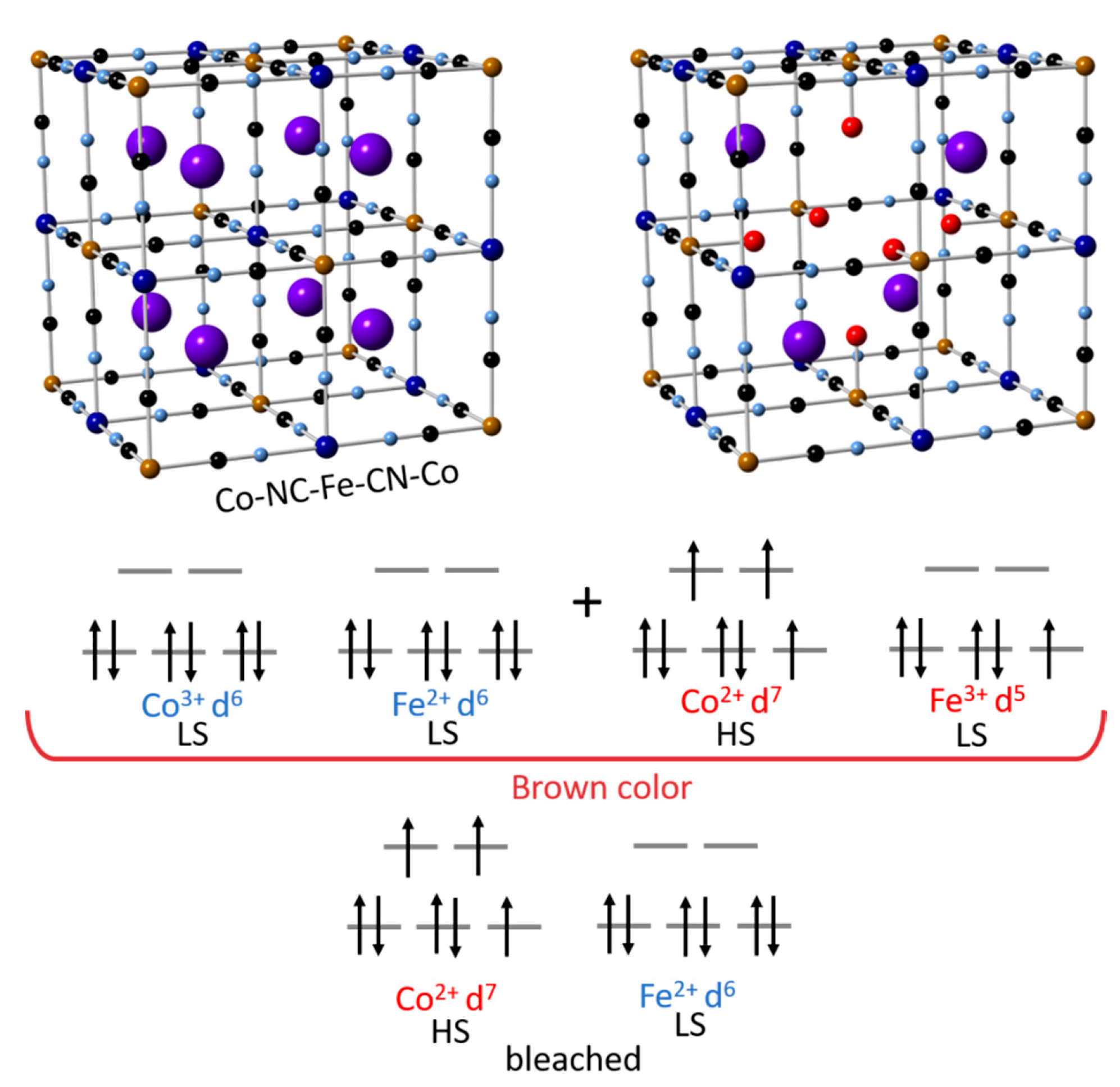
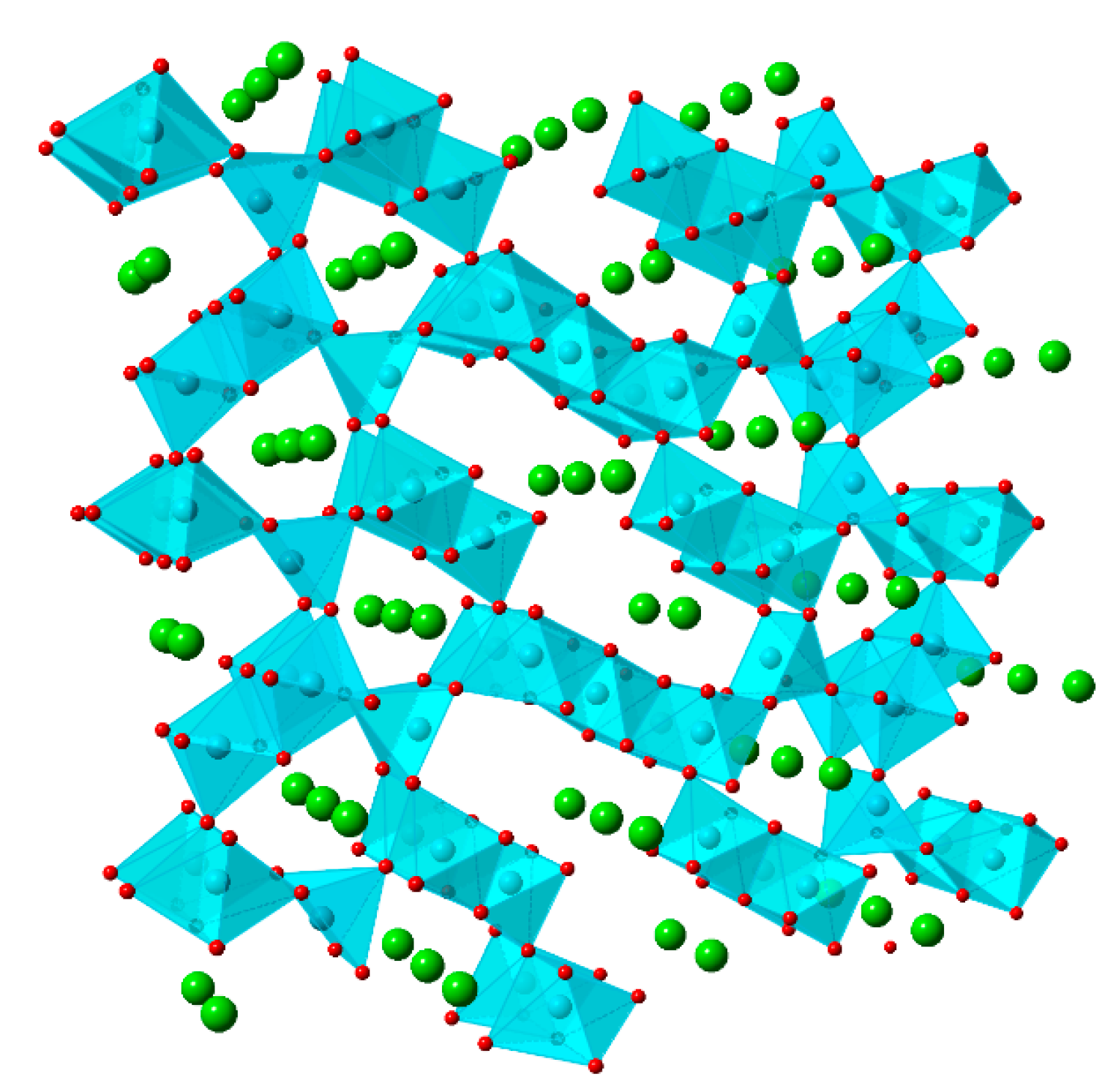
5.1. Cobalt Hexacyanoferrate (CoHCF)
5.2. Nickel Hexacyanoferrate (NiHCF)
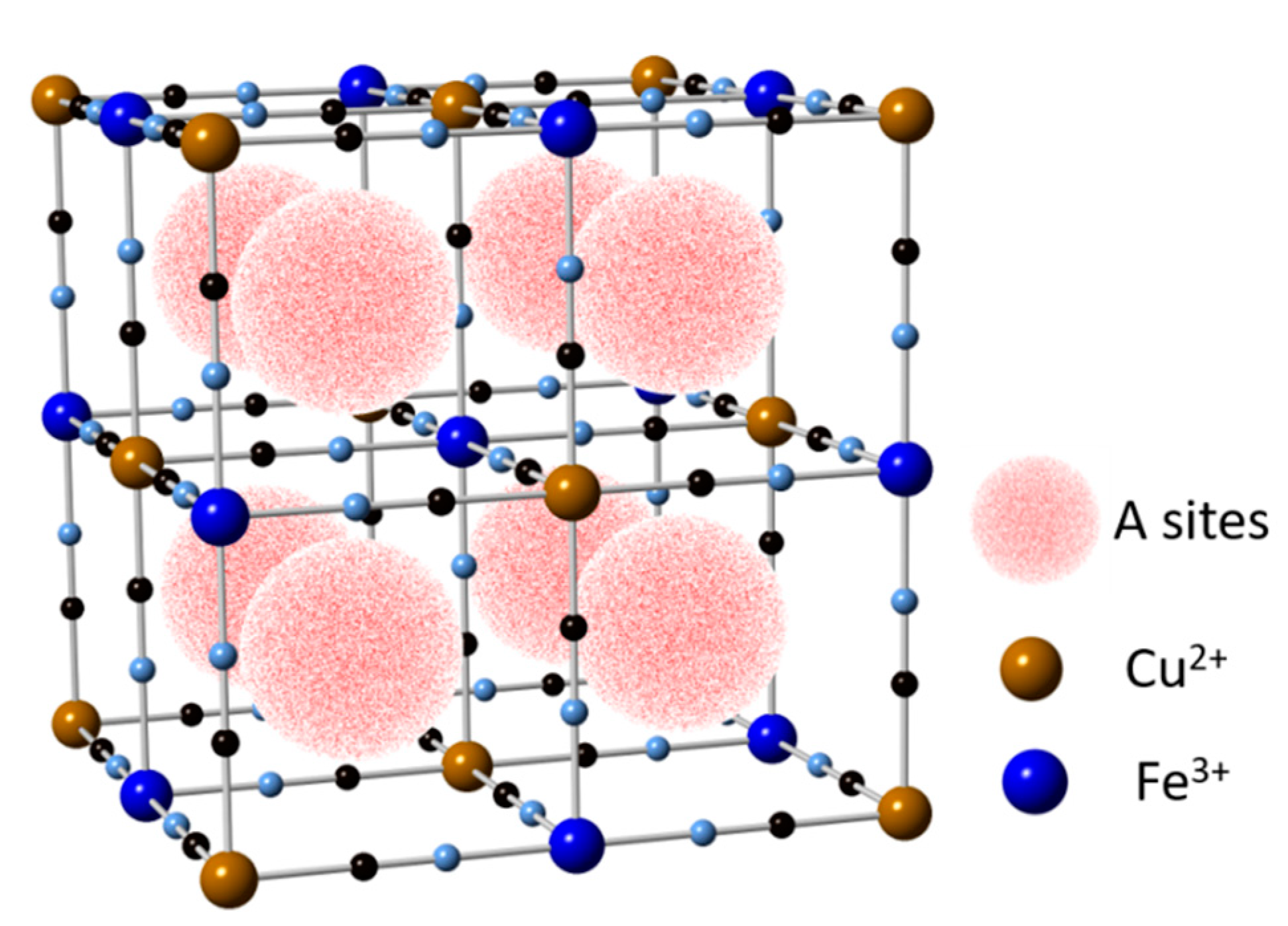
5.3. Copper Hexacyanoferrate (CuHCF)
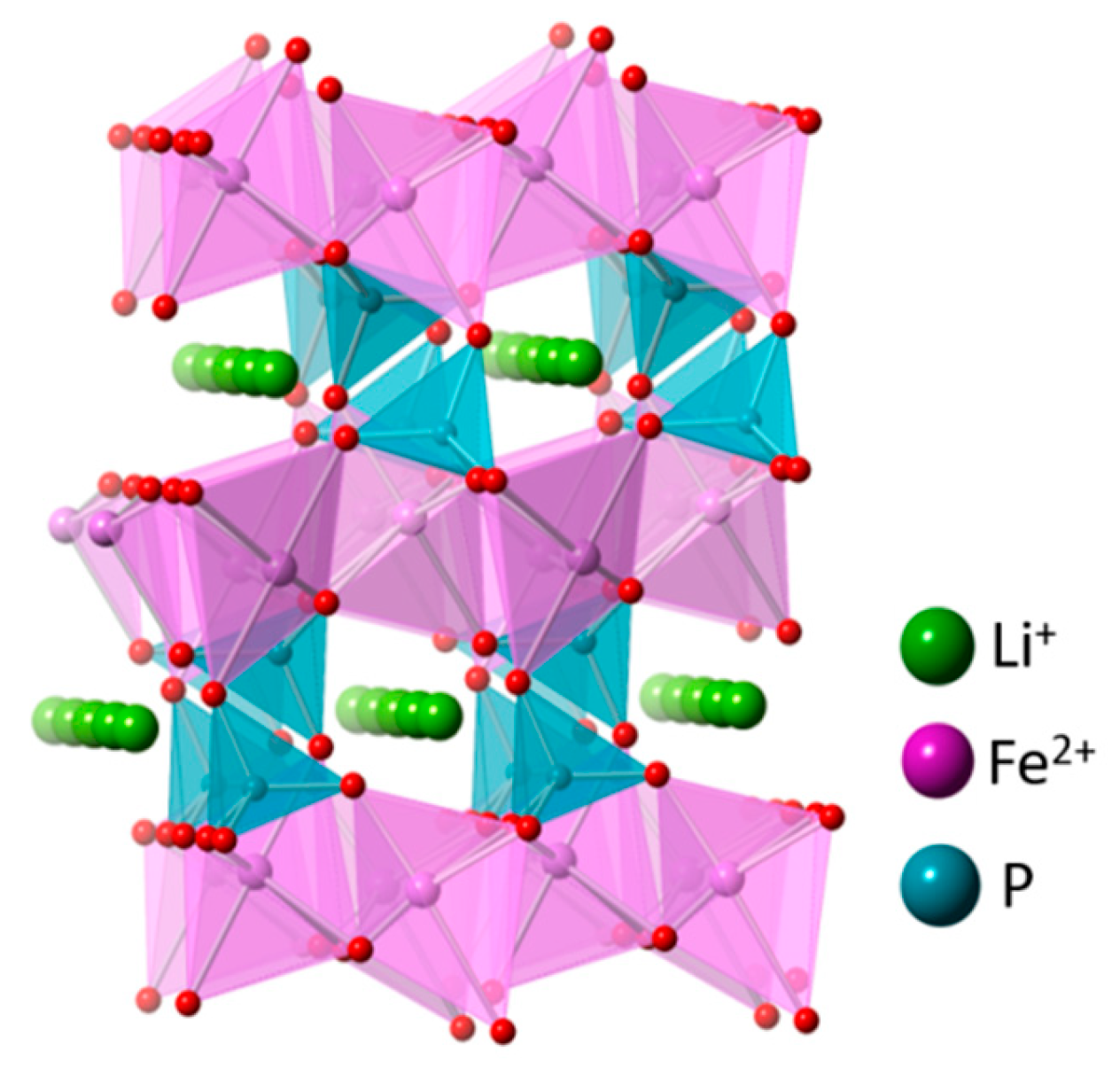
5.4. LiFePO4
5.5. Na2Mn5O10
5.6. Na4Mn9O18
6. Anode Materials Used in MEB
6.1. Silver
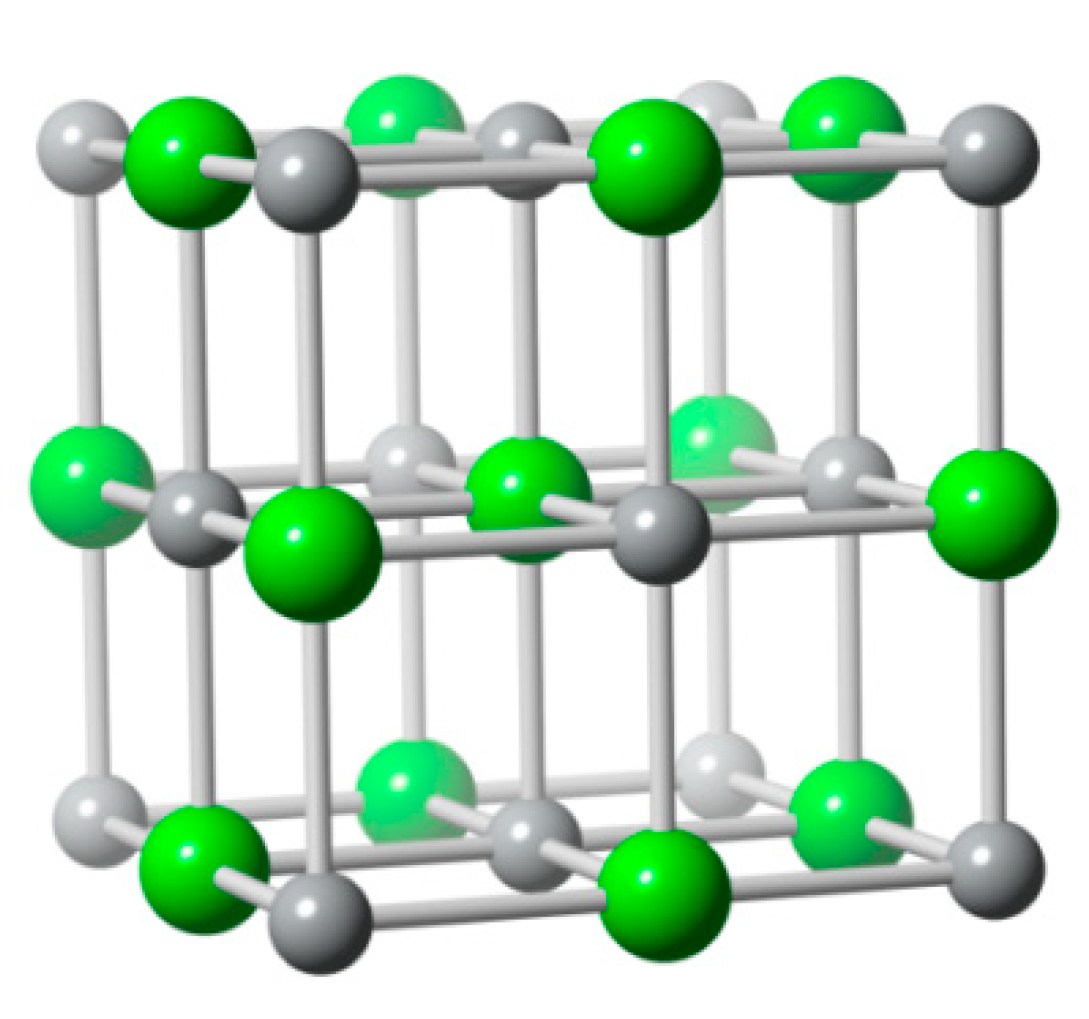
6.2. Bismuth Oxychloride (BiOCl)
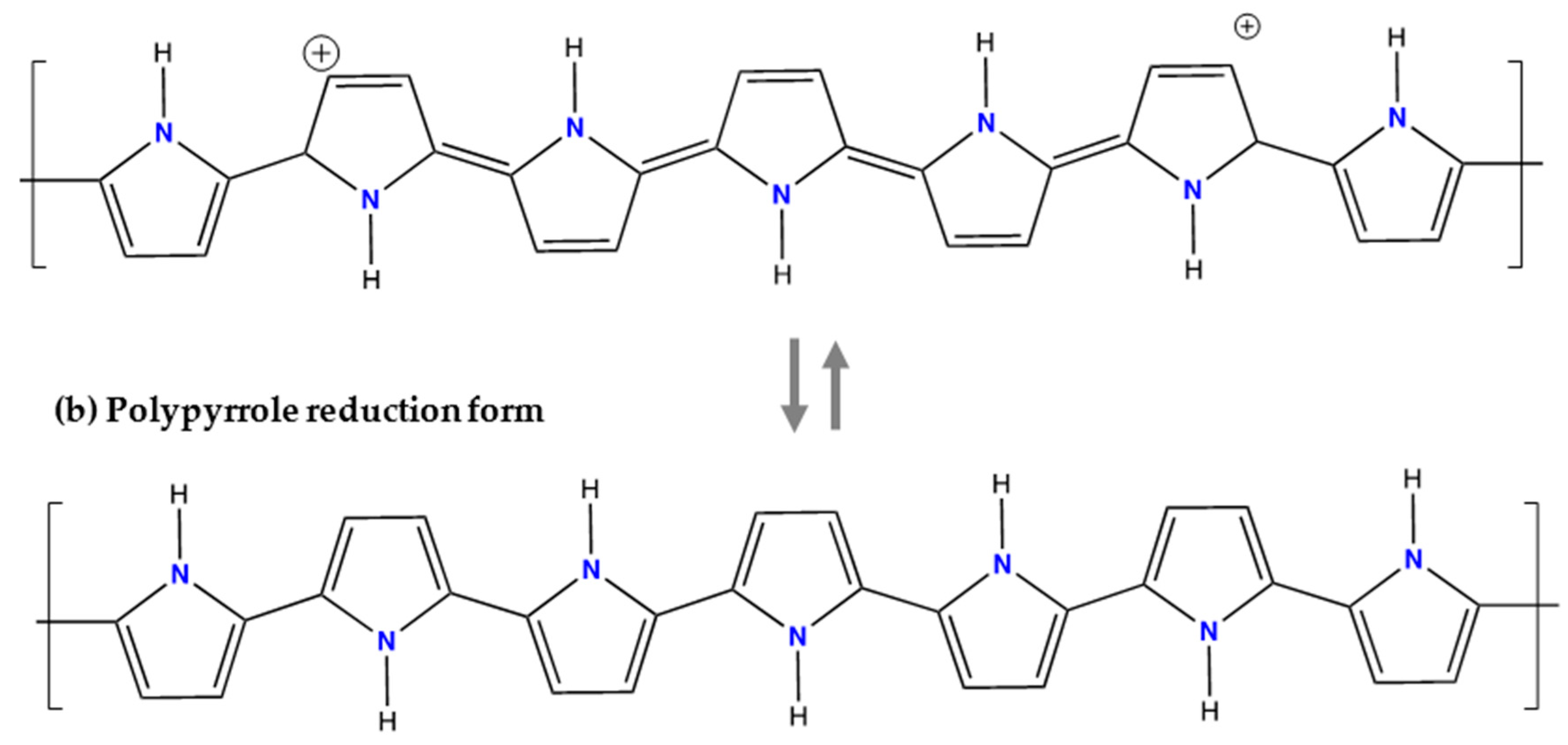
6.3. Polypyrrole (PPy)
7. Conclusion and Outlooks
- (a)
- Synthetic or natural saline solutions such as altiplanic brines in South America can serve as electrolytes in areas where no rivers with considerable flows are available. This alternative opens the possibility for this technology to be used in nearby coastal areas, or high altitude salt lake brines, as is the case for many altiplanic places in South America.
- (b)
- Another possibility would be to consider rejection flows from reverse osmosis plants as concentrated NaCl solution and seawater as the diluted solution in arid places where there are no rivers or treatment plants that flow into the sea.
- (c)
- It is interesting to note that fresh water availability, which is the main restrictive factor to implement MEB technologies in arid places, can be overcome by using solar energy to obtain distilled water with complete recycling of the electrolyte to achieve conversion of solar energy into electrical energy. The solar distillation technologies are well known and have a historical record of applications in Northern Chile as far back as 1842 and worked effectively for 40 years [68]. This fresh water collection alternative would make harvesting energy from salinity gradient possible at lower costs and greater efficiency.
- (d)
- The use of wastewater effluents as a source of fresh water could be useful for obtaining electrical energy through MEBs. However, more research related to the behavior of electrodes in the presence of biological contaminants and free chlorine is needed.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Marino, M.; Misuri, L.; Ruffo, R.; Brogioli, D. Electrode kinetics in the “capacitive mixing” and “battery mixing” techniques for energy production from salinity differences. Electrochim. Acta 2015, 176, 1065–1073. [Google Scholar] [CrossRef]
- Marino, M.; Misuri, L.; Carati, A.; Brogioli, D. Proof-of-Concept of a Zinc-Silver Battery for the Extraction of Energy from a Concentration Difference. Energies 2014, 7, 3664–3683. [Google Scholar] [CrossRef]
- Lee, J.; Yoon, H.; Lee, J.; Kim, T.; Yoon, J. Extraction of Salinity Gradient Energy using a Hybrid Capacitive-mixing System. ChemSusChem 2017, 10, 1600–1606. [Google Scholar] [CrossRef] [PubMed]
- Brogioli, D. Extracting renewable energy from a salinity difference using a capacitor. Physcal Rev. Lett. 2009, 058501, 31–34. [Google Scholar] [CrossRef]
- La Mantia, F.; Pasta, M.; Deshazer, H.D.; Logan, B.E.; Cui, Y. Batteries for Efficient Energy Extraction from a Water Salinity Difference. Nanoletters 2011, 11, 1810–1813. [Google Scholar] [CrossRef]
- Brogioli, D.; Ziano, R.; Rica, R.A.; Salerno, D.; Kozynchenko, O.; Hamelers, H.V.M.; Mantegazza, F. Exploiting the spontaneous potential of the electrodes used in the capacitive mixing technique for the extraction of energy from salinity difference. Energy Environ. Sci. 2012, 5, 9870–9880. [Google Scholar] [CrossRef]
- Kim, T.; Rahimi, M.; Logan, B.E.; Gorski, C.A. Evaluating Battery-like Reactions to Harvest Energy from Salinity Differences using Ammonium Bicarbonate Salt Solutions. ChemSusChem 2016, 9, 981–988. [Google Scholar] [CrossRef]
- Kim, T.; Rahimi, M.; Logan, B.E.; Gorski, C.A. Harvesting energy from salinity differences using battery electrodes in a concentration flow cell Harvesting energy from salinity differences using battery electrodes in a concentration flow cell. Environ. Sci. Technol. 2016. [Google Scholar] [CrossRef]
- Morais, W.G.; Gomes, W.J.A.S.; Huguenin, F. Neutralization Pseudocapacitors: An Acid-Base Machine. J. Phys. Chem. C 2016, 120, 17872–17877. [Google Scholar] [CrossRef]
- Hasan, K.N.; Khai, T.X.; Kannan, R.; Zakaria, Z.A. Harnessing Blue energy: A Review Techniques and Preliminary Analysis. MATEC Web Conf. 2017, 131, 04013. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, B.; Song, S.; Fan, Y. A membrane-less Na ion battery-based CAPMIX cell for energy extraction using water salinity gradients. RSC 2013, 3, 26205–26209. [Google Scholar] [CrossRef]
- Sharma, K.; Kim, Y.H.; Yiacoumi, S.; Gabitto, J.; Bilheux, H.Z.; Santodonato, L.J.; Mayes, R.T.; Dai, S.; Tsouris, C. Analysis and simulation of a blue energy cycle. Renew. Energy 2016, 91, 249–260. [Google Scholar] [CrossRef]
- Tan, G.; Zhu, X. Polyelectrolyte-coated CuHCF and BiOCl electrodes for efficient salinity gradient energy recovery in capacitive mixing. Energy Technol. 2019, 8. [Google Scholar] [CrossRef]
- Logan, B.E.; Elimelech, M.; States, U. Membrane-based processes for sustainable power generation using water. Nature 2012. [Google Scholar] [CrossRef]
- Sales, B.B.; Liu, F.; Schaetzle, O.; Buisman, C.J.N.; Hamelers, H.V.M. Electrochemical characterization of a supercapacitor flow cell for power production from salinity gradients. Electrochim. Acta 2012, 86, 298–304. [Google Scholar] [CrossRef]
- Pattle, R.E. Production of Electric Power by Mixing Fresh and Salt Water in the Hydroelectric Pile. Nature 1954, 174, 660. [Google Scholar] [CrossRef]
- Clampitt, B.H.; Kiviat, F.E. Energy recovery from saline water by means of electrochemical cells. Sci. New Ser. 1976, 194, 719–720. [Google Scholar] [CrossRef]
- Jia, Z.; Wang, B.; Song, S.; Fan, Y. Blue energy: Current technologies for sustainable power generation from water salinity gradient. Renew. Sustain. Energy Rev. 2014, 31, 91–100. [Google Scholar] [CrossRef]
- Marino, M.; Kozynchenko, O.; Tennison, S.; Brogioli, D. Capacitive mixing with electrodes of the same kind for energy production from salinity differences. J. Phys. Condens. Matter 2016, 28, 114004. [Google Scholar] [CrossRef]
- Post, J.W.; Hamelers, H.V.M.; Buisman, C.J.N. Energy Recovery from Controlled Mixing Salt and Fresh Water with a Reverse Electrodialysis System. Environ. Sci. Technol. 2008, 42, 5785–5790. [Google Scholar] [CrossRef]
- Pasta, M.; Battistel, A.; Mantia, F. La Batteries for lithium recovery from brines. Energy Environ. Sci. 2012, 5, 9487–9491. [Google Scholar] [CrossRef]
- Ye, M.; Pasta, M.; Xie, X.; Cui, Y.; Criddle, S. Performance of a Mixing Entropy Battery Alternately Flushed with Wastewater Effluent and Seawater for Recovery of Salinity-gradient Energy - Suplementary Information. Energy Environ. Sci. 2014, 1–8. [Google Scholar]
- Tehrani, S.H.M.H.; Seyedsadjadi, S.A.; Ghaffarinejad, A. Application of electrodeposited cobalt hexacyanoferrate fi lm to extract energy from water salinity gradients. RSC Adv. 2015, 5, 30032–30037. [Google Scholar] [CrossRef]
- Nie, P.; Yuan, J.; Wang, J.; Le, Z.; Xu, G.; Hao, L.; Pang, G.; Wu, Y.; Yan, X.; Zhang, X. Prussian Blue Analogue with Fast Kinetics Through Electronic Coupling for Sodium Ion Batteries. Appl. Mater. Interfaces 2017, 9, 20306–20312. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Hu, Q.; Yu, Y.; Wang, H.; Tang, Z.; Wen, Z.; Chen, C. Potassium-rich iron hexacyanoferrate/dipotassium terephthalate @carbon nanotube composite for K-ion full cells with optimized electrolyte. J. Mater. Chem. A 2017, 0, 19017–19024. [Google Scholar] [CrossRef]
- Rica, R.A.; Ziano, R.; Salerno, D.; Mantegazza, F.; van Roij, R.; Brogioli, D. Capacitive Mixing for Harvesting the Free Energy of Solutions at Different Concentrations. Entropy 2013, 15, 1388–1407. [Google Scholar] [CrossRef]
- Trocoli, R.; Bidhendi, G.K.; Mantia, F. La Lithium recovery by means of electrochemical ion pumping: A comparison between salt capturing and selective exchange. J. Phys. Condens. Matter 2016, 28, 114005. [Google Scholar] [CrossRef]
- Pasta, M.; Wessells, C.D.; Cui, Y.; La Mantia, F. A Desalination Battery. Nanoletters 2012, 12, 839–843. [Google Scholar] [CrossRef]
- Senthilkumar, S.T.; Go, W.; Han, J.; Thuy, L.P.T.; Kishor, K.; Kim, Y.; Kim, Y. Emergence of rechargeable seawater batteries. J. Mater. Chem. A 2019, 7, 22803–22825. [Google Scholar] [CrossRef]
- Gude, G. Geothermal Source for Water Desalination—Challenges and Opportunities; Butterworth-Heinemann: Oxford, UK, 2018; Volume 1, ISBN 9780128152447. [Google Scholar]
- Tapia, J.; González, R.; Townley, B.; Oliveros, V.; Álvarez, F.; Aguilar, G.; Menzies, A.; Calderón, M. Geology and geochemistry of the Atacama Desert. Antonie van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2018, 111, 1273–1291. [Google Scholar] [CrossRef]
- Bardi, U. Extracting Minerals from Seawater: An Energy Analysis. Sustainability 2010, 2, 980–992. [Google Scholar] [CrossRef]
- Cortecci, G.; Boschetti, T.; Mussi, M.; Lameli, C.H.; Mucchino, C.; Barbieri, M. New chemical and original isotopic data on waters from El Tatio geothermal field, northern Chile. Geochem. J. 2005, 39, 547–571. [Google Scholar] [CrossRef]
- Taylor, P.; Park, J.; Sato, H.; Nishihama, S. Solvent Extraction and Ion Exchange Lithium Recovery from Geothermal Water by Combined Adsorption Methods. Solvent Extr. Ion Exch. 2012, 30, 398–404. [Google Scholar] [CrossRef]
- Ogawa, Y.; Koibuchi, H.; Suto, K.; Inoue, C. Effects of the Chemical Compositions of Salars de Uyuni and Atacama Brines on Lithium Concentration during Evaporation. Resour. Geol. 2014, 64, 91–101. [Google Scholar] [CrossRef]
- Guduru, R.K.; Icaza, J.C. A Brief Review on Multivalent Intercalation Batteries with Aqueous Electrolytes. Nanomaterials 2016, 6, 41. [Google Scholar] [CrossRef]
- Yang, N.; Sun, H. Biocoordination chemistry of bismuth: Recent advances. Coord. Chem. Rev. 2007, 251, 2354–2366. [Google Scholar] [CrossRef]
- Ma, F.; Li, Q.; Wang, T.; Zhang, H.; Wu, G. Energy storage materials derived from Prussian blue analogues. Sci. Bull. 2017, 62, 358–368. [Google Scholar] [CrossRef]
- Gomes, W.J.A.S.; De Oliveira, C.; Huguenin, F. Energy Harvesting by Nickel Prussian Blue Analogue Electrode in Neutralization and Mixing Entropy Batteries. Langmuir 2015, 31, 8710–8717. [Google Scholar] [CrossRef]
- Ye, M.; Pasta, M.; Xie, X.; Dubrawski, K.L.; Xu, J.; Liu, C.; Cui, Y.; Criddle, C.S. Charge-Free Mixing Entropy Battery Enabled by Low-Cost Electrode Materials. ACS Omega 2019, 4, 11785–11790. [Google Scholar] [CrossRef]
- Berrettoni, M.; Giorgetti, M.; Zamponi, S.; Conti, P.; Ranganathan, D.; Zanotto, A.; Saladino, M.L.; Caponetti, E. Synthesis and Characterization of Nanostructured Cobalt Hexacyanoferrate. J. Phys. Chem. C 2010, 114, 6401–6407. [Google Scholar] [CrossRef]
- Kholoud, E.; Watanabe, H.; Takahashi, A.; Emara, M.M.; Abd-El-Nabey, B.A.; Kurihara, M.; Tajima, K.; Kawamoto, T. Cobalt haxacyanoferrate nanoparticles for wet-processed brown-bleached electrochromic devices with hydridization of high spin/low-spin phases. J. Mater. Chem. C 2017, 5, 8921–8926. [Google Scholar] [CrossRef]
- Martínez-García, R.; Knobel, M.; Balmaseda, J.; Yee-Madeira, H.; Reguera, E. Mixed valence states in cobalt iron cyanide. J. Phys. Chem. Solids 2007, 68, 290–298. [Google Scholar] [CrossRef]
- Wessells, C.D.; Peddada, S.V.; Huggins, R.A.; Cui, Y. Nickel Hexacyanoferrate Nanoparticle Electrodes For Aqueous Sodium and Potassium Ion Batteries. Nanoletters 2011, 11, 5421–5425. [Google Scholar] [CrossRef]
- Malecki, G.; Ratuszna, A. Crystal structure of cyanometallates Me3[Co(CN)6]2 and KMe[Fe(CN)6] with Me=Mn2+, Ni2+, Cu2+. Powder Diffr. 1999, 14, 25–30. [Google Scholar] [CrossRef]
- Loos-Neskovic, C.; Ayrault, S.; Badillo, V.; Jimenez, B.; Garnier, E.; Fedoroff, M.; Jones, D.J.; Merinov, B. Structure of copper-potassium hexacyanoferrate ( II ) and sorption mechanisms of cesium. J. Solid State Chem. 2004, 177, 1817–1828. [Google Scholar] [CrossRef]
- Intaranont, N.; Garcia-Araez, N.; Hector, A.L.; Milton, J.A.; Owen, J.R. Selective lithium extraction from brine by chemical reaction with battery materials. J. Mater. Chem. A 2014, 2, 6374–6377. [Google Scholar] [CrossRef]
- Biendicho, J.J.; West, A.R. Thermally-induced cation disorder in LiFePO4. Solid State Ionics 2011, 203, 33–36. [Google Scholar] [CrossRef]
- Liu, S.; Fan, C.; Zhang, Y.; Li, C.; You, X. Low-temperature synthesis of Na 2 Mn 5 O 10 for supercapacitor applications. J. Power Sources 2011, 196, 10502–10506. [Google Scholar] [CrossRef]
- Ghodbane, O.; Pascal, J.; Charles, I.; Montpellier, G.; Cnrs, U.M.R.; Aime, E. Microstructural Effects on Charge-Storage Properties in MnO 2 -Based Electrochemical. ACS Appl. Mater. Interfaces 2009, 1, 1130–1139. [Google Scholar] [CrossRef]
- Liu, X.; Zhang, N.; Ni, J.; Gao, L. Improved electrochemical performance of sol – gel method prepared Na 4 Mn 9 O 18 in aqueous hybrid Na-ion supercapacitor. J. Solid State Electrochem. 2013, 1, 1939–1944. [Google Scholar] [CrossRef]
- Jiang, X.; Sha, Y.; Cai, R.; Shao, Z. The solid-state chelation synthesis of LiNi1/3Co1/3Mn1/3O2 as a cathode material for lithium-ion batteries. J. Mater. Chem. A Mater. Energy Sustain. 2015, 00, 1–9. [Google Scholar] [CrossRef]
- Palacín, M.R. Recent advances in rechargeable battery materials: A chemist’s perspective. Chem. Soc. Rev. 2009. [Google Scholar] [CrossRef]
- Meng, J.; Guo, H.; Niu, C.; Zhao, Y.; Xu, L.; Li, Q.; Mai, L. Advances in Structure and Property Optimizations of Battery Electrode Materials. Joule 2017. [Google Scholar] [CrossRef]
- Bro, P.; Marincic, N.; Mallory, P.R.; Science, P. The High Rate Oxidation of Silver Electrodes in Chloride Solutions. J. Eletrochemical Soc. 1970, 116, 1338–1341. [Google Scholar] [CrossRef]
- Jaya, S.; Rao, T.P.; Rao, G.P. Mono- and multilayer formation studies of silver chloride on silver electrodes from chloride-containing solutions. J. Appl. Electrochem. 1987, 17, 635–640. [Google Scholar] [CrossRef]
- Margalit, N. Cathodes for Seawater Activated Cells. J. Electrochem. Soc. 1975, 122, 1005–1008. [Google Scholar] [CrossRef]
- Birss, V.I.; Smith, C.K. The anodic behavior the formation and reduction thin silver chloride films. Electrochim. Acta 1987, 32, 259–268. [Google Scholar] [CrossRef]
- Ha, H.; Payer, J. Electrochimica Acta The effect of silver chloride formation on the kinetics of silver dissolution in chloride solution. Electrochim. Acta 2011, 56, 2781–2791. [Google Scholar] [CrossRef]
- Jin, X.; Lu, J.; Liu, P.; Tong, H. The electrochemical formation and reduction of a thick AgCl deposition layer on a sil v er substrate. J. Electroanal. Chem. 2003, 542, 85–96. [Google Scholar] [CrossRef]
- Keramidas, G.; Voutsas, G.P.; Rentzeperis, P.I. The crystal structure of BiOCl. Zeitschrift für Krist. 1993, 205, 35–40. [Google Scholar] [CrossRef]
- Su, X.; Hatton, T.A. Redox-electrodes for selective electrochemical separations. Adv. Colloid Interface Sci. 2017, 244, 6–20. [Google Scholar] [CrossRef] [PubMed]
- Pasta, M.; Wessells, C.D.; Huggins, R.A.; Cui, Y. A high-rate and long cycle life aqueous electrolyte battery for grid-scale energy storage. Nat. Commun. 2012, 3, 1147–1149. [Google Scholar] [CrossRef] [PubMed]
- Morais, W.G.; Lima, G.; Gomes, W.J.A.S.; Huguenin, F. Electrochemical Systems for Renewable Energy Conversion from Salinity and Proton Gradients. J. Brazil Chem. Soc. 2018, 29, 934–947. [Google Scholar] [CrossRef]
- Stejskal, J.; Trchova, M.; Bober, P.; Moravkova, Z.; Kopecky, D.; Vrñata, M.; Prokes, J.; Varga, M.; Watzlova, E. Polypyrrole salts and bases: Superior conductivity of nanotubes and their stability towards the loss of conductivity by deprotonation. RSC Adv. 2016, 6, 88382–88391. [Google Scholar] [CrossRef]
- Nam, D.; Choi, K. Bismuth as a New Chloride-Storage Electrode Enabling the Construction of a Practical High Capacity Desalination Battery. J. Am. Chem. Soc. 2017, 139, 11055–11063. [Google Scholar] [CrossRef]
- Musabikha, S.; Ketut Aria Pria Utama, I. Mukhtasor Corrosion in the Marine Renewable Energy: A Review. Proc. Ocean. Mech. Aerosp. 2016, 3, 121–128. [Google Scholar]
- Kristoferson, L.A.; Bokalders, V. Solar Water Distillation. In Renewable Energy Technologies; Pergamon: Oxford, UK, 1986; pp. 215–218. [Google Scholar]
- Miyai, Y.; Ooi, K.; Katoh, S. Recovery of Lithium from Seawater Using a New Type of Ion-Sieve Adsorbent Based on MgMn2O4. Sep. Sci. Technol. 1988, 23, 179–191. [Google Scholar] [CrossRef]
- Liu, M.; Rong, Z.; Malik, R.; Canepa, P.; Jain, A.; Ceder, G.; Persson, K.A. Spinel compounds as multivalent battery cathodes: A systematic evaluation based on ab initio calculations. Energy Environ. Sci. 2015, 8, 964–974. [Google Scholar] [CrossRef]
- Mao, M.; Gao, T.; Wang, C. A critical review of cathodes for rechargeable Mg batteries. Chem. Soc. Rev. 2018, 47, 8804–8841. [Google Scholar] [CrossRef]
- Kim, S.; Lee, J.; Kim, C.; Yoon, J. Na2FeP2O7 as Novel Material for Hybrid Capacitive Deionization. Electrochim. Acta 2016, 255, 369–378. [Google Scholar] [CrossRef]
- Huang, Y.; Cheng, F.; Guo, L.; Yang, H.Y. Ultrahigh Performance of Novel Electrochemical Deionization System based on NaTi2(PO4)3/rGo Nanocomposite. J. Mater. Chem. A 2017, 5, 18157–18165. [Google Scholar] [CrossRef]
- Chen, F.; Huang, Y.; Guo, L.; Ding, M.; Yang, H.Y. A dual-ion electrochemistry deionization system based on AgCl-Na0.44MnO2 electrodes. Nanoscale 2017, 9, 10101–10108. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Mo, R.; Shi, W.; Huang, Y.; Leong, Z.Y.; Ding, M.; Chen, F.; Yang, H.Y. Prussian blue anode for high performance electrochemical deionization promoted by faradic mechanism. Nanoscale 2017, 9, 13305–13312. [Google Scholar] [CrossRef] [PubMed]
- Desai, D.; Beh, E.S.; Sahu, S.K.; Vedharathinam, V.; Van, Q.; De Lannoy, C.; Jose, A.P.; Volkel, A.R.; Rivest, J. Electrochemical Desalination of Seawater and Hypersaline Brines with Coupled Electricity Storage. ACS Energy Lett. 2018, 3, 375–379. [Google Scholar] [CrossRef]
- Choi, S.; Chang, B.; Kim, S.; Lee, J.; Yoon, J.; Choi, J.W. Battery Electrode Materials with Omnivalent Cation Storage for Fast and Charge-Efficient Ion Removal of Asymmetric Capacitive Deionization. Adv. Funct. Mater. 2018, 1802665, 1–9. [Google Scholar] [CrossRef]
- Lee, J.; Kim, S.; Kim, C.; Yoon, J. Hybrid capacitive deionization to enhance the desalination performance of capacitive techniques. Energy Environ. Sci. 2017, 00, 1–7. [Google Scholar] [CrossRef]
- Zhu, X.; Xu, W.; Tan, G.; Wang, Y. Concentration Flow Cells for Efficient Salinity Gradient Energy Recovery with Nanostructured Open Framework Hexacyanoferrate Electrodes. Energy Technol. Environ. Sci. 2018, 3, 5571–5580. [Google Scholar] [CrossRef]
- Li, G.; Yang, Z.; Jiang, Y.; Jin, C.; Huang, W.; Ding, X. Towards polyvalent ion batteries: A zinc-ion battery based on NASICON. Nano Energy 2016, 25, 211–217. [Google Scholar] [CrossRef]
- Qu, Q.T.; Shi, Y.; Tian, S.; Chen, Y.H.; Wu, Y.P.; Holze, R. Short communication A new cheap asymmetric aqueous supercapacitor: Activated carbon // NaMnO 2. J. Power Sources 2009, 194, 1222–1225. [Google Scholar] [CrossRef]
- Shaz, M.; Van Smaalen, S.; Palatinus, L.; Hoinkis, M.; Klemm, M.; Horn, S.; Claessen, R. Spin-Peierls transition in TiOCl. Physcal Rev. Lett. 2005, B71, 100405. [Google Scholar] [CrossRef]
- Gao, P.; Zhao, X.; Zhao-karger, Z.; Diemant, T.; Behm, R.J.; Fichtner, M. Vanadium Oxychloride / Magnesium Electrode Systems for Chloride Ion Batteries. Appl. Mater. Interfaces 2014, 6, 22430–22435. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Reddy, M.A.; Mu, X.; Diemant, T.; Zhang, L.; Zhao-karger, Z.; Sai, V.; Chakravadhanula, K.; Clemens, O.; Behm, R.J.; et al. Rechargeable Batteries VOCl as a Cathode for Rechargeable Chloride Ion Batteries Angewandte. Angew. Chem. Int. Ed. 2016, 55, 4285–4290. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Zhao-karger, Z.; Wang, D.; Fichtner, M. Metal Oxychlorides as Cathode Materials for Chloride Ion Batteries. Angew. Chem. Int. Ed. 2013, 52, 13621–13624. [Google Scholar] [CrossRef]
- Maguire, J.A.; Banewicz, J.J. Direct Intercalation of Alkali Metal Ions in FeOCl. Mat. Res. Bull 1984, 19, 1573–1580. [Google Scholar] [CrossRef]
- Zhao, X.; Li, Q.; Yu, T.; Yang, M.; Fink, K.; Shen, X. Carbon incorporation effects and reaction mechanism of FeOCl cathode materials for chloride ion batteries. Nat. Publ. Gr. 2016, 6, 1–8. [Google Scholar] [CrossRef]
- Lind, M.D. Refinement of the Crystal Structure of Iron Oxychloride. Acta Cryst 1970, B26, 1058–1062. [Google Scholar] [CrossRef]
- El-Halim, A.M.A.; Fawzy, M.H.; Stay, A. Saty Cyclic voltammetric behaviour and some surface characteristics of the lead electrode in aqueous NaCl solutions. J. Electroanal. Chem. 1991, 316, 275–292. [Google Scholar] [CrossRef]
- Zuo, W.; Zhu, W.; Zhao, D.; Sun, Y.; Li, Y.; Liu, J.; Lou, X.W. Bismuth oxide: A versatile high-capacity electrode material for rechargeable aqueous metal-ion batteries. Energy Environ. Sci. 2016, 9, 2881–2891. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | Natural Source | Cl− [ppm] | Na+ [ppm] | Mg2+ [ppm] | Ca2+ [ppm] | Li+ [ppm] |
|---|---|---|---|---|---|---|
| [29,32] | Seawater | 19.345 | 10.752 | 1.195 | 0.416 | 0.179 |
| [33,34] | Geothermal | 7.899 | 4.459 | 1.700 | 250 | 34,27 |
| [35] | Atacama Brine | 198.000 | 42.700 | 40.500 | 368 | 1.820 |
| Ref. | PBA | Electrode Area [cm²] | Range Potential [V] | Range Density Current [A/cm²] | N° Cycle | Gain Voltage [V] |
|---|---|---|---|---|---|---|
| [11] | CuHCF | 5 | 0.2–0.65 | ±0.1 | 10 | 0.102 |
| [7] | CuHCF | 7 | 0–0.8 | ±0.2 | 20 | 0.172 |
| [8] | CuHCF | 3 | 0–0.9 | ±0.5 | - | 0.172 |
| [13] | CuHCF | 1 | - | ±0.2 | 50 | - |
| [23] | CoHCF | 1.3 | 0–1.1 | ±0.01 | 30 | 0.153 |
| [39] | NiHCF | 1 | 0–0.6 | ±0.01 | - | - |
| Ref. | BM | Electrode Area [cm²] | Volume MEB | Range Potential [V] | Electrode Distance [mm] | Range Density Current [A/cm²] | N° Cycle |
|---|---|---|---|---|---|---|---|
| [cm³] | |||||||
| [5] | LiFePO4 | 2 | 0.35 | 0–0.5 | 10 | ± 0.5 | 100 |
| [5] | Na2Mn5O10 | 2 | 0.35 | - | 10 | ± 0.25 | 100 |
| [1] | Na2Mn5O10 | 1 | - | - | 0.6 | - | - |
| [22] | Na4Mn9O18 | 9 | 1.5 | - | 1.7 | ± 0.03 | 12 |
| [3] | Na4Mn9O18 | 3.01 | 0.06 | 0–0.6 | 0.2 | ± 0.5 | 3 |
| Ref. | Active Material | Synthesis Method | Covering Method | Active Material [%] | Substrate | Electrode Mass [mg /cm²] |
|---|---|---|---|---|---|---|
| [5] | LiFePO4 | Polymeric Synthesis | Drop Casting | 80 | Carbon Cloth | 25 |
| [22] | Na4Mn9O18 | Solid State Reaction | Ink Coating | 85 | Carbon Cloth | 14 |
| [1] | Na2Mn5O10 | Pachini Method | Doctor Blade | 80 | Graphite | 8 |
| [7] | MnO2 | Co-Precipitation | Doctor Blade | 70 | Graphite Sheet | 34 |
| [11] | CuHCF | Dropwise | Spread | 90 | Carbon Plate | 5 |
| [7] | CuHCF | Co-Precipitation | Doctor Blade | 70 | Graphite Sheet | - |
| [8] | CuHCF | Co-Precipitation | Doctor Blade | 70 | Graphite Foil | 4.4 |
| [13] | CuHCF | Co-Precipitation | Spread | 80 | Carbon Paper | 4.8 |
| [23] | CoHCF | Co-Precipitation | Electrodeposition | - | CPE | 0.193 |
| [39] | NiHCF | Co-Precipitation | Hand Painting | 80 | Carbon Cloth | - |
| Ref. | Active Material | Relevant Characteristics | Reversible Capacity [mAh/g] | Efficiency [%] | Blue Energy Recovery | |
|---|---|---|---|---|---|---|
| Advantage | Disadvantage | |||||
| [5] | LiFePO4 | High Stability | Poor E.C | 120 | - | 13.5 µW/cm2 |
| [22] | Na4Mn9O18 | High Capacity Retention | Low Charge Capacity | 113 | 68 | 0.65 kW/m3 |
| [5] | Na2Mn5O10 | High Surface Area | Low Charge Capacity | - | 75 | 10.5 µW/cm2 |
| [11] | CuHCF | High Capacity | High Irreversible Reaction | - | 69 | 13990 µW/cm2 |
| [7] | CuHCF | - | - | 411 mW/m2 | ||
| [8] | CuHCF | - | - | 6.3 mW/m2 | ||
| [13] | CuHCF | - | - | 87 mW/m2 | ||
| [23] | CoHCF | High Energy Density | Cell Voltage Limited (<1 V) | 130 | 65 | 4632 µW/cm2 |
| [39] | NiHCF | Efficient Energy Consumption | Low water recovery | 60 | - | 16.8 kJ/mol |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galleguillos, F.; Cáceres, L.; Maxwell, L.; Soliz, Á. Electrochemical Ion Pumping Device for Blue Energy Recovery: Mixing Entropy Battery. Appl. Sci. 2020, 10, 5537. https://doi.org/10.3390/app10165537
Galleguillos F, Cáceres L, Maxwell L, Soliz Á. Electrochemical Ion Pumping Device for Blue Energy Recovery: Mixing Entropy Battery. Applied Sciences. 2020; 10(16):5537. https://doi.org/10.3390/app10165537
Chicago/Turabian StyleGalleguillos, Felipe, Luis Cáceres, Lindley Maxwell, and Álvaro Soliz. 2020. "Electrochemical Ion Pumping Device for Blue Energy Recovery: Mixing Entropy Battery" Applied Sciences 10, no. 16: 5537. https://doi.org/10.3390/app10165537
APA StyleGalleguillos, F., Cáceres, L., Maxwell, L., & Soliz, Á. (2020). Electrochemical Ion Pumping Device for Blue Energy Recovery: Mixing Entropy Battery. Applied Sciences, 10(16), 5537. https://doi.org/10.3390/app10165537