1. Introduction
The use of stimuli-responsive or “smart” materials is a promising approach in drug delivery to improve the pharmacokinetic parameters and selectivity of active pharmaceutical ingredients (API) while reducing the dosage and side effects [
1,
2]. The use of stimuli-responsive materials to develop carriers for drug delivery has been found to be of particular interest in cancer therapy, in which these materials take advantage of the different features of the tumoral environment compared to the healthy one, e.g., lowered interstitial pH [
3], high intracellular concentration of glutathione [
4,
5] and expression of different enzymes [
6,
7], which can be used as triggers. Besides the physiological triggers, the use of external stimuli can also be applied to stimulate and control the release of the payload from the carrier. In cancer therapy, ultrasonic and magnetic fields are the most attractive external stimuli; in fact, their application can increase the temperature of a selected area, providing the “on-demand” release of the therapeutic and simultaneously causing cell death by hyperthermia [
8]. Hybrid carriers made by combining organic and inorganic materials have demonstrated the ability to respond simultaneously to physical, chemical or biological stimuli and provide a release profile suitable for the API to improve therapeutic efficacy and reduce side effects.
Core–shell hybrid systems, where the core is composed of inorganic material and the shell is composed of organic material, generally polymers, are attracting great attention [
9]. Iron- and iron-oxide-based particles are good candidates, and they have been extensively investigated in biomedical field as delivery systems and contrast agents and in hyperthermia therapy [
10,
11,
12]. However, their main drawback is related to their low stability in solution and their accompanying tendency to form large aggregates and precipitate. Several strategies aiming to overcome this drawback have been reported, in particular dealing with the surface modification by introduction of, e.g., amino, hydroxy and carboxylic functional groups. Polymer coating represents another widely used approach, as it provides a steric barrier improving the colloidal stability and preventing the agglomeration [
13,
14]. The properties of the resulting carrier depend on the nature of the coating agent, such as its chemical structure (e.g., hydrophilicity/hydrophobicity and biodegradation characteristics), molecular weight and mechanism of anchoring or attachment (e.g., electrostatic or covalent bonding) [
15,
16,
17,
18].
To develop core–shell carriers, natural or synthetic polymers have been considered for the shell [
19], among which polysaccharides have received considerable attention due to peculiarities like biodegradability, biocompatibility, low toxicity and presence of different functional groups which allow chemical modification [
20].
Among the polysaccharides, chitosan has received great attention due to the high versatility provided by its chemical structure. Chitosan is made of D-glucosamine and N-acetyl D-glucosamine units. The two hydroxyl groups in C3 and C6 and the amino group in the deacetylated D-glucosamine unit are the active sites for further conjugation with inorganic core, therapeutics, targeting ligands and imaging agents.
Following the positive results obtained by our group in a previous study [
12] in which the aim was to enhance the stability of iron nanoparticles in solution by surface modification and coating, the main aim in the present study was to make the system sensitive to magnetic and ultrasonic fields and variation in the surrounding pH to control the release rate and intensity of a chemotherapeutic drug.
Doxorubicin (DOX), has been considered as a model drug. It is an anthracycline antibiotic, and it is currently used in the treatment of leukemia, breast cancer, ovarian cancer and lymphoma [
21,
22] Besides the therapeutic efficacy, DOX exhibits severe side effects, such as cardiotoxicity, related to the low selectivity. Additionally, multidrug resistance usually develops after DOX treatment, preventing further therapy with DOX. Therefore, considerable attention has been paid to developing suitable carriers to improve the cytotoxicity of DOX, which would maximize the therapeutic value while minimizing any side effects [
12,
23].
As a result, the reported methodologies of preparation and modification (via arene diazonium tosylate with the addition of the amphiphilic coat) of iron microparticles allow preparing stimuli-responsive drug carriers with high stability, drug loading capacity and the possibility to control the release rate. The use of the carrier improved the DOX cytotoxicity compared to that of the free formulation, with a decrease in the required dosage.
2. Materials and Methods
2.1. Materials
The following reagents were supplied by Sigma Aldrich at reagent grade: low-molecular-weight chitosan (CS) (Mw < 104 g/mol, D.D 75–85%); N-hydroxysuccinimide (NHS); N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC), powder; methanesulfonic acid (MSA); N,N-dimethylformamide 99%; tert-butyl nitrite; 4-toluenesulfonic acid (p-TsOH); 4-aminobenzoic acid; iron trichloride (FeCl3); sodium borohydride; and doxorubicin hydrochloride (drug reference standard). C3H6O3 L-Lactic acid, 80% water solution, was purchased from Lachner Neratovice, Czech Republic. Laboratory-grade sodium chloride, potassium dihydrogen phosphate, sodium carbonate and sodium hydroxide were acquired from Penta, Prague, Czech Republic. The C3H6O solvent acetone, sodium hydroxide, sodium chloride, sodium phosphate, and potassium phosphate (all reagent grade) were bought from IPL Lukes, Uhresky Brod, Czech Republic. Chloroform CHCl3 (HPLC grade), acetic acid CH3CO2H (HPLC grade), and hydrochloric acid (reagent grade) were purchased from Chromservis, Prague, Czech Republic.
2.2. Synthesis of Amphiphilic Chitosan Derivative
The amphiphilic chitosan derivative (PLACA-g-CS) was obtained by grafting carboxy-enriched low-molecular-weight poly(lactic acid) (M
w 10 kDa) to the chitosan (CS) backbone by carbodiimide chemistry involving amino and carboxylic groups. The carboxy-enriched polylactic acid (PLACA) was synthesized as described elsewhere [
24,
25] The amphiphilic chitosan (PLACA-g-CS) was prepared by soaking 0.5 g of CS in dimethylformamide overnight and then dissolving it in aqueous solution containing 1% v/v of acetic acid at the concentration of 2 mg/mL. Afterwards, 0.5 g of PLACA (M
w 10,000 g/mol), EDC and N-hydroxysuccinimide (NHS) (at a 2:3:6 molar ratio of PLACA:EDC:NHS) were dissolved in 50 mL of chloroform, then the solution was mixed with that containing CS and stirred for 48 h at room temperature. The reaction was stopped, and the final product was precipitated by adding NaOH 0.1 M and then recovered by centrifugation at 14,000 rpm for 20 min. The pellet was washed with chloroform and then water (containing 1% v/v of acetic acid) to remove any unreacted components, and then it was frozen at −20°C for 24 h and dried under vacuum.
The
1H-NMR analysis was performed to prove that grafting had taken place. Spectra were recorded on a Varian Unity Inova 400 MHz unit; 16–32 scans were collected with a recycle delay time of 4 to 6 s between the scans, and deuterium oxide supplemented with 5% deuterium hydrochloric acid and 1 µL (CH
3)
3COD was used for signal referencing (δ 1.23 ppm). The spectra were evaluated using first-order spectral analysis.
1H-NMR spectra can be found in the
Supplementary Materials.
The degree of deacetylation and the number of free amino groups in the PLACA-g-CS conjugate were determined by conductometric titration, in adherence with a well-established procedure [
26].
2.3. Preparation and Surface Modification of Zero-Valent Iron Microparticles
The zero-valent surface-modified iron oxide (ZV) microparticles were prepared in two steps: (i) preparation of the iron oxide (IO) microparticles and (ii) surface modification by diazonium chemistry [
12,
24]. In brief, FeCl
3 × 6H
2O (0.406 g, 1.5 mmol) and NaBH
4 (0.171 g, 4.5 mmol) were dissolved in deionized water (10 mL). Then, 5 mL of the FeCl
3 × 6H
2O solution and 5 mL of the NaBH
4 solution were combined in a triple-neck flask and stirred for 10 min under an argon atmosphere, forming a black-colored precipitate. Next, an additional 5 mL of the FeCl
3 × 6H
2O solution and 5 mL of the NaBH
4 solution were added simultaneously to the reaction mixture via syringe for the growth of microparticles. The resulting suspension was stirred for 10 min in an argon atmosphere. Then, 20 mL of the aqueous solution of 4-carboxybenzenediazonium tosylate (0.3 g) was added in the prepared suspension of microparticles in one portion and stirred for 40 min. The particles were isolated using a permanent magnet and washed with distilled water, ethanol and acetone in order to obtain a clear solution above the particles. To remove the traces of solvents, microparticles were freeze-dried.
FTIR-ATR (Attenuated total reflection-infrared spectroscopy) determined the presence of an organic layer FTIR Spectrometer (Nicolet iS5, Thermo Fisher Scienific) equipped with an iD5 ATR accessory and ZnSe crystal; resolution 4 cm−1; 64 scans).
2.4. Preparation of Core–Shell Iron Microparticles
The core–shell microparticles were prepared by linking PLACA-g-CS to the surface of the ZV microparticles through amide bonding between the free CS amino groups and the ZV carboxyl groups by carbodiimide chemistry [
12]. In brief, 4.6 mg of
N-hydroxysuccinimide (NHS), 3.8 mg N-(3-dimethylaminopropyl)-N’-ethylcarbodiimide hydrochloride (EDC) and 20 mg of iron microparticles were added to 20 mL of distilled water and stirred mechanically for 6 h. The obtained suspension was added to 30 mL of chloroform solution containing PLACA-g-CS (200 mg). The mixture was stirred for 48 h at 7500 rpm. Afterward, the particles were recovered by a magnet, accurately washed by distilled water to remove all the unreacted components and then freeze-dried.
The formation of the amide bond was detected by ATR-FTIR (Nicolet iS5 FTIR Spectrometer equipped with an iD5 ATR accessory and Ge crystal; resolution 4 cm−1; 64 scans). Microparticle morphology and size in solution were investigated by transmission electronic microscopy (TEM- JEOL JEM 2100) and by laser light scattering (Mastersize 3000, Malven Panalytic). Particle dispersions were measured under the following specifications: data acquisition rate of 10 kHz; measurement time 10 s; temperature 25 °C. Data were processed by software according to Fraunhofer diffraction and Mie scattering theory.
The influence of the surface modification and the presence of the polymer coat on the magnetic properties of the microparticles was measured by vibrating-sample magnetometer (VSM 7407 LakeShore Cryotronics) at room temperature.Applied field strength 2.17 Tesla (T).
2.5. Encapsulation Efficiency and Release Studies
Following the positive results obtained in our previous work [
12], where the optimal conditions for maximum encapsulation efficiency of DOX in a similar system were found, the initial drug concentration of 1 mg/mL (total volume 5 mL) was used, with a weight ratio between drug and the carrier of 1:10 and preparation pH of 5.5.
An aqueous solution of DOX (1 mg/mL) was added to the microparticle solution (10 mg/mL; volume of 5 mL) and stirred mechanically for 24 h in the dark. Afterward, the mixture was centrifuged at 14,000 rpm for 15 min, and the pellet was recovered and washed with distilled water. The supernatant was analyzed by UV-Vis (Perkins-Cary 300 Ultraviolet-Visible) at 480 nm to determine the amount of DOX unloaded. To calculate the encapsulation efficiency, a standard curve was used; the encapsulation efficiency (EE) was calculated using the following equation:
where
Dt represents the total theoretical amount of the drug added (mg/mL), and
Df equals the concentration after the encapsulation process (mg/mL).
2.5.1. Passive Release
The effect of pH (3, 5 and 7.4) on the DOX release was investigated under thermostable conditions (37 °C) and orbital shaking (150 rpm, on a Stuart Incubator Orbital Shaker, type GFL 3033).
Release studies were carried out in the following media: phosphate buffer solution, pH 3.0; citrate-phosphate buffer, pH 5.0; and phosphate buffer, pH 7.4.
Briefly, 50 mg of the sample was suspended in 50 mL of the medium; at predetermined time intervals, an aliquot (3 mL) was withdrawn and analyzed by a UV-Vis spectrophotometer (Cary UV 300), with the absorbance at 480 nm being recorded. The dissolution medium was replaced with a fresh one to maintain the total volume constant. The amount of drug released (DR) was determined by the following equation:
where
D(t) (mg) represents the drug released at time
t, and
D(0) (mg) represents the amount of loaded drug. All studies were conducted in triplicates.
2.5.2. Ultrasound-Triggered Release
The sensitivity of the microparticles to ultrasound and the response in terms of doxorubicin release rate were evaluated using the same setup as for the passive release (pH 3; pH5 and 7.4). Two sets of solutions containing nanoparticles (1 mg/mL, volume of 50 mL) were transferred into plastic tubes and placed into a water bath (37 °C). The experimental conditions were as follows: US power (2 and 20 W/cm2), application time of 10 s, and 30 s per cycle. After each cycle and between two consecutive cycles, 3 mL of the medium was withdrawn and replaced by a fresh 3 mL to maintain the total volume constant. The amount of DOX in the release medium was evaluated by the UV-Vis spectrophotometer, as described above.
2.6. Cytotoxicity and Hemolysis Assay
Cytotoxicity was determined by MTT assay using a fibroblast NIH/3T3 cell line. The cells were cultured in the medium (2.5 × 10
5 cells/mL) containing the samples for 24, 48 and 72 h. Culture medium without microparticles, bare microparticles and DOX were used as controls to evaluate the benefits of the microcarriers. The microparticle suspensions were prepared in the cell growth medium at 50 µg/mL concentration. The cells were washed with 200 μL of PBS per well, and then 50 µL of 1 mg/mL MTT solution was added to each well. After 2 h incubation, the MTT solution was removed by aspiration, and 50 µL of isopropanol was added to dissolve the formazan crystals. A Tecan multiplate reader was used to evaluate the optical density at 595 nm. Cell viability percentage was calculated as the ratio of mean absorbance of triplicate readings of sample wells (
I sample) compared to the mean absorbance of control wells (
Icontrol):
The cytotoxicity, in combination with ultrasound, was evaluated. The formulations were seeded in 96-well plate containing 2.5 × 105 cells/mL and exposed to 1 MHz ultrasound (2 and 20 W/cm2) using the same timing adopted in the in vitro release studies. Evaluation after 48 and 72 h was conducted to understand the effect of ultrasound treatment over time.
Hemocompatibility of bare microparticles was evaluated by the hemolysis assay, following a reported protocol [
27,
28], where first 1.5 mL of acid citrate dextrose (ACD) was added to 10 mL of a human blood sample. Afterwards, 100 µL of the formulation containing particles (concentration ranging from 5 to 100 µg/mL) was added to 1 mL of the treated sample and incubated for 2 h at 37 °C under orbital shaking. The samples were centrifuged at 6000 rpm for 15 min; the plasma was collected (100 µL plasma + 1 mL Na
2CO
3); and the OD values at 450, 380 and 415 nm were recorded. The obtained values were compared with the controls. In order to achieve 100% hemolysis, the cells were lysed using the nonionic detergent Triton X-100 as positive control, while PEG (1000 µg/mL) was used as a negative control. Each assay was performed in triplicate.
2.7. Statistical Analysis
All the reported analyses were performed in triplicate, and all data were processed by one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test in GraphPad Prism software (Version 6.04, San Diego, CA, USA). Probability values (p) of less than 0.05 were considered statistically significant.
3. Results and Discussions
3.1. Amphiphilic Chitosan Characterization
The success of the conjugation of PLACA to the CS backbone has been evaluated by
1H-NMR spectroscopy (
Figure S1). The relative spectra and the evaluation of free amino groups along the CS backbone after modification are reported in the
Supplementary Materials.
3.2. Microparticles Characterization
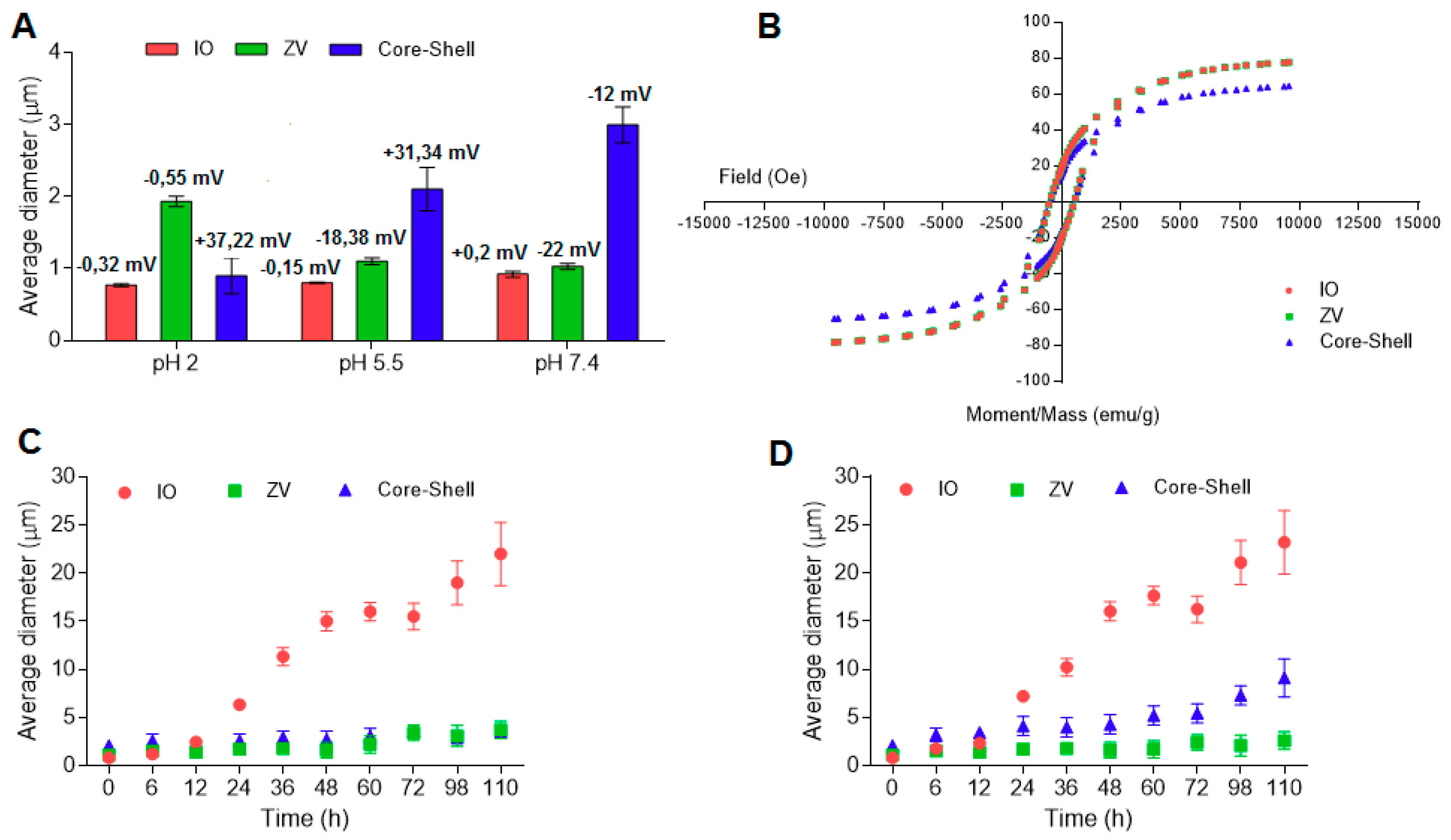
Figure 1 shows the effects of surface modification, the conjugation with the amphiphilic PLACA-g-CS and the media pH on the average diameter and the ζ-potential of the microparticles in solution.
In the preparation medium (phosphate buffer pH 5.5), the introduction of the organic layer shifts the microparticle size from 800 nm to 1 µm, while it increases 2-fold, reaching the dimension of 2 μm, after coating. Surface modification and coating strongly affect the ζ-potential value. IO particles have ζ-potential close to zero, as no ionizable groups are displaced on the surface. Following the modification with 4-carboxybenzenediazonium tosylate, the presence of carboxylic groups in the ionic state (pH 5.5 and 7.4) cause a drop in the ζ-potential, which assumes a negative value. Further conjugation with the amphiphilic chitosan increases the ζ-potential to positive values (pH 2 and 5.5), ensuring stability in the solution. At pH 7.4, the ζ-potential drops to a negative value due to the deprotonation of the amino groups.
The influence of the surface modification and the further conjugation with the PLACA-g-CS polymer on the magnetic properties of the microparticles was investigated by vibrating-sample magnetometer, using IO as control [
12]. The saturation magnetization value (M
s) was 78 emu/g in IO, and no significant variation in M
s was detected after the introduction of the organic layer. Conversely, addition of the polymer shell caused a 20% reduction in the magnetic response (
Figure 1B). As previously described [
27,
29], an Ms value of 16.3 emu/g is necessary to separate IO microparticles in aqueous solution by employing an external magnetic field [
30]. With this supposition, the adopted set-up can be considered optimal for preserving the magnetic response.
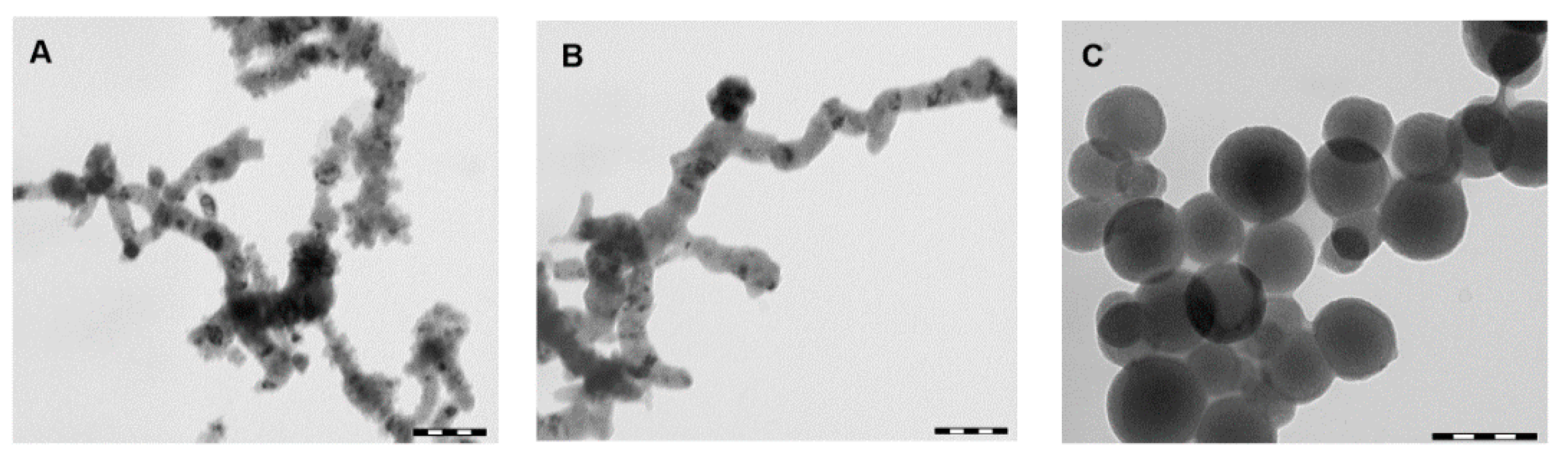
Figure 2 shows TEM micrographs of IO, ZV and core-shell microparticles.
As can be seen in
Figure 2A,B, IO and ZV microparticles tend to aggregate in a string-like structure, which means low stability in solution [
31,
32,
33]. The TEM micrograph reported in
Figure 2B shows that not enough carboxylic groups are present on the surface of the microparticles in order to stabilize them and avoid the aggregation phenomena. Conversely, after conjugation with PLACA-g-CS (
Figure 2C), no string-like structure is observed; this is due to an increase in the microparticles’ stability in solution, which can be ascribed to electrostatic repulsion between the positively charged microparticles.
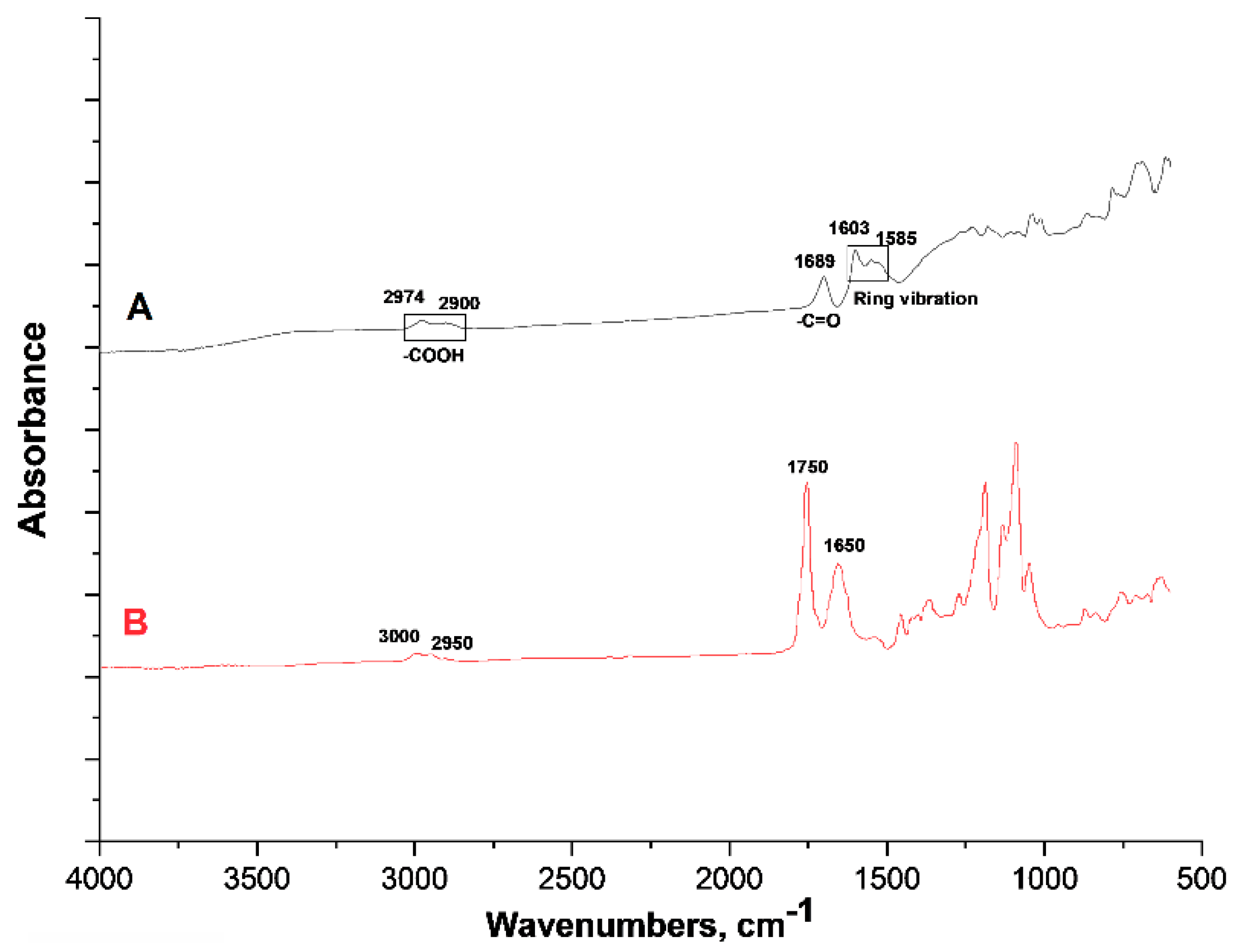
The presence of the PLACA-g-CS has been proved by ATR-FTIR spectroscopy, using ZV microparticles as control to demonstrate the formation of the new amide bond.
Figure 3 shows the FTIR-ATR spectra relative to ZV and core-shell microparticles.
In the spectra of ZV microparticles (
Figure 3, Track A), the peaks at 2974, 2900 and 1689 cm
−1 are ascribed to the presence of carboxylic groups, while those at 1603 and 1585 cm
−1 to the benzene ring [
26,
27]. In the spectra referring to the core–shell particles (
Figure 3, Track B), the peaks at 3000 and 2950 cm
−1 are related to carboxylic groups and methyl groups (-CH
3) belonging to the PLACA structure. The peak at 1650 cm
−1 might refer to the amide bonds between PLACA and CS and between the carboxylic groups in the ZV particles and CS amino groups. The peak at 1750 cm
−1 refers to the carbonyl group of PLACA, confirming its presence in the structure as reported previously [
19]. The peaks in the range between 1000 and 1400 cm
−1 are related to chitosan, as they are located at the frequency that is typical for the polysaccharide ring.
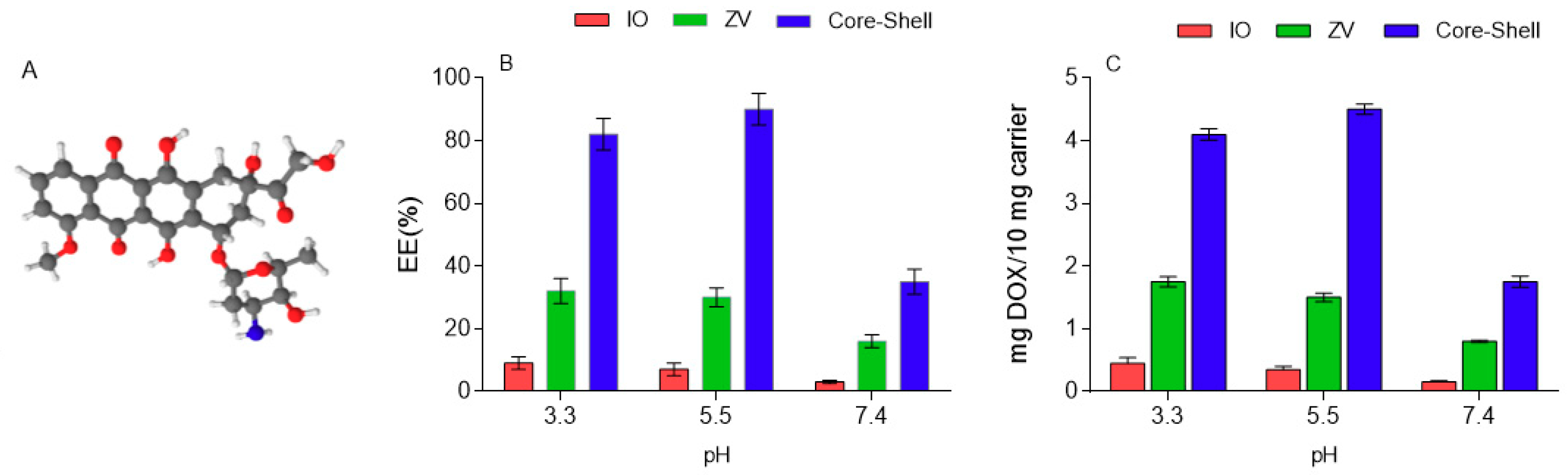
3.3. DOX Encapsulation Efficiency
Figure 4 shows the effect of surface modification and polymer coating on the EE of DOX under different pH levels. IO microparticles are considered as control, and a low EE (less than 10%) is observable, tending to decrease with the increase of the pH. In IO particles, the DOX is physically adsorbed but then rapidly desorbed from the surface. In ZV particles, the presence of carboxylic groups provides a stronger interaction with DOX, consequently allowing a greater amount to be held on the surface. The best results are obtained when particles are coated with PLACA-g-CS, as steric entrapment also takes place along with electrostatic and H-bond interactions, enhancing the ability to entrap the drug. The surrounding pH also plays a certain role, particularly in the core–shell formulations, as it influences the ionization state of the functional groups along the polymers, consequently influencing how they are displaced on the space.
3.4. Passive and US-Triggered DOX Release Kinetics
The non-invasiveness and the ability to regulate the depth of the penetration by tuning frequency, duty cycles, and time of exposure represent some of the advantages of using ultrasound (US) as an external trigger to cause a burst in the release of DOX [
34]. For in vivo application, ultrasonic waves can reach deeper sites compared to light radiation, and the associated sonoporation phenomenon facilitates the diffusion of therapeutic molecules through the cell membrane [
35].
In vitro ultrasound-induced drug delivery mechanisms have been described in detail [
36,
37]. Herein, the enhancement in the DOX release after application of US cycles could be ascribed to the oscillatory motion of the surrounding fluid occurring in the absence of cavitation. Herein, two main mechanisms could be responsible for the intensification of DOX release after US application. The first is due to the oscillation of the surrounding fluid, which increases the diffusivity of the molecules; the second is related to the rupture of the carrier structure. However, the former plays a marginal role, while a complete disruption of the particles leads to an intense burst. In
Figure 5 the effect of US power, application time and pH on the DOX release is resumed.
The trend illustrated in
Figure 5 shows how the exposure to ultrasound increases the release between 10 and 50% with a maximum release occurring at 20 W/cm
2 applied for 30 s.
In
Table 1, the amounts (in μg) of DOX released from 10 mg of carrier after 110 min in the medium and exposure to US cycles in different media are reported. In the brackets, the percentage released refers to the total amount (in mg) loaded.
The data shown in
Table 1 demonstrate how the pH affects the overall amount of drug released, with or without the application of ultrasound cycles. In the absence of US, the amount of DOX released increases when the pH increases from 3 to 7.4, reaching the maximum, 37%, after 2 h of exposure. The application of US drastically increases the percentage of drug released, reaching 100%. The synergic effect of pH and US is observable considering the amount of DOX released at the same US power and exposure time but at different pH levels. When increasing the pH, the amount of released drug increases simultaneously. An exception is noted after application of US at 20 W/cm
2 for 30 s where, independently from the pH, the whole amount of the loaded drug is released. The effect of pH can be mostly ascribed to the ionization state of the functional groups on the surface of the particles (ZV) and on those displaced along the polymer (core–shell) affecting the electrostatic interactions and formation of H-bonds between particles and drug. Conversely, the ultrasound tends to physically alter the structure, with a consequent increase, or burst, in the release.
3.5. Cytotoxicity and Hemolysis Assessment of Microparticles
The MTT has been performed to determine (i) the cytotoxicity of the bare microparticles and the impact of the surface properties on the in vitro biocompatibility; (ii) the advantages of using a microcarrier in the delivery of DOX compared to the free drug, considering the in vitro cytotoxicity enhancement in particular; and (iii) DOX cytotoxicity, extended over time. IO microparticles have been used as the control to evaluate the effect of the surface modification and the polymer coat on the cytocompatibility of the system.
Figure 6 shows cell viability evaluation for all formulations.
NIH/3T3 cells were treated with a solution containing 50 µg of microparticles, and the viability was determined after 24, 48 and 72 h (
Figure 6). The toxic effect was taken into consideration when the viability was below 80%. The control, containing only cells, was treated with ultrasound to determine any possible contribution of US on the cell viability.
Results reported in
Figure 6 demonstrate the advantages of the core–shell design in improving DOX cytotoxicity. According to the results, the presence of the polymer shell could also play a certain role by interacting with the cell membrane and favoring the uptake. In fact, DOX cytotoxicity is higher when loaded in the core–shell structure than in ZV; in the case of ZV, the release begins immediately after contact with the medium, leading to the release of a certain amount of the drug in the solution before the internalization of the particles. The cell viability tends to decrease over time when exposed to core–shell microparticles, conversely to the observed increase in viability 48 and 72 h after exposure to free DOX, even if the core–shell viability value is maintained as much lower in comparison.
The 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) test was performed in the presence of US using the same power, number of cycles and application time used in the release studies. The application of US at 2 and 20 W/cm2 power did not affect the cell viability but might reversibly alter the structure of the membrane of the cell by sonoporation, favoring the entry of the microparticles and free DOX. In the case of core–shell microparticles, the positive ζ-potential represents an additional advantage, as it promotes the interactions with the negatively charged cell membrane. The use of 5 × 10 s and 5 × 30 s US cycles did not cause any significant increase in temperature, with an observed variation of less than 1 °C. This is important to determine, as the temperature affects the shell structure and, consequently, the release rate and mechanism. Moreover, an increase in temperature affects the cell viability.
The graph in
Figure 6 shows how the cytotoxicity of the free DOX and DOX-loaded ZV particles is lower than that of the DOX loaded in the core–shell particles due to the slower release of the drug. As reported in the release studies subsection, the US cycles cause a burst in the DOX release, meaning that a large amount of drug is released in a short period of time; such an event, in combination with sonoporation, leads to a higher permeation of the drug into the cell, with a decrease in the viability. The increased in vitro cytotoxicity of DOX resulting from the use the US is provided by the synergic effects of US and DOX. US acts either directly and indirectly, by sonoporation and by causing a burst in the DOX release. Moreover, the possibility to turn the burst on and off is only provided by the core–shell systems. In the ZV, the burst cannot be controlled, and the application of a single cycle of US causes an intensive release of DOX which is equal to the total amount adsorbed. The effect of US exposure on the cell viability can be described as direct and indirect: (i) it causes an improvement in the ability of the microparticles to permeate through the cell membrane by sonoporation (direct) [
22] and (ii) it causes a release burst of DOX (indirect), reaching high concentrations which are highly toxic for the cells. As can be seen from
Figure 6, in the case of bare ZV and core–shell microparticles in absence of US and coating, the cell viability is higher than 80 %, and this persists up to 72 h, indicating the absence of in vitro cytotoxicity for the selected cell lines at the tested concentration. However, due to the nature of the material used, a non-cytotoxic profile was expected. In agreement with our previous studies and others already published [
38,
39,
40], cells treated with free DOX solution demonstrate viability of around 40%, confirming the well-known cytotoxicity of the drug.
Results reported in
Figure 6 prove the beneficial role played by the core–shell carrier in driving the drug inside the cells. According to these results, the presence of the polymer shell could play a specific role in the cell uptake and in controlling the release of the drug. The cell viability tends to decrease over time when exposed to core–shell microparticles, conversely to the observed increase in viability 48 and 72 h after exposure to free DOX, even if the core–shell viability value is maintained as much lower in comparison.
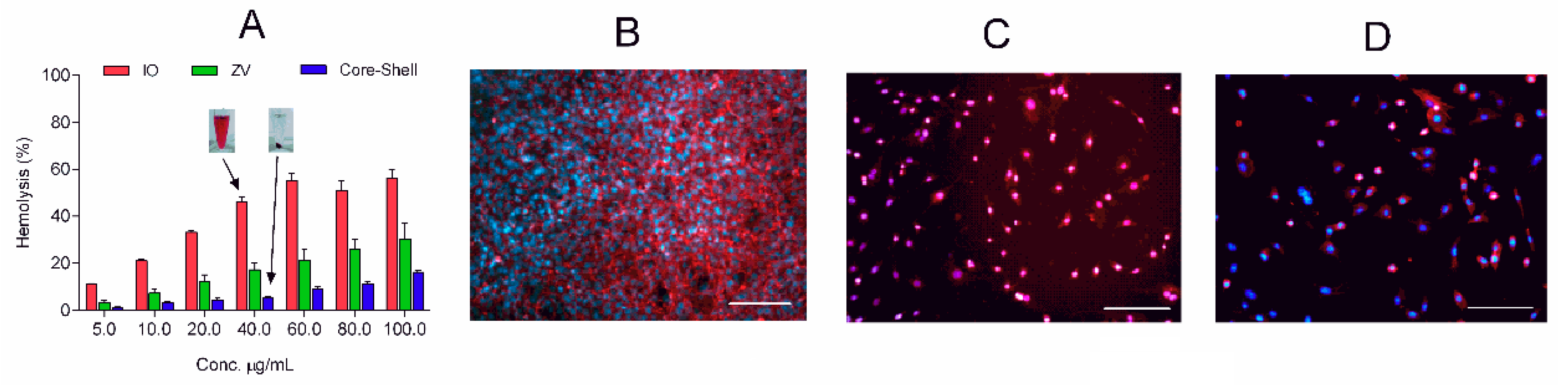
Blood compatibility represents a critical parameter for evaluating biological safety when materials are intended for intravenous injection. The hemocompatibility of the prepared carriers was evaluated by hemolysis test as shown in
Figure 7. Results provide information about the influence of the surface properties on the interaction with the red blood cells. The presence of the organic layer on the IO reduced the hemotoxicity to lower than 5% for concentrations up to 10 µg/mL. A more interesting observation is that of the hemocompatibility in the core–shell microparticles reaching 60 µg/mL, 6-fold greater than ZV. The obtained values are in contrast with other studies reported in the literature; previous works have shown that positively charged particles are more reactive than neutral or negatively charged ones, damaging blood cells [
41,
42]. Additional experiments are required to better understand the hemocompatibility of the prepared particles and their interactions with the red blood cells.
4. Conclusions
In the present work, hybrid microparticles with an iron core and amphiphilic polymer shell able to respond to variation in the surrounding pH, the presence of magnetic field, and ultrasound treatment were developed for smart delivery of bioactive compounds. Doxorubicin hydrochloride has been used as a model drug, as it is well known and widely used in the treatment of different cancers. The core–shell microparticles were prepared in four steps: (i) preparation of iron oxide microparticles; (ii) surface modification by the introduction of phenyl carboxyl groups; (iii) conjugation with the amphiphilic CS derivative (PLACA-g-CS); and (iv) drug loading. The core–shell microparticles show a size of around 3 µm, a positive ζ-potential of + 34 mV and excellent stability in the physiological environment.
The PLACA-g-CS shell allows a higher encapsulation efficiency and a pH-dependent controlled delivery, preserving the magnetic response of the iron core under a magnetic field and limiting the decrease in this response to 20%. The core–shell system demonstrates US-responsive behavior characterized by a reversible change in the spatial displacement of the polymeric chains around the core, causing a release burst with intensity dependent on the power and application time of the US. The in vitro test performed on NIH/3T3 cells demonstrated that the hybrid microparticles in combination with US improve the cytotoxicity of doxorubicin and allow it to last for a longer time compared to the that of the free drug formulation. Moreover, the prepared hybrid system has shown good hemocompatibility, 6-fold greater than that of simple iron oxide microparticles, for concentrations up to 60 µg/mL.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}