Phase Equalization, Charge Transfer, Information Flows and Electron Communications in Donor–Acceptor Systems

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Molecular States and Their Phases
= 〈Ψ(N)|Q(N)〉 〈Q(N)|Ψ(N)〉 ≡ 〈Ψ(N)|PQ(N)|Ψ(N)〉,
3. Electronic Communications
≡ ∫M(ζ|θ) exp{iΦ(ζ|θ)] dr.
= 〈θ|ζ〉 〈ζ|θ〉 ≡ 〈θ|Pζ|θ〉,
Pϑ = |ϑ〉 〈ϑ|, Pϑ2 = Pϑ, ϑ = (θ, ζ).
= Σs |Ψs(Ψ)〉 〈Ψs(Ψ)| ≡ Σs Ps(Ψ),
= Σs 〈i|Ψs(Ψ)〉 〈Ψs(Ψ)|j〉 = Σs Ci,s(Ψ) Cj,s(Ψ)*≡ γi,j(Ψ)
= 〈i|Pb(Ψ)|j〉 〈j|Pb(Ψ)|i〉 = 〈i|Pb(Ψ)PjPb(Ψ)|i〉 ≡ 〈i|Pjb(Ψ)|i〉
= 〈j|Pb(Ψ)|i〉 〈i|Pb(Ψ)|j〉 = 〈j|Pb(Ψ)PiPb(Ψ)|j〉 ≡ 〈j|Pib(Ψ)|j〉.
4. Phase–Current Relation
= (ħ/m) p(r) ∇ϕ(r) ≡ p(r) V(r).
= (2m/ħ)2 ∫p(r) V(r)2 dr = (2m/ħ)2 ∫p(r)−1 j(r)2 dr.
5. Current Coherence in Donor–Acceptor Systems
= (2m/ħ)2 ∫pλ(r) Vλ(r)2 dr
= (2m/ħ)2 ∫pλ(r)−1 jλ(r)2 dr.
≡ Σκ ∫wκ(r; X) kκ(r; X) dr ≡ ∫k(r; X) dr,
≡ Σκ ∫wκ(r; X) Vκ(r; X) dr ≡ ∫V(r; X) dr,
≡ Σκ ∫wκ(r) jκ(r; X) dr ≡ ∫j(r; X) dr.
6. Overall Charge Transfer
μα(B+) = μα(aB+) = μα(bB+) = −AB+,
= ηα(A+,A+) − ηα(A+,B+) + ηα(B+,B+) − ηα(B+,A+) > 0,
= [∂μα(X+)/∂Nα(Y+)]v(α), X+, Y+ ∈ (A+, B+).
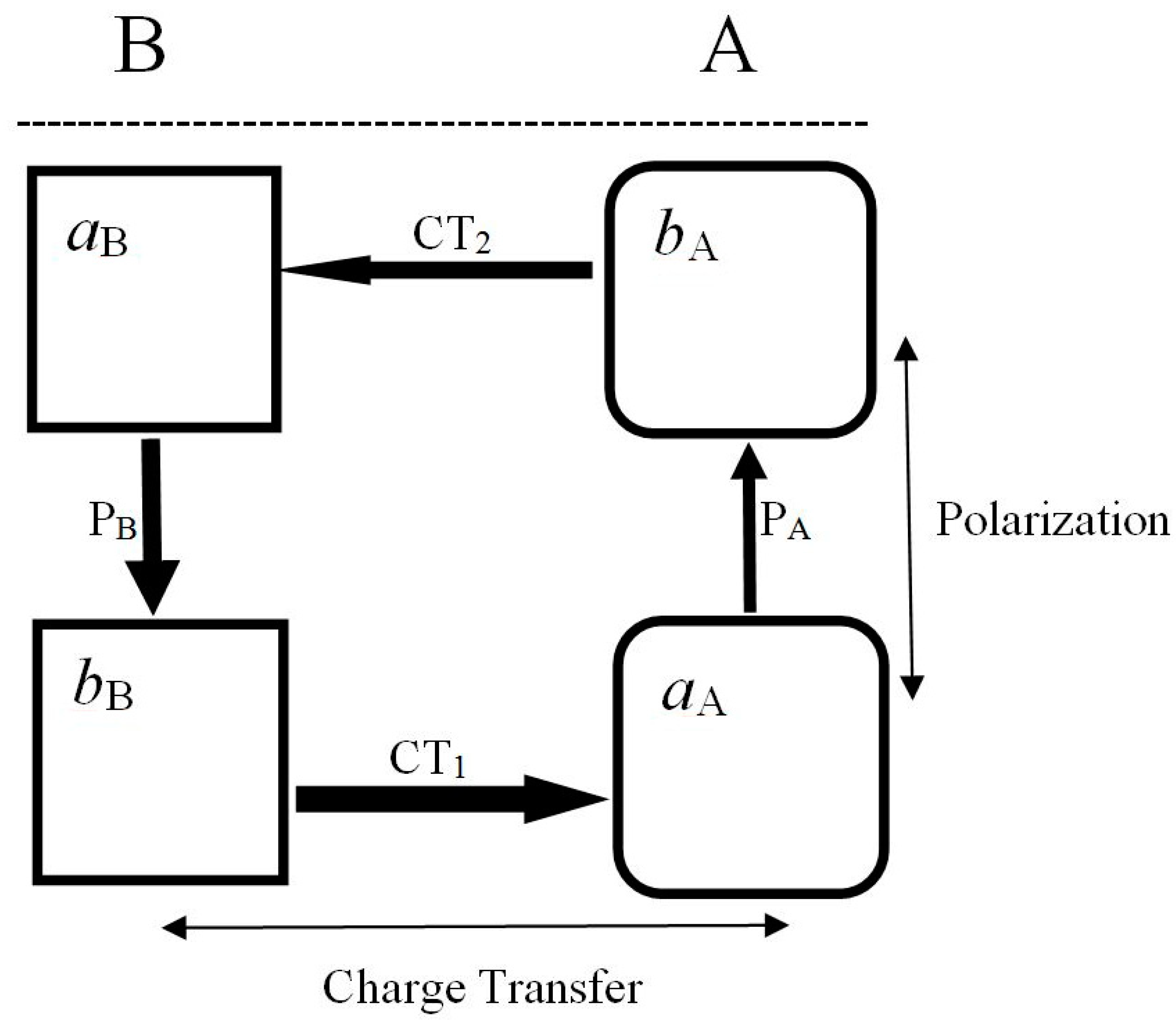
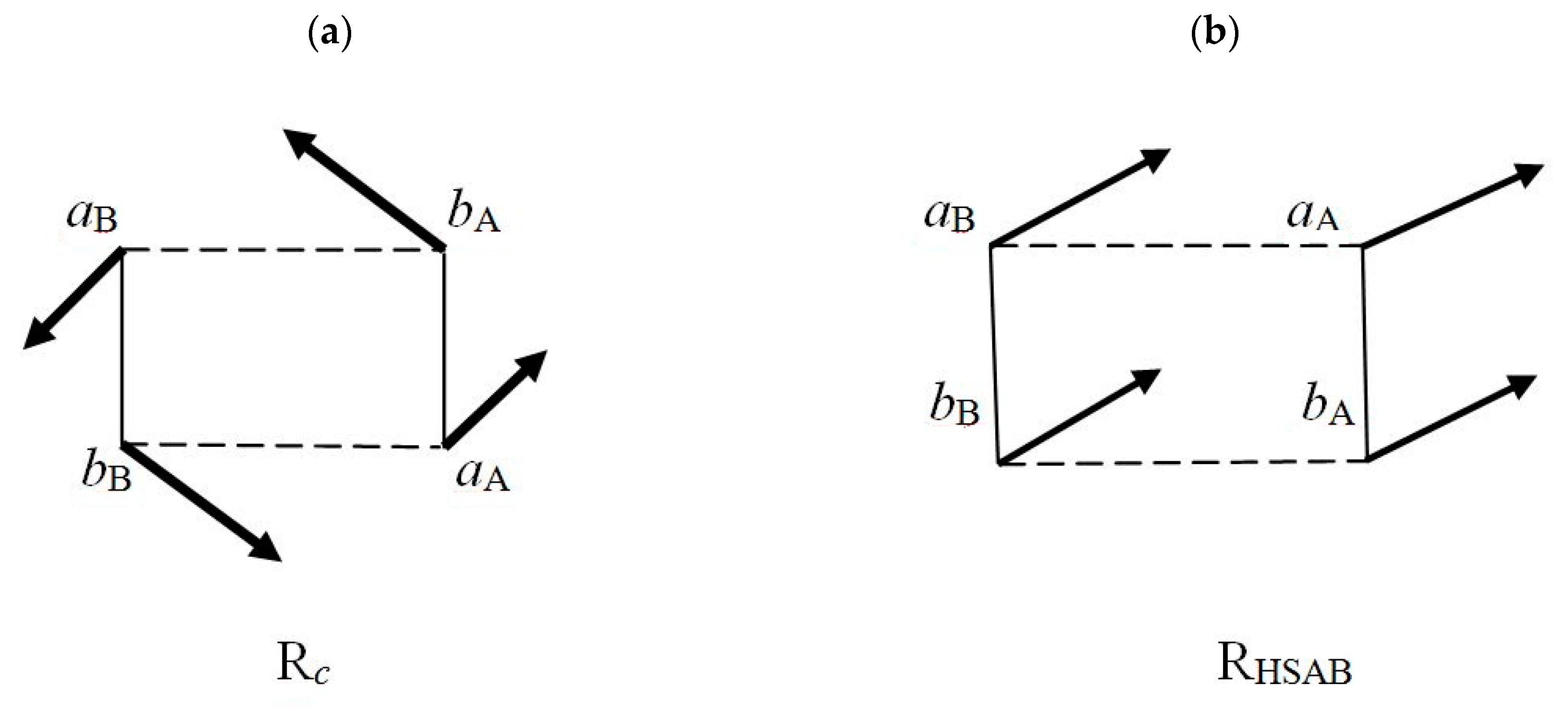
7. Partial Electronic Flows
≡ {aA(α), bA(α), aB(α), bB(α)} ≡ {λ1, λ2, λ3, λ4},
(2) As internal and external acceptor, e.g., aA site in RHSAB;
(3) As flow-through fragment, e.g., all sites in Rc and (aB, bA) parts of RHSAB.
hc(CT1) = h1,1 − h1,4 + h4,4 − h4,1.
hc(CT2) = h3,3(c) − h2,3(c) + h2,2(c) − h3,2(c).
hHSAB(CT1) = h2,2(HSAB) − h2,4(HSAB) + h4,4(HSAB) − h4,2(HSAB).
EHSAB(CT1) = −[uHSAB(CT1)]2/[2hHSAB(CT1)].
λ2: δuc(bA) = [h2,1(c) − h2,4(c)] Nc(CT1) + [h2,3(c) − h2,2(c)] Nc(CT2),
λ3: δuc(aB) = [h3,1(c) − h3,4(c)] Nc(CT1) + [h3,3(c) − h3,2(c)] Nc(CT2),
λ4: δuc(bB) = [h4,1(c) − h4,4(c)] Nc(CT1) + [h4,3(c) − h4,2(c)] Nc(CT2).
+ [h1,1(HSAB) − h1,3(HSAB)] NHSAB(CT2),
λ2: δuHSAB(bA) = [h2,2(HSAB) − h2,4(HSAB)] NHSAB(CT1)
+ [h2,1(HSAB) − h2,3(HSAB)] NHSAB(CT2),
λ3: δuHSAB(aB) = [h3,2(HSAB) − h3,4(HSAB)] NHSAB(CT1)
+ [h3,1(HSAB) − h3,3(HSAB)] NHSAB(CT2),
λ4: δuHSAB(bB) = [h4,2(HSAB) − h4,4(HSAB)] NHSAB(CT1)
+ [h4,1(HSAB) − h4,3(HSAB)] NHSAB(CT2).
uHSAB(PB) = δuHSAB(aB) − δuHSAB(bB).
hα(PB) = h3,3(α) − h3,4(α) + h4,4(α) − h4,3(α), α = (c, HSAB).
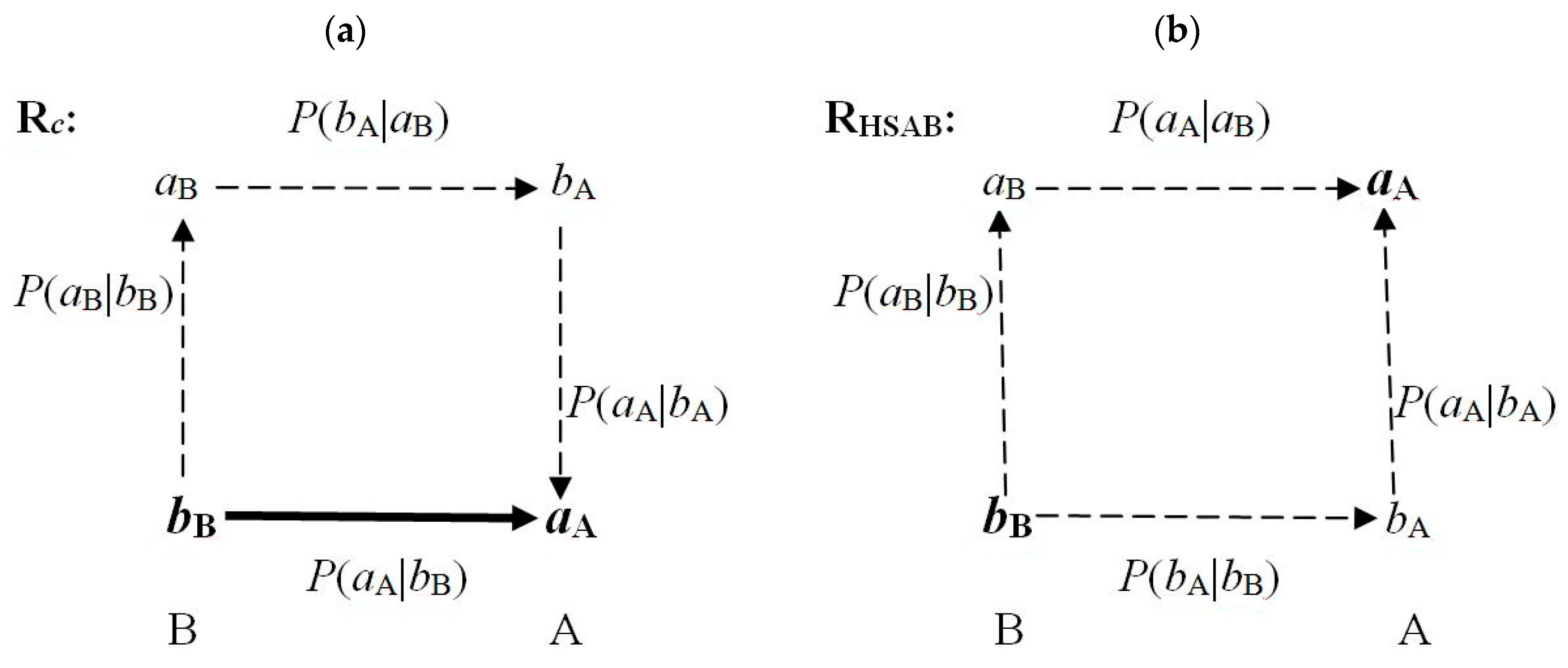
8. Communication Considerations
PHSAB(bB→bA→aA) = PHSAB(bB→bA) PHSAB(bA→aA).
9. Conclusions
Funding
Conflicts of Interest
Appendix A. Continuity Relations Revisited
= (ħ/m) p(r, t) ∇ϕ(r, t)
≡ p(r, t) V(r, t).
≡ S[p] + i S[ϕ],
M = 4[(∇lnR)2 + (i∇ϕ)2],
= ∫p[(∇lnp)2 + 4(∇ϕ)2] dr = ∫p−1(∇p)2 dr + 4∫p (∇ϕ)2 dr ≡ I[p] + I[ϕ],
= 4∫[(∇R)2 − (∇ϕ)2] dr ≡ M[R] + M[ϕ]
= ∫p[(∇lnp)2 − 4(∇ϕ)2] dr = M[p] + M[ϕ].
σp ≡ dp/dt = ∂p/∂t + ∇· j = ∂p/∂t + ∇p · V = 0.
= ∂p(r, t)/∂t + V(r, t) ·∇p(r, t) = ∂p(r, t)/∂t + ∇· j(r, t) = 0.
Appendix B. Schrödinger Equation and Wavefunction Components
= Hψ = {−[ħ2/(2m)] [ΔR + 2i∇R·∇ϕ − R(∇ϕ)2] + vR} exp(iϕ),
= [ħ2/(2m)] ∫[(∇R)2 + R2(∇ϕ)2]dr + ∫R2 v dr ≡ 〈T〉ψ + 〈Vne〉ψ,
T(N) = Σk T(k), Vne(N) = Σk v(k), Uee(N) = Σk<l g(k, l).
≡ Σs Cs(t0) Ψs(N, τ),
UA(NA0) = Σ(k<l)∈ A g(k, l), UAB(NA0, NB0) = Σk∈ AΣl∈ B g(k, l), etc.
= Σu Cu(X+, t0) {exp[iϕu(X+, τ)] φu(NX0)}
≡ Σu Cu(X+, t0) Φu(NX0, τ),
= φu(NA0) φw(NB0) exp{i[ϕu(A+,τ) + ϕw(B+,τ)]}
≡ ϑu,w(NA0, NB0) exp[iθu,w(τ)]},
= Σu,w Du,v(t0) Φu(NA0, τ) Φw(NB0, τ)
= Σu,w Du,v(t0) Θu,w(A+, B+; τ),
Du,v(t0) = 〈ϑu,w(NA0, NB0)|ΨR+(N, t0)〉.
Appendix C. Information Principle
Appendix D. Reactant Entanglement
Appendix E. Density Matrices for Interacting Subsystems
= ΣsΣs’ [∫Φs’*(ξ) Φs(ξ) dξ] [∫φs’*(x) Lx φs(x) dx]
≡ Σs Σs’ ρs,s’(A) Ls’,s(A) = trA[ρ(A) L(A)].
≡ ΣsΣs’ ρs,s’(A) Ωs’,s(x, x’) = trA[ρ(A) ΩA(x, x’)].
References
- Fisher, R.A. Theory of statistical estimation. Proc. Camb. Phil. Soc. 1925, 22, 700–725. [Google Scholar] [CrossRef]
- Frieden, B.R. Physics from the Fisher Information—A Unification; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Shannon, C.E. The mathematical theory of communication. Bell Syst. Tech. J. 1948, 27, 379–493, 623–656. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois: Urbana, IL, USA, 1949. [Google Scholar]
- Kullback, S.; Leibler, R.A. On information and sufficiency. Ann. Math. Stat. 1951, 22, 79–86. [Google Scholar] [CrossRef]
- Kullback, S. Information Theory and Statistics; Wiley: New York, NY, USA, 1959. [Google Scholar]
- Abramson, N. Information Theory and Coding; McGraw-Hill: New York, NY, USA, 1963. [Google Scholar]
- Pfeifer, P.E. Concepts of Probability Theory; Dover: New York, NY, USA, 1978. [Google Scholar]
- Nalewajski, R.F. Information Theory of Molecular Systems; Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Nalewajski, R.F. Information Origins of the Chemical Bond; Nova Science Publishers: New York, NY, USA, 2010. [Google Scholar]
- Nalewajski, R.F. Perspectives in Electronic Structure Theory; Springer: Heidelberg, Germany, 2012. [Google Scholar]
- Nalewajski, R.F.; Parr, R.G. Information theory, atoms-in-molecules and molecular similarity. Proc. Natl. Acad. Sci. USA 2000, 97, 8879–8882. [Google Scholar] [CrossRef] [PubMed]
- Nalewajski, R.F. Information principles in the theory of electronic structure. Chem. Phys. Lett. 2003, 272, 28–34. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Information principles in the Loge Theory. Chem. Phys. Lett. 2003, 375, 196–203. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Broniatowska, E. Information distance approach to Hammond Postulate. Chem. Phys. Lett. 2003, 376, 33–39. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Parr, R.G. Information-theoretic thermodynamics of molecules and their Hirshfeld fragments. J. Phys. Chem. A 2001, 105, 7391–7400. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Hirshfeld analysis of molecular densities: Subsystem probabilities and charge sensitivities. Phys. Chem. Chem. Phys. 2002, 4, 1710–1721. [Google Scholar] [CrossRef]
- Parr, R.G.; Ayers, P.W.; Nalewajski, R.F. What is an atom in a molecule? J. Phys. Chem. A. 2005, 109, 3957–3959. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Broniatowska, E. Atoms-in-Molecules from the stockholder partition of molecular two-electron distribution. Theoret. Chem. Acc. 2007, 117, 7–27. [Google Scholar] [CrossRef]
- Heidar-Zadeh, F.; Ayers, P.W.; Verstraelen, T.; Vinogradov, I.; Vöhringer-Martinez, E.; Bultinck, P. Information-theoretic approaches to Atoms-in-Molecules: Hirshfeld family of partitioning schemes. J. Phys. Chem. A 2018, 122, 4219–4245. [Google Scholar] [CrossRef] [PubMed]
- Hirshfeld, F.L. Bonded-atom fragments for describing molecular charge densities. Theoret. Chim. Acta 1977, 44, 129–138. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Entropic measures of bond multiplicity from the information theory. J. Phys. Chem. A 2000, 104, 11940–11951. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Entropy descriptors of the chemical bond in Information Theory: I. Basic concepts and relations. Mol Phys 102:531-546; II. Application to simple orbital models. Mol. Phys. 2004, 102, 547–566. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Entropic and difference bond multiplicities from the two-electron probabilities in orbital resolution. Chem. Phys. Lett. 2004, 386, 265–271. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Reduced communication channels of molecular fragments and their entropy/information bond indices. Theoret. Chem. Acc. 2005, 114, 4–18. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Partial communication channels of molecular fragments and their entropy/information indices. Mol. Phys. 2005, 103, 451–470. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Entropy/information descriptors of the chemical bond revisited. J. Math. Chem. 2011, 49, 2308–2329. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Quantum information descriptors and communications in molecules. J. Math. Chem. 2014, 52, 1292–1323. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Multiple, localized and delocalized/conjugated bonds in the orbital-communication theory of molecular systems. Adv. Quantum Chem. 2009, 56, 217–250. [Google Scholar]
- Nalewajski, R.F.; Szczepanik, D.; Mrozek, J. Bond differentiation and orbital decoupling in the orbital communication theory of the chemical bond. Adv. Quantum Chem. 2011, 61, 1–48. [Google Scholar]
- Nalewajski, R.F.; Szczepanik, D.; Mrozek, J. Basis set dependence of molecular information channels and their entropic bond descriptors. J. Math. Chem. 2012, 50, 1437–1457. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Electron communications and chemical bonds. In Frontiers of Quantum Chemistry; Wójcik, M., Nakatsuji, H., Kirtman, B., Ozaki, Y., Eds.; Springer: Singapore, 2017; pp. 315–351. [Google Scholar]
- Nalewajski, R.F.; Świtka, E.; Michalak, A. Information distance analysis of molecular electron densities. Int. J. Quantum Chem. 2002, 87, 198–213. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Broniatowska, E. Entropy displacement analysis of electron distributions in molecules and their Hirshfeld atoms. J. Phys. Chem. A 2003, 107, 6270–6280. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Use of Fisher information in quantum chemistry. Int. J. Quantum Chem. 2008, 108, 2230–2252. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Köster, A.M.; Escalante, S. Electron localization function as information measure. J. Phys. Chem. A 2005, 109, 10038–10043. [Google Scholar] [CrossRef]
- Becke, A.D.; Edgecombe, K.E. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys. 1990, 92, 5397–5403. [Google Scholar] [CrossRef]
- Silvi, B.; Savin, A. Classification of chemical bonds based on topological analysis of electron localization functions. Nature 1994, 371, 683–686. [Google Scholar] [CrossRef]
- Savin, A.; Nesper, R.; Wengert, S.; Fässler, T.F. ELF: The electron localization function. Angew. Chem. Int. Ed. Engl. 1997, 36, 1808–1832. [Google Scholar] [CrossRef]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136B, 864–971. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140A, 133–1138. [Google Scholar] [CrossRef]
- Levy, M. Universal variational functionals of electron densities, first-order density matrices, and natural spin-orbitals and solution of the v-representability problem. Proc. Natl. Acad. Sci. USA 1979, 76, 6062–6065. [Google Scholar] [CrossRef] [PubMed]
- Parr, R.G.; Yang, W. Density-Functional Theory of Atoms and Molecules; Oxford University Press: New York, NY, USA, 1989. [Google Scholar]
- Dreizler, R.M.; Gross, E.K.U. Density Functional Theory: An Approach to the Quantum Many-Body Problem; Springer: Berlin, Germany, 1990. [Google Scholar]
- Nalewajski, R.F. (Ed.) Density Functional Theory I-IV, Topics in Current Chemistry; Springer: Berlin, Germany, 1966; Volume 180–183. [Google Scholar]
- Nalewajski, R.F.; de Silva, P.; Mrozek, J. Use of nonadditive Fisher information in probing the chemical bonds. J. Mol. Struct. 2010, 954, 57–74. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Through-space and through-bridge components of chemical bonds. J. Math. Chem. 2011, 49, 371–392. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Chemical bonds from through-bridge orbital communica-tions in prototype molecular systems. J. Math. Chem. 2011, 49, 546–561. [Google Scholar] [CrossRef]
- Nalewajski, R.F. On interference of orbital communications in molecular systems. J. Math. Chem. 2011, 49, 806–815. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Gurdek, P. On the implicit bond-dependency origins of bridge interactions. J. Math. Chem. 2011, 49, 1226–1237. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Direct (through-space) and indirect (through-bridge) components of molecular bond multiplicities. Int. J. Quantum Chem. 2012, 112, 2355–2370. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Gurdek, P. Bond-order and entropic probes of the chemical bonds. Struct. Chem. 2012, 23, 1383–1398. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Exploring molecular equilibria using quantum information measures. Ann. Phys. 2013, 525, 256–268. [Google Scholar] [CrossRef]
- Nalewajski, R.F. On phase equilibria in molecules. J. Math. Chem. 2014, 52, 588–612. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Quantum information approach to electronic equilibria: Molecular fragments and elements of non-equilibrium thermodynamic description. J. Math. Chem. 2014, 52, 1921–1948. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Phase/current information descriptors and equilibrium states in molecules. Int. J. Quantum Chem. 2015, 115, 1274–1288. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Quantum information measures and molecular phase equilibria. In Advances in Mathematics Research; Baswell, A.R., Ed.; Nova Science Publishers: New York, NY, USA, 2015; Volume 19, pp. 53–86. [Google Scholar]
- Nalewajski, R.F. On phase/current components of entropy/information descriptors of molecular states. Mol. Phys. 2014, 112, 2587–2601. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Complex entropy and resultant information measures. J. Math. Chem. 2016, 54, 1777–1782. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Phase description of reactive systems. In Conceptual Density Functional Theory; Islam, N., Kaya, S., Eds.; Apple Academic Press: Waretown, NJ, USA, 2018; pp. 217–249. [Google Scholar]
- Nalewajski, R.F. Quantum information measures and their use in chemistry. Curr. Phys. Chem. 2017, 7, 94–117. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Quantum Information Theory of Molecular States; Nova Science Publishers: New York, NY, USA, 2016. [Google Scholar]
- Nalewajski, R.F. Resultant information approach to donor-acceptor systems. In An Introduction to Electronic Structure Theory; Nova Science Publishers: New York, NY, USA, 2020; in press. [Google Scholar]
- Nalewajski, R.F. Information equilibria, subsystem entanglement and dynamics of overall entropic descriptors of molecular electronic structure. J. Mol. Model. 2018, 24, 212–227. [Google Scholar] [CrossRef]
- Nalewajski, R.F. On entangled states of molecular fragments. Trends Phys. Chem. 2016, 16, 71–85. [Google Scholar]
- Nalewajski, R.F. Virial theorem implications for the minimum energy reaction paths. Chem. Phys. 1980, 50, 127–136. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Understanding electronic structure and chemical reactivity: Quantum-information perspective. Appl. Sci. 2019, 9, 1262–1292, In The Application of Quantum Mechanics to the Reactivity of Molecules. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Overall Entropy/Information Descriptors of Electronic States and Chemical Reactivity; Islam, N., Bir Singh, S., Ranjan, P., Haghi, A.K., Eds.; Apple Academic Press: Waretown, NJ, USA, 2020; in press. [Google Scholar]
- Nalewajski, R.F. On entropy/information description of reactivity phenomena. In Advances in Mathematics Research; Baswell, A.R., Ed.; Nova Science Publishers: New York, NY, USA, 2019; Volume 26, pp. 97–157. [Google Scholar]
- Nalewajski, R.F. Role of electronic kinetic energy (resultant gradient information) in chemical reactivity. J. Mol. Model. 2019, 25, 259–278, Berski, S., Sokalski, W.A. (Eds.). [Google Scholar] [CrossRef] [PubMed]
- Nalewajski, R.F. On classical and quantum entropy/information descriptors of molecular electronic states. In Research Methodologies and Practical Applications of Chemistry; Pogliani, L., Haghi, A.K., Islam, N., Eds.; Apple Academic Press: Waretown, NJ, USA, 2019; in press. [Google Scholar]
- Callen, H.B. Thermodynamics: An Introduction to the Physical Theories of Equilibrium Thermostatics and Irreversible Thermodynamics; Wiley: New York, NY, USA, 1962. [Google Scholar]
- Nalewajski, R.F. Equidensity orbitals in resultant-information description of electronic states. Theoret. Chem. Acc. 2019, 138, 108–123, In Chemical Concepts from Theory and Computation; Liu, S. (Ed.). [Google Scholar] [CrossRef]
- Nalewajski, R.F. Resultant information description of electronic states and chemical processes. J. Phys. Chem. A 2019, 123, 45–60. [Google Scholar] [CrossRef] [PubMed]
- Harriman, J.E. Orthonormal orbitals for the representation of an arbitrary density. Phys. Rev. A 1980, 24, 680–682. [Google Scholar] [CrossRef]
- Zumbach, G.; Maschke, K. New approach to the calculation of density functionals. Phys. Rev. A 1983, 28, 544–554, Erratum, Phys Rev A 29, 1585–1587. [Google Scholar] [CrossRef]
- Macke, W. Zur wellermechanischen behandlung von vielkoeperproblemen. Ann. Phys. 1955, 17, 1–9. [Google Scholar] [CrossRef]
- Gilbert, T.L. Hohenberg-Kohn theorem for nonlocal external potentials. Phys. Rev. B 1975, 12, 2111–2120. [Google Scholar] [CrossRef]
- Nalewajski, R.F.; Korchowiec, J.; Michalak, A. Reactivity criteria in charge sensitivity analysis. Top. Curr. Chem. 1996, 183, 25–141, In Density Functional Theory IV; Nalewajski, R.F., Ed. [Google Scholar]
- Nalewajski, R.F.; Korchowiec, J. Charge Sensitivity Approach to Electronic Structure and Chemical Reactivity; World Scientific: Singapore, 1997. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Sensitivity analysis of charge transfer systems: In situ quantities, intersecting state model and its implications. Int. J. Quantum Chem. 1994, 49, 675–703. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Charge sensitivity analysis as diagnostic tool for predicting trends in chemical reactivity. In Proceedings of the NATO ASI on Density Functional Theory; (Il Ciocco, 1993); Dreizler, R.M., Gross, E.K.U., Eds.; Plenum: New York, NY, USA, 1995; pp. 339–389. [Google Scholar]
- Chattaraj, P.K. (Ed.) Chemical Reactivity Theory: A Density Functional View; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Nalewajski, R.F. Chemical reactivity description in density-functional and information theories. Acta Phys. Chim. Sin. 2017, 33, 2491–2509, In Chemical Concepts from Density Functional Theory; Liu, S.(Ed.). [Google Scholar]
- Gyftopoulos, E.P.; Hatsopoulos, G.N. Quantum-thermodynamic definition of electronegativity. Proc. Natl. Acad. Sci. USA 1965, 60, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Parr, R.G.; Levy, M.; Balduz, J.L. Density functional theory for fractional particle number: Derivative discontinuities of the energy. Phys. Rev. Lett. 1982, 49, 1691–1694. [Google Scholar] [CrossRef]
- Mulliken, R.S. A new electronegativity scale: Together with data on valence states and on ionization potentials and electron affinities. J. Chem. Phys. 1934, 2, 782–793. [Google Scholar] [CrossRef]
- Iczkowski, R.P.; Margrave, J.L. Electronegativity. J. Am. Chem. Soc. 1961, 83, 3547–3551. [Google Scholar] [CrossRef]
- Parr, R.G.; Donnelly, R.A.; Levy, M.; Palke, W.E. Electronegativity: The density functional viewpoint. J. Chem. Phys. 1978, 69, 4431–4439. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute hardness: Companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Dirac, P.A.M. The Principles of Quantum Mechanics; Oxford University Press: Oxford, UK, 1967. [Google Scholar]
- Chandra, A.K.; Michalak, A.; Nguyen, M.T.; Nalewajski, R.F. On regional matching of atomic softnesses in chemical reactions: Two-reactant charge sensitivity study. J. Phys. Chem. A 1998, 102, 10182–10188. [Google Scholar] [CrossRef]
- Nalewajski, R.F. Manifestations of the maximum complementarity principle for matching atomic softnesses in model chemisorption systems. Top. Catal. 2000, 11, 469–485. [Google Scholar] [CrossRef]
- Prigogine, I. From Being to Becoming: Time and Complexity in the Physical Sciences; Freeman WH & Co.: San Francisco, CA, USA, 1980. [Google Scholar]
- Flores-Gallegos, N.; Esquivel, R.O. Von Neumann entropies analysis in Hilbert space for the dissociation processes of homonuclear and heteronuclear diatomic molecules. J. Mex. Chem. Soc. 2008, 52, 19–30. [Google Scholar]
- López-Rosa, S. Information Theoretic Measures of Atomic and Molecular Systems. Ph.D. Thesis, University of Granada, Granada, Spain, 2010. [Google Scholar]
- López-Rosa, S.; Esquivel, R.O.; Angulo, J.C.; Antolín, J.; Dehesa, J.S.; Flores-Gallegos, N. Fisher information study in position and momentum spaces for elementary chemical reactions. J. Chem. Theory Comput. 2010, 6, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Esquivel, R.O.; Liu, S.; Angulo, J.C.; Dehesa, J.S.; Antolín, J.; Molina-Espíritu, M. Fisher information a steric effect: Study of internal rotation barrier in ethane. J. Phys. Chem. A 2011, 115, 4406–4415. [Google Scholar] [CrossRef] [PubMed]
- Lieb, E. Topics in Quantum Entropy and Entanglement; Three lectures at Priceton Condensed Matter Summer School: Princeton, NJ, USA, 30–31 July 2014. [Google Scholar]
- Davydov, A.S. Quantum Mechanics; Pergamon Press: Oxford, UK, 1965. [Google Scholar]
- Cohen-Tannoudji Diu, B.; Laloë, F. Quantum Mechanics; Wiley: New York, NY, USA, 1977. [Google Scholar]
- Bader, R.F.W. Atoms in Molecules; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]



© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nalewajski, R.F. Phase Equalization, Charge Transfer, Information Flows and Electron Communications in Donor–Acceptor Systems. Appl. Sci. 2020, 10, 3615. https://doi.org/10.3390/app10103615
Nalewajski RF. Phase Equalization, Charge Transfer, Information Flows and Electron Communications in Donor–Acceptor Systems. Applied Sciences. 2020; 10(10):3615. https://doi.org/10.3390/app10103615
Chicago/Turabian StyleNalewajski, Roman F. 2020. "Phase Equalization, Charge Transfer, Information Flows and Electron Communications in Donor–Acceptor Systems" Applied Sciences 10, no. 10: 3615. https://doi.org/10.3390/app10103615
APA StyleNalewajski, R. F. (2020). Phase Equalization, Charge Transfer, Information Flows and Electron Communications in Donor–Acceptor Systems. Applied Sciences, 10(10), 3615. https://doi.org/10.3390/app10103615