Abstract
Due to the adverse effect of atmospheric aerosols on public health and their ability to affect climate, extensive research has been undertaken in recent decades to understand their sources and sinks, as well as to study their physical and chemical properties. Atmospheric aerosols are important players in the Earth’s radiative budget, affecting incoming and outgoing solar radiation through absorption and scattering by direct and indirect means. While the cooling properties of pure inorganic aerosols are relatively well understood, the impact of organic aerosols on the radiative budget is unclear. Additionally, organic aerosols are transformed through chemical reactions during atmospheric transport. The resulting complex mixture of organic aerosol has variable physical and chemical properties that contribute further to the uncertainty of these species modifying the radiative budget. Correlations between oxidative processing and increased absorptivity, hygroscopicity, and cloud condensation nuclei activity have been observed, but the mechanisms behind these phenomena have remained unexplored. Herein, we review environmentally relevant heterogeneous mechanisms occurring on interfaces that contribute to the processing of aerosols. Recent laboratory studies exploring processes at the aerosol–air interface are highlighted as capable of generating the complexity observed in the environment. Furthermore, a variety of laboratory methods developed specifically to study these processes under environmentally relevant conditions are introduced. Remarkably, the heterogeneous mechanisms presented might neither be feasible in the gas phase nor in the bulk particle phase of aerosols at the fast rates enabled on interfaces. In conclusion, these surface mechanisms are important to better understand how organic aerosols are transformed in the atmosphere affecting the environment.
Keywords:
organic aerosol; climate; human health; atmospheric aging; interface; oxidation; ozone; pollutant; aromatic 1. Atmospheric Aerosols: Classification and Importance to Societies
Atmospheric aerosols, or the liquid and solid particulates suspended in air, can be emitted to the atmosphere or be generated in situ. Particles emitted directly to the atmosphere are called primary aerosols, and can originate from either natural or anthropogenic processes [1]. Wind-blown mineral, dust, sea spray ejected from breaking waves, volcanic ash, pollen, and transported soil particles, among others, are all examples of natural particulates found in the atmosphere [2]. Smoke and soot from industry, controlled agricultural burns, and fires from burning wood, dominate anthropogenic primary aerosol emissions. Secondary aerosols are mainly generated through the condensation or coagulation of gaseous species or ultrafine particulate into larger particles or onto preexisting aerosols, such as black carbon or sea spray aerosols [3]. Secondary aerosols can also be formed through chemical reactions between volatile organics emitted from plants and a wide host of anthropogenic sources, and trace gas species such as nitrogen oxides (NOx) emissions from fossil fuel combustion, dimethyl sulfide emissions over the oceans, and sulfur dioxide emissions from fossil fuel combustion and volcanoes [3,4,5,6]. These physical and chemical processes can alter the residence time of species in the atmosphere as well as change their optical and cloud condensation nuclei (CCN) properties.
It has been proposed that the natural aerosol loading of the planet can be modulated directly through anthropogenic emissions, or indirectly by influencing biogeochemical cycles [1,7,8]. Human-driven changes to the climate system, such as increased temperatures, and higher carbon dioxide (CO2) or ozone (O3) levels [9,10], can cause fluctuations in the aerosol emissions budget, as well. For instance, rising global temperature and CO2 concentrations cause an increase in biogenic volatile organic emissions (BVOCs) [11,12]. However, expected emissions of BVOCs could be diminished because of deforestation, land usage changes, and losses of viable crop land due to aridification and salinification of soils [13,14]. Human activities, such as fossil fuel burning, industrial development, and controlled agricultural burns, can increase the amounts of sulfur, black carbon, and organic aerosols in the atmosphere [15]. Surface O3 concentrations should reasonably increase with growing BVOC emissions [16]. However, increasing atmospheric moisture, driven by rising global temperatures, can partially counteract this trend by converting O3 into hydroxyl radical (HO•) [16,17]. Reductions in greenhouse gas emissions have, likewise, cut the amounts of anthropogenic aerosols released to the atmosphere [1]. In fact, some of the warming predicted by the end of this century might be attributable to smaller magnitudes of negative radiative forcing from aerosol–cloud and aerosol–radiation effects, due to a reduction in the total atmospheric aerosol loading [18,19,20]. Therefore, accounting for the impact of feedback loops on total aerosol loadings and trace gas species is important for predicting the effect of aerosols in the future climate.
Particle size highly affects the fate of atmospheric particles because it dictates the residence time of aerosols against natural removal from the atmosphere through conversion or deposition. In the atmosphere, existing particles typically have diameters from 1 nm to 100 μm. These particles can generally be grouped into four classes. In the first class, called nucleation mode (1–10 nm), aerosol physical processes—such as gas to particle conversion and the condensation of particles with water vapor—occur, resulting in the generation of larger particles. In the second class, named Aitken mode, particles are defined in the range of 10 to 100 nm in diameter, and can undergo further coagulation and condensation to form accumulation mode particles (the third class), which range from 0.1 to 1 μm in diameter. In the fourth class are windblown mineral dust, sea spray, pollen, and volcanic ash, as well as cloud-scavenged accumulation mode particles, that can range from 1 to 100 μm in size, and are termed coarse mode aerosols [3,21].
The size regime of atmospheric aerosols plays a key role in determining for how long the particulates reside in the atmosphere. Nucleation and Aitken-sized aerosols have short lifespans, on the order of a few hours to a few days, because they tend to coagulate into larger particles. Studies have demonstrated that these nucleation events have strong diurnal and seasonal patterns that are driven by BVOCs from forests and other vegetation. For example, the concentration of nanoparticles is the highest in the morning and early afternoon, when sunlight-driven gas-phase chemistry between trace gases and BVOCs promotes new particle formation [22,23]. By mid-afternoon, the mass concentration of the smallest particles decreases as they coagulate into larger particles [24]. This behavior was confirmed by observing slight increases in the mass concentration of accumulation and coarse-sized aerosols during the evening and overnight hours [22].
Coarse mode particulates have been demonstrated to have lifetimes shorter than a few days, which is two orders of magnitude smaller than the lifespan of accumulation mode particulates [25]. This is primarily due to gravity-driven deposition being the main route of atmospheric removal for coarse particles, regardless of the type of deposition surface [26]. Other studies have demonstrated that the time scales of formation and deposition of accumulation mode aerosols over vegetated areas are on the same order of magnitude [27]. The previous finding agrees with measurements collected over a botanical garden that demonstrated the amplitude of diurnal changes in the particle number concentration of accumulation mode particulates was quite small comparing to nucleation and Aitken-sized aerosols [22]. This is why the 0.1–1 μm particles are given the name “accumulation mode”, because they tend to collect and linger in the atmosphere for 1–3 weeks before they are removed via deposition [3,25,28]. Therefore, when reviewing below important heterogeneous atmospheric mechanisms, processes relevant to these accumulation mode aerosols are central for understanding the changes in composition and associated properties during aerosol aging.
Regardless of their source or fate, atmospheric aerosols have many direct and indirect impacts on the health of the Earth’s inhabitants and atmosphere [29]. The most obvious of these explicit effects are the deleterious health impacts observed from the inhalation of particulate matter (PM) with diameters less than 10 μm (PM10). As the main component of urban smog, these small and ubiquitous particles pass into the lungs after being inhaled, leading to respiratory complications. Fine particulate matter with diameters smaller than 2.5 μm (PM2.5) often enters the bloodstream, and contributes to systemic inflammation that can lead to hypertension, even after short exposure times [30]. Long term exposure to PM2.5 has been linked to cardiovascular disease, respiratory diseases, asthma, low birth weight, lung cancer, stroke, and even diabetes [31,32]. These complications arising from PM2.5 exposure have been estimated to cause 3.2 million premature deaths per year [33]. Consequently, the World Health Organization has set exposure guidelines used to shape regulatory policies based on total mass concentrations of PM2.5. However, recent studies have suggested that the toxicity of PM can vary greatly based on composition and that using non-differentiated PM mass may result in an underestimation of its impact on mortality [34]. For instance, carbonaceous aerosols were determined to be up to 5 times more harmful than their inorganic counterparts and, thus, responsible for up to a quarter of all air pollution-related deaths annually [35,36]. Furthermore, it was determined that emissions from residential and commercial energy sources, such as cooking and heating fires, generators, and waste incineration, could represent 60–70% of PM-related mortalities in the populous areas of South Asia [36].
2. The Effect of Anthropogenic Aerosols on the Earth’s Radiative Balance
In addition to their implications on human health, aerosols can directly alter the radiation balance of the Earthsecondary by interacting with sunlight [37]. The Earth’s radiative balance describes the energetic equilibrium between the incoming solar radiation, and what is absorbed or reflected to space by the atmosphere and the surface. In this regard, radiative forcing is a measure of how much a particular atmospheric constituent traps (warms) or reflects (cools) incoming solar and outgoing terrestrial radiation [28]. The warming effect of methane, carbon dioxide, chlorofluorocarbons, and other greenhouse gases, is generally well-understood and documented at all but very low altitudes [15]. Thus, current efforts are focused in the utilization of small unmanned aerial systems (sUAS) capable of providing high spatiotemporal resolution of the level of greenhouse gases at low altitudes, to characterize emissions and provide information of the boundary layer needed to calibrate instruments in satellites [38]. Additionally, there is still a large degree of uncertainty in the net magnitude and direction (positive or negative) of the contribution of aerosols toward the radiative forcing budget, mainly attributable to their variable composition, size, optical properties, and hygroscopicity (Figure 1) [15,39,40,41].
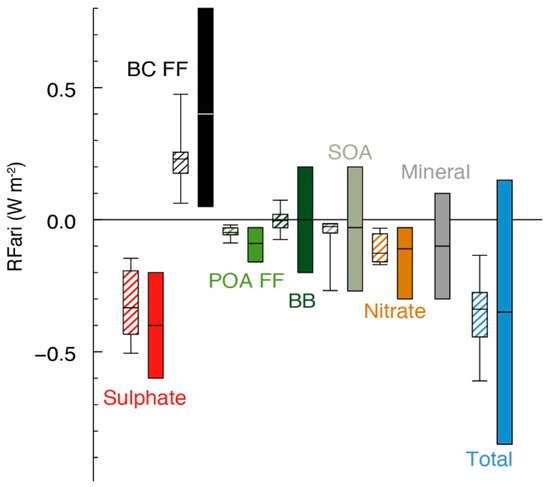
Figure 1.
Annual mean top of the atmosphere radiative forcing due to aerosol–radiation interactions (RFari, in W m−2) due to different anthropogenic aerosol types, for the 1750–2010 period. Hatched whisker boxes show median (line), 5th to 95th percentile ranges (box) and min/max values (whiskers) from AeroCom II models [37] corrected for the 1750–2010 period. Solid colored boxes show the AR5 best estimates and 90% uncertainty ranges. BC FF is for black carbon from fossil fuel and biofuel, POA FF is for primary organic aerosol from fossil fuel and biofuel, BB is for biomass-burning aerosols and SOA is for secondary organic aerosols. Reproduced with permission from reference [15].
In addition to directly modulating solar radiation, aerosols can indirectly impact the radiative balance by altering cloud properties and formation dynamics. Studies have demonstrated that increases in aerosol concentrations often translates to increased cloud droplet number concentrations, resulting in higher cloud albedo due to enhanced cloud formation [42,43]. Elevated aerosol concentrations also decrease the size of cloud droplets. These clouds do not produce precipitation efficiently and, thus, have longer lifetimes and increased horizontal and vertical extension that enhance the albedo as well [44]. However, it is still poorly understood how aerosol composition impacts aerosol-driven cloud enhancement.
Inorganic aerosols, such as sulfates, nitrates, and sea salt aerosols, are known to be excellent drivers of a net cooling radiative forcing, and act as excellent CCN by increasing the amount of water associated with the particles. This promotes enhanced cloud formation and thus heightens atmospheric reflectivity [39,45,46]. However, water is a potent infrared absorber and, thus, the magnitude of cooling effects exerted on the atmosphere by inorganic aerosols may be diminished [15]. Mineral dust, carried by winds from deserts, such as the Sahara, has been demonstrated to scatter incoming radiation to assist in cooling the planet [47]. Nevertheless, particles, including photoactive components, such as iron and titanium oxides, can increase the absorptive power of sand and dust particles, resulting in a net neutral or slightly positive radiative forcing [48]. Another important atmospheric aerosol constituent is black carbon (BC), which is produced from the incomplete combustion of fossil fuels, biofuels, and vegetation. BC is an efficient absorber of radiation and, thus, contributes a net positive effect on aerosol radiative forcing (Figure 1) [49]. This warming effect also continues once BC is deposited onto the Earth’s surface, reducing the amount of radiation reflected back to outer space via absorption. This phenomenon is thought to be contributing to the acceleration of ice and snow melting in the polar regions through heat trapping in BC deposits [50,51].
Unlike the extensive knowledge about the sources and properties of inorganic and BC aerosols, far less information is available regarding the fates of organic aerosols (OA) in the atmosphere [4,52,53,54]. Models presently underestimate the OA loading in the troposphere, particularly from biomass-burning sources, by 24% relative to field observations [55]. The OA loading and other key aerosol properties, such as optical absorptivity and hygroscopicity, seem to be driven by seasonal and regional variabilities [55,56]. Most previous attempts to constrain uncertainties from OA have focused on the formation of secondary organic aerosol (SOA) from biogenic sources, such as isoprene and several terpenes [57]. To increase the certainty in future global temperature and climate forecasts made by climate models, knowledge of the fate of anthropogenically driven organic compounds in the atmosphere is needed [58]. In addition, better inventories of sources and sinks of atmospheric oxidizers are necessary to predict how oxidation contributes to aerosol aging [1,59]. The study of the transformations of overlooked sources of SOA, such as biomass-burning aerosols and polycyclic aromatic hydrocarbons under atmospherically relevant conditions, provided information needed to constrain the uncertainties that OA contribute to radiative forcing [60,61,62]. Correlations between the oxidative processing and increased absorptivity, hygroscopicity, and CCN activity of aerosols have been proposed [63]. Oxidation also amplifies the ability of OA to absorb radiation in the near-ultraviolet range. These competing factors make the impact of OA on the net radiative forcing budget complicated [40,41]. Thus, the continuous transformation of OA into aged secondary organic aerosol during atmospheric transport [64] changes their physical and chemical properties. As a result of these changes, there is a high level of uncertainty associated to the radiative forcing due to aerosol–radiation interactions (RFari) [15]; see Figure 1.
Recent studies have also demonstrated that as SOA undergoes subsequent stages of aging, it becomes increasingly water soluble and absorbs more light [65,66]. This aged absorptive organic aerosol fraction is often called brown carbon, after the light brown color of collected samples [67]. Field observations suggest that reactions taking place in clouds are critical for the secondary formation of brown carbon, which is thought to be an underrepresented source of positive radiative forcing [41,68]. To sum up, the brown carbon contribution from SOA has been identified as one of the key uncertainties of the anthropogenic effect of aerosols on climate [15]. Moreover, heterogeneous reactions have been proposed as important for the aging of aerosols, increasing the complexity of the problem [69]. Therefore, in order to better constrain the impacts of SOA properties on climate, a better understanding needs to be gained on the heterogeneous processing of aerosols, the products of these reactions, and how these products transform the optical and hygroscopic properties of SOA.
3. Processing at Atmospheric Interfaces
Much of the chemical processing of aerosols occurs at the air–particle interface, which can be the region between the gas and either a solid, a semisolid, or a liquid. Different products may result from reactions on each kind of interface [70]. The interaction and uptake of a gas molecule by an aerosol particle occurs in a similar manner regardless of the phase, but additional parameters are needed to describe the processes on solid or semisolid interfaces, which are summarized next, and illustrated in Figure 2. In a general model, the aerosol surface can be seen as interacting with a number of gas molecules colliding on it. This collisional flux can be defined as
where [X]g is the concentration of the species X in the gas phase, and is the mean thermal velocity of X [71]. As gas molecules collide with the aerosol surface, a portion of these equilibrate with the surface. This accommodation process is dictated by the temperature, composition, and pressure of the gas, as well as the phase and state of the surface [72].
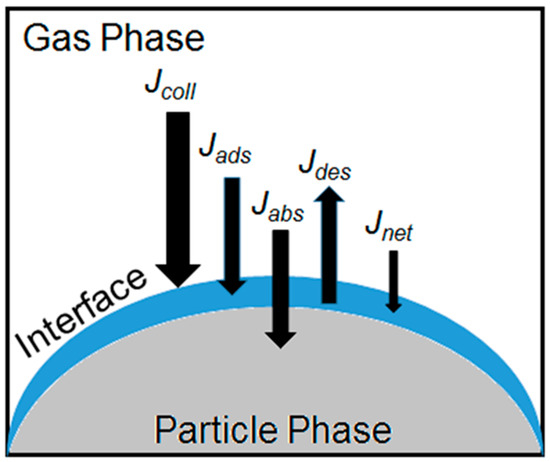
Figure 2.
Illustration of key processes occurring at the interface between aerosols and the atmosphere, including the collisional flux (Jcoll), the adsorption flux (Jads), the absorption flux (Jabs), the desorption flux (Jdes), and the next uptake (Jnet) of molecules into the particle.
Experimental observations are often corrected with a thermal accommodation coefficient, , which can be described by the relationship
where Ein is the incident energy flux, Ere is the reflected energy flux, and Ew is the flux of emitted molecules if they were in thermal equilibrium with the surface [72,73,74]. Molecules that do not immediately bounce off the surface can be held by van der Waals interactions or hydrogen bonds. This process, known as adsorption, is often termed physisorption because it represents the contribution of physical processes to the sticking of gaseous species to the interface. The adsorption flux can be estimated by the following equation:
with the surface accommodation coefficient αs, which is the ratio of accommodated molecules to the number of molecular collisions, and θ is the fractional surface coverage by the species of interest [71,75].
The fate of the adsorbed molecule can be one of the next three processes: (1) absorption, (2) chemisorption, and (3) desorption. A portion of the adsorbed molecules are taken up and accommodated into the aerosol in a process called absorption. The absorption flux can be defined as
where is the bulk accommodation coefficient [71] that is determined by the relationship
In Equation (5), are the surface to bulk transfer rate constant and the desorption rate constants, respectively [75]. Alternatively, adsorbed molecules can become chemisorbed to the aerosol if they undergo chemical reactions with other adsorbed species or with aerosol constituents. Chemisorption processes, such as these, are apparent through a hyperbolic relationship between the concentration of the gas and the reaction rate. This is often described by the Langmuir–Hinshelwood adsorption mechanism, which provides information regarding the partitioning of the reactant gas between the gas phase and the particle phase, followed by the reaction between the adsorbed gas and species in and on the aerosol [75,76].
Finally, a portion of the adsorbed molecules can detach or desorb from the surface back into the gas phase. The desorption flux can be expressed as
where is the desorption lifetime [71]. The net uptake of the molecules into the aerosols, Jnet, describes the difference between the adsorption (Jads) and desorption (Jdes) fluxes. However, in practice, it is extremely challenging to isolate and measure any of the individual processes needed to define the adsorption and desorption fluxes. Therefore, many studies have focused on determining the reactive uptake of gases by aerosol surfaces (γ). This quantity describes the overall uptake for all surface processes combined, regardless if they are physical or chemical in nature [77,78]. While γ cannot be measured directly, it can be described using the experimentally observed reactive uptake of a gas into the interfacial region, Equation (7):
where is a correction factor defined as
which includes the gas-phase diffusion coefficient for the compound of interest (Dg) and the particle diameter (dp) [71]. Therefore, the net flux between a gas and the surface of an aerosol, Jnet [78], can also be defined as
In order to better comprehend the oxidative aging processes that aerosols undergo, it is imperative to understand how oxidants interact with aerosol surfaces [78,79]. Competition between chemical reactions during adsorption and absorption can lead to radical chemistry in both the bulk and the interface [70]. This complex interaction between aerosols and the atmosphere gives rise to multiple reaction pathways “in” and “on” aerosols, leading to the complex compositions observed in field observations [70]. With a deeper understanding of how the processes occur at the surface of aerosols, the resulting physical and chemical outcomes affecting optical and chemical transformations can be revealed.
4. Aging of Aerosols in the Atmosphere
Understanding the atmospheric aging of aerosol altering its properties is a topic of current interest to atmospheric chemists and climate scientists [69]. Gas-phase reactions between atmospheric oxidizers and volatile organic species have been studied extensively. In general, common atmospheric oxidants, like ozone, nitric oxide, and peroxides, react with gas-phase organic molecules to generate less volatile and more polar products that partition to the particle phase, forming SOA [4,63,79]. Studying how SOA behaves as it is transported through the atmosphere is an important matter to describe the way urban pollution becomes more oxygenated (higher O/C ratio) during the aging process [63]. Despite the varied sources of organic aerosols, oxalic acid has been identified as a pervasive component of aged SOA, representing about 50% of the total OA mass in aerosols collected in a rural location downwind of agricultural and urban areas [80]. Thus, there must be a common mechanism of oxidative chemical processing that generates oxalic acid as an end product in the atmosphere [60,61,62,80,81,82,83]. In order to elucidate such a mechanism of oxidative aging, it is imperative to understand the major routes atmospheric aerosol processing can take, and how they can contribute to aerosol complexity. Even though physical processing (Section 3) drives the formation of new aerosols from the emission of natural and anthropogenic particles, the fate of these newborn particles during transport can be extremely complex, due to the many forces acting on them.
The chemical processing of SOA generating aged SOA can proceed through oxidative pathways, non-oxidative pathways, or photochemical pathways [64]. During oxidative processing, trace gases react with organic species on the surface of the aerosol to produce alkyl radicals R• (Figure 3). Molecular oxygen (O2) then adds to the radical to form the highly reactive alkylperoxy radical RO2•, which then can undergo reactions with a host of atmospheric radicals (NO, NO2, RO2•, HO2•, NO3•) [64]. These reactions result in the addition of polar functional groups, such as hydroperoxyl (HOO–), nitrate (–NO3), nitro (–NO2), hydroxyl (–OH), carboxylic acid (–COOH), carbonyls (>C=O), among others [84]. These added polar groups, in addition to increasing hygroscopicity, decrease molecule volatility [85]. These oxidation processes enhance further SOA formation via condensation and coagulation.
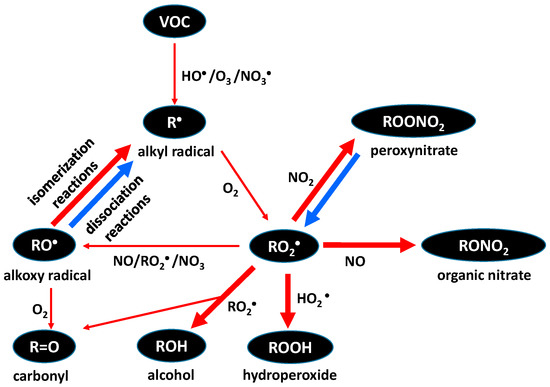
Figure 3.
A simple non-photochemical oxidation mechanism for a typical volatile organic compound (VOC). Thick red arrows describe reactions leading to a large volatility drop, while blue arrows describe reactions leading to a significant rise in volatility.
Semi-volatile species produced from oxidation reactions can undergo accretion reactions in aerosols to form heavier species after partitioning into the particle phase [64,86]. This non-oxidative pathway can take many forms, but most, if not all, accretion reactions involve polymerization or radical recombination of organic compounds. The resulting products are heavier and nonvolatile complex oligomeric species unlikely to be formed by oxidation in the gas phase. Based on observations from field studies, it has been inferred that accretion reactions are enhanced in acidic aerosols by acid-catalyzed nucleophilic additions to aldehydes, carbonyls, and alcohols [86]. Additionally, alkenes, such as limonene, have also been shown to more readily undergo accretion reactions such as dimerization in the presence of strong acids [87,88]. Two pathways dominated the oxidation of β-pinene via nitrate radicals (NO3•), the reactions RO2• + NO3• and RO2• + HO2•, with similar SOA yields from both channels [89]. As many atmospheric aerosols are comprised of acidic components, these reactions represent a common pathway for the formation of non-volatile species in aerosols [90].
In addition to chemical reactions, sunlight can also facilitate the processing, accretion, and aging chemistry in and on atmospheric particles [82,83,91,92,93,94,95,96,97]. Noteworthy, when gaseous carboxylic acid partition from air into the particle phase, they exert enhanced acidities to the water interface that are at least larger than 1.7 pH-units than predicted by bulk pKa values [95]. Organic and inorganic molecules can act as chromophores, or light absorbing moieties, that absorb visible and ultraviolet radiation. These compounds, such as conjugated double bonds or carbonyls, lead to the formation of excited state molecules that can undergo homolytic bond cleavage, radical recombination, electron transfer, decomposition, hydrogen abstraction, photosensitization, and proton coupled electron transfer [82,83,93,94,96,97,98,99,100,101]. The resulting products, which are similar to what is observed with particle-phase accretion chemistry, dehydrate or cyclize during the night to form oligomers or supramacromolecular structures. When the sun rises the next day, photons are absorbed by these complexes, leading to their dissociation. This pattern of diurnal cycling is commonly seen with α-dicarbonyls, which are prevalent in aged OA, and is a possible source of positive radiative forcing from OA [82,83,92,97].
Sunlight can also accomplish aerosol processing indirectly via the photochemical production of hydroxyl radical (HO•), which is one of the most abundant tropospheric oxidizers [102]. Volatile organics often first undergo gas-phase oxidation by HO•, decreasing volatility and encouraging coagulation to form SOA. Photochemistry can also alter the balance of available trace oxidizers, such as ozone and nitrogen dioxide, through the cycling of halogen radicals [103]. Therefore, photochemical processes are an important factor in the direct and indirect production of SOA through oxidative and non-oxidative means.
All the aerosol processes discussed above can change the physiochemical properties of SOA. It has been demonstrated that higher O/C ratios in aerosols enhances hygroscopicity due to the greater hydrophilic character of the newly formed polar functional groups [4]. Highly oxygenated SOA can serve as CCN at lower relative humidity, whereas more hydrophobic OA can only do this in very moist environments. Diminished compound volatility has also been observed relative to an elevated O/C ratio. As aging proceeds, volatility can be altered further via oligomerization and fragmentation [104]. All of these processes can amplify the number of light absorbing functional groups, which increase the absorbing power of aged SOA [91]. Oligomerization is also thought to be a contributing factor to the creation of light absorbing brown carbon in humic-like substances (HULIS) [105,106]. Through understanding the interplay between chemical processing and aerosol properties, more complete mechanistic models can be developed to describe the evolution of aerosols, including their composition and absorptivity [64,91].
5. Environmental Information from Laboratory Studies of Heterogeneous Reactions
While the physical and chemical processes that lead to aerosol formation in the environment are a current focus of study, it remains very unclear how the compositional complexity observed in field measurements is generated. Gas phase and aqueous phase (bulk) studies can guide the initial prediction of the products expected during interfacial reactions, but are physically limited and often underestimate the variety of possible products and pathways of atmospheric significance [60,61,107]. Cloud chambers [108,109,110], Knudsen cells [111,112], droplet train flow reactors [113,114,115], flow tube reactors [116,117,118,119], and molecular dynamics simulations [120] have been used to investigate different properties of aerosols and the results of atmospheric processing. Multiple studies utilizing the methods outlined above indicated that a Langmuir–Hinshelwood adsorption mechanism generally controls the reactive uptake of oxidants by the surface of aerosols [115,116,118]. Additionally, it was determined that ozonolysis on the particle surfaces could be a key source of gas-phase aldehyde production [117]. However, the previous methods do not always provide atmospherically relevant results reflecting aerosol surface to volume ratios, interfacial diffusion rates and mass accommodation values, the presence of organic coatings and water associated with particles, or the hydrophobicity of the components [70].
X-ray Photoelectron Spectroscopy (XPS) has been used to probe the composition of ambient aerosols at the surface of collected particles, demonstrating its strong correlation with size [121]. For instance, carbon content is elevated in accumulation mode aerosols [121], which suggests that these particulates are likely dominated by OA born in nucleation events. Furthermore, chemical species associated with mineral dusts and soil (e.g., silicates, carbonates, and iron oxides) were only observed in coarse mode particulates [121]. XPS was also applied to analyze BC aerosol generated in a laboratory chamber showing that sulfur associated with diesel particles was present only as sulfate [122]. Further efforts to couple XPS with mass spectrometry (MS) enabled a better understanding of the differences in chemical composition and speciation of aerosol surfaces with respect to the bulk particle [123]. Moreover, the XPS MS technique previously demonstrated how deposition of inorganic species on soot and BC aerosols could decrease their overall hydrophobicity [124]. Overall, XPS analysis of aerosols provided relevant information to describe how changes in composition at the interface can improve CCN activity or increased absorptivity of aerosols.
Collected aerosols from field studies have been characterized using aerosol time-of-flight mass spectrometers [125,126,127], electron microscopy [128,129], X-ray diffraction [130,131], and many other techniques to determine the outcome of atmospheric aging. These techniques provide insight into the composition of the aerosol, but generally do not permit tracking of how the particles arrived to that state. Recently, the combination of multiple MS techniques was used to examine several generations of organic aerosols derived from the gas-phase oxidation of α-pinene, showing a drop in the volatility of OA constituents during aging [132]. However, no mechanism was proposed as to how these reactions occur. Therefore, new instrumental methods should be also capable of providing mechanistic insights of the aging process (Section 4), e.g., by probing the heterogeneous oxidation of aerosol proxies such as iodide and substituted catechols [60,62,107]. The main outcome of this new surface sensitive MS method, developed with proxies, was to have identified reaction pathways that predominate in the interface of air with water [60,62,107] and, in addition, to have compared the surface oxidation of solid catechol in air under variable relative humidity [61]. The last findings showed the mechanistic pathways [60,61,62] by which organic precursors are created to change the optical properties of aerosols [82,83,91,92].
The oxidation of aqueous particles has been investigated using droplet generation systems paired with inline detection systems, to allow for the observation of intermediates that might be lost in traditional collection and offline processing methods. Nebulizers coupled to flow tubes interfaced with mass spectrometers, particle sizers, and infrared spectrometers, have been employed in the laboratory to examine how transformations might occur in clouds [116,133]. However, interfacial reactions often proceed too quickly to capture reactive intermediates in these systems. To bridge this gap, online electrospray ionization mass spectrometry (OESI-MS) has been developed (Figure 4) [60,62,95,107]. In OESI-MS, a nebulizing system interfaced with an electrically grounded ESI system provided aqueous microdroplets that can probe reactions at the air–water interface with shortened analysis times, as the contact time of the colliding gas with the aqueous surface only lasts for a few microseconds [60,107].
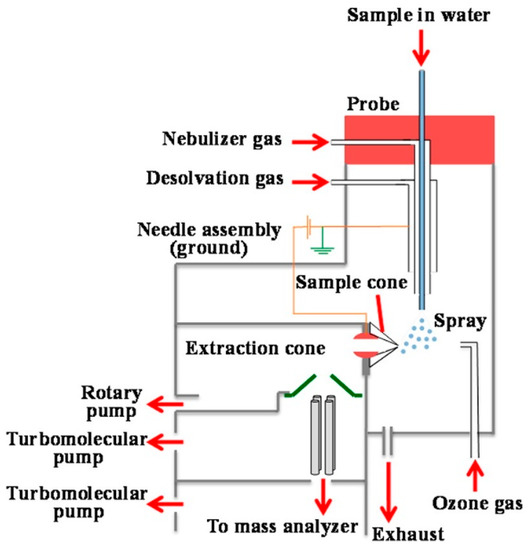
Figure 4.
Schematic diagram of the pneumatically assisted electrically grounded online electrospray ionization mass spectrometry (OESI-MS) set-up, utilized to probe reactions at the air–water interface. Reproduced with permission from reference [107].
The major characteristic of these pneumatically assisted aerosolization systems has been described to be the high ratio of nebulizing gas to liquid flow (1.2 × 103) [134]. Ions formed after the microsecond-lasting reaction are ejected off the droplet surfaces in a series of cascade desolvation steps [135], to be detected by a mass spectrometer within 1 millisecond. As a result, this analytical method allows for selectively sampling the reactants and products in the microdroplets interface. The customized instrument has allowed for the delivery of gaseous ozone to examine the in situ oxidation of inorganic and organic species on the surface of aqueous microdroplets under atmospheric relevant conditions [60,62,107].
The interfacial conversion of iodide to hypoiodous acid and molecular iodine on the surface of aqueous microdroplets exposed to O3 was explored in detail by OESI-MS [107]. This study proposed the important role of reactions occurring on the surface of sea spray aerosols and their implications in tropospheric ozone depletion [107]. A related study demonstrated that iodide and bromide can be respectively enriched by 7.3 × 103 and 7.5 × 102 times at the air–water interface relative to bulk seawater (Figure 5), as it happens when sea spray aerosol is formed during bubble bursting and wave breaking processes [134]. In turn, tropospheric ozone can react with the surface available iodide to produce reactive halogen species (RHS), molecular iodine, and hypoiodous acid. This mechanism of RHS production was proposed as important to provide the missing light sensitive precursors needed to deplete ozone over the open ocean [107].
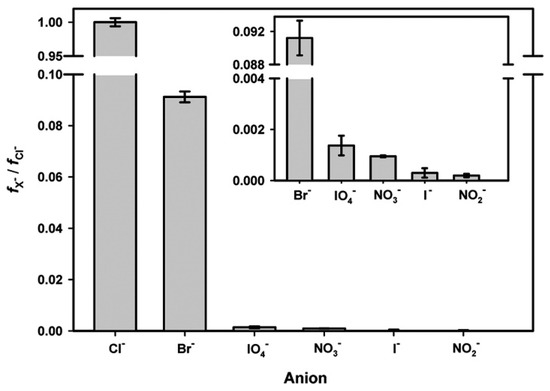
Figure 5.
Anion fractionation factor, fX−, normalized relative to the fractionation of chloride, fCl−, for molar ratios of anions found in seawater. A comparison to bulk seawater value indicates that at the air–water interface, iodide and bromide can be respectively enriched by 7.3 × 103 and 7.5 × 102 times. Reproduced with permission from reference [134].
The fast oxidation by ozone and in situ-produced hydroxyl radicals of important hydroxylated aromatics, such as catechol, was studied in detail at the air–water interface by OESI-MS [60]. Catechol, 1,2-diihydroxybenzene, is widely found in biomass-burning emissions. While catechol was previously thought to primarily undergo ring-opening reactions when exposed to ozone [136], the application of OESI-MS resolved the simultaneous contribution to oxidation by an indirect electron transfer-initiated oxidation, providing HO• radicals [60]. In addition to observing common catechol ozonolysis products [137], it was proposed, for the first time, that oxidation by HO• caused the production of polyhydroxylated aromatic compounds and quinones in atmospheric particles [60]. This mechanism provided an explanation for the production of light absorbing brown carbon, along with how complex mixtures of organic compounds are generated [60,61]. Carefully performed analysis spiking standards indicated that when microdroplets containing 100 µM catechol at pH 8.0 were exposed to 3.18 ppmv O3 (g) during a contact time of 1 µs, the concentration of the main low molecular weight carboxylic acids produced at the interface was [glyoxylic acid] = 200 nM > [oxalic acid] = 174 nM > [cis,cis-muconic acid] = 129 nM > [3-oxopropanoic acid] = 109 nM [60].
Furthermore, the previous concepts were expanded to a series of substituted catechols that served as proxies to explore how anthropogenically emitted phenols (or oxy-aromatics) derived from benzene, anisole, and toluene, are oxidized at the air–water interface [62]. A variety of some important polyphenols and quinones produced during the oxidation of substituted catechols at the air–water interface are included in Figure 6 [62], as an example of the many different species accounted by these mechanisms [60,61,62]. The work predicted, thermodynamically, the trend of favorable electron transfer reactions of the substituted catechols with ozone, and concluded that stronger electron donors to the aromatic system lead to an enhancement in the rate of product formation during oxidation [62]. The interfacial oxidation of catechol [60,61,62] has also been proposed to be affected by Baeyer–Villiger (BV) oxidation of the unsaturated dicarboxylic acids, generating unsaturated, oxo-, and dicarboxylic acids [60,61]. A pioneer constrain for the interfacial production of HO• of (1.1 ± 0.3) × 1013 radicals cm−3 was determined when microdroplets with 100 μM of substituted catechol, at pH 7.8, reacted with 2.7 ppmv O3(g) [62].
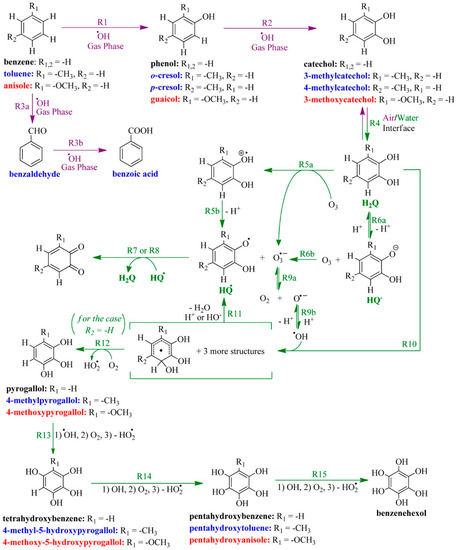
Figure 6.
Sequential oxidation of (black bold font) benzene, (blue bold font) toluene, and (red bold font) anisole in the (purple font and reactions) gas phase by hydroxyl radicals, which continue at the (green font and reactions) air–water interface, and in aqueous particles after partitioning of the corresponding substituted catechols. The names of the products are color-coded to the corresponding precursors. Complete lists of the produced polyphenols and their corresponding quinone redox pairs are available in the original paper with the observed mass-to-charge m/z values registered in the mass spectrometer. Reproduced with permission from reference [62].
For the study of reactions on solid surfaces in the laboratory, the preparation of thin films with proxy species found in solid atmospheric particles has been useful to investigate the uptake of reactive gases and characterize the transformations that alter the initial composition. Studies in aerosol chambers and flow tubes [138,139,140,141,142] have demonstrated how to use gas-phase reactions to generate SOA in controlled environments. The resulting aerosols are analyzed by different techniques, e.g., optical spectroscopy, before they are collected for further characterization by chromatography and mass spectrometry [139]. Variations in the physical state of the aerosol, reaction time, size, and the procedure for measuring rate of loss, can lead to the dissimilar reaction rates and uptake coefficients reported by different groups studying the same molecules [70]. These approaches typically capture the variations in composition in the early stages of aging, but are not necessarily representative of the extended aging periods experienced during transport [61].
More recently, thin films have been used to simulate relevant reactions on the aerosol surface modeling the air–particle interface [61,116,136,143]. This approach has proven quite useful to monitor the degradation of the proxy materials and evolution of condensed and gas phase products in gas cells or environmental chambers [144]. While fast processes in the microsecond timescale at the air–water interface were studied in the customized OESI-MS set-up described above [60], most likely, some products that form over longer times could have remained hidden. Therefore, an additional system was developed to study oxidative processing on surfaces that could pair the real-time monitoring from OESI-MS [60,62] with offline compositional analysis by multiple techniques [61]. A clear picture of reactions on solid surfaces was provided by this new flow-through reaction chamber (Figure 7) [61]. Oxidation resistant infrared holders, with optical windows supporting the thin films of proxy species, were placed into a reaction chamber. Ozone under variable relative humidity (0–90%) could be circulated through the reaction chamber at a constant flow rate. In such a system, samples could be regularly scanned, spectroscopically, in the infrared, to monitor the oxidation process [61]. Of note is that protocols were also developed to extract the reacted films and analyze their products by chromatography and mass spectrometry [61].
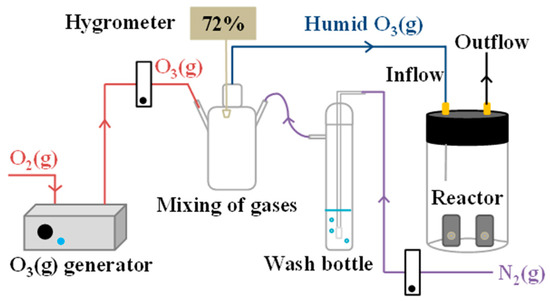
Figure 7.
Schematic diagram of the flow-through reactor system with mixing of dry and wet gases. Reproduced with permission from reference [61].
This flow-through reaction chamber was used to examine the heterogeneous oxidation of catechol at the air–solid interface. The work employed infrared spectroscopy, UV–visible spectroscopy, chromatography, and mass spectrometry to analyze the reaction products. Not only did this experimental design validate findings using the air–water interface technique, but also provided new details of intermediates and products that escaped detection in the microsecond contact time of OESI-MS [60,61]. The longer exposure times in the air–solid interface study enabled the observation of organic acids produced from direct ozonolysis, and polyhydroxylated aromatics, bi- and terphenyls products via the crosslinking of reactive semiquinone intermediates [61]. Relative humidity was found to be a key driver in the reaction rate at the air–solid interface [61], suggesting that adsorbed water facilitates ring cleavage and electron transfer as observed in the air–water interface study [60].
6. Outcomes Learned from Recent Interfacial Studies
This review has discussed recent important heterogeneous atmospheric mechanisms that contribute to the understanding of the composition, processing, and properties of aerosols. The first important conclusion from these recent heterogeneous oxidation studies is that ozone is an important player in tropospheric chemistry, which enables the cycling of radicals and other trace atmospheric species by previously unconsidered mechanisms. This secondary pollutant formed through a series of gas phase reactions [29,145,146] is widely available to oxidize many atmospheric species as described above. When ozone reacts either with iodide or a substituted catechol, a wide range of reactive intermediates are produced alongside the oxidized products [60,61,62,107]. The generation of radicals can further propagate chain reactions through previously unexpected channels, as evidenced by the hydroxylation pathway observed during the oxidation catechol by ozone under humid conditions [60,61,62]. Both inorganic and organic systems revealed that tropospheric pollution can impact atmospheric composition. Elevated emissions of volatile organic compounds (VOCs) and NOx in urban areas favor high O3 levels and, thus, impact the oxidation mechanism on aerosols surfaces [146].
A second important takeaway from examining heterogeneous reactions at the atmosphere–aerosol interface is that the oxidation of organic aerosols by trace species can dramatically alter their composition, while increasing their hygroscopicity and UV–visible absorptivity. It was determined that SOA and some primary OA are prone to undergo multistep chemical reactions with trace atmospheric oxidizers (e.g., HO•, O3, NOx) and sunlight, that alter their composition [60,61,91,92,93,107]. Each successive step of oxidation has the ability to add polar functional groups, decreasing the volatility of the constituents [4]. This process leads to the partitioning of organics into the aqueous phase of particles, where further processing can occur [147,148]. Additionally, with the incorporation of polar functional groups, new chromophores can be generated in these secondary organic aerosols along with organic aerosols generated from combustion or biomass-burning events [60,61,62]. As a consequence, the processed organics can absorb ultraviolet and visible radiation [149].
The third remark is that investment in designing, developing, and testing advanced instrumentation and reactors in the laboratory is a key aspect to make progress in this area of research. Without such efforts, it would be impossible to reveal the mechanistic information needed to predict the effect of aerosols. The use of the advanced and creative methods, summarized above, has pushed the boundaries of current knowledge about how aerosols undergo oxidation reactions at atmospheric interfaces. OESI-MS has been a fundamental tool for studying surface chemistry by actually examining the structure of the interfacial region and reactions occurring in this boundary region [60,62,95,107,134,150,151]. The experiments studying the oxidation of iodide [107], catechol [60], and substituted catechols [62] constitute the forefront measurements to observe the products of chemical aging at the air–water interface. The combined use of FTIR and UV–visible spectroscopies, mass spectrometry, and chromatography to characterize aerosol proxies reacting at the air–solid interface under variable humidity has been demonstrated [61]. The setup utilized a flow-through reactor to provide the reactive uptake coefficient of ozone on catechol films [61] under variable humidity conditions, and elucidated the formation of quinones as well as biphenyl and terphenyl rings [61]. Present and future laboratory, chamber, and field studies to examine the product mixtures resulting from these mechanisms are being inspired [152,153,154,155,156,157,158,159,160,161,162,163] by this work, to gather information for inclusion in the next generation of computational models to predict the effects of aerosols on climate.
Finally, this review has provided examples describing how to design laboratory studies to collect and interpret the data of oxidative aging of aerosols. Aerosol and cloud water samples collected at different geographical locations often display similarities in composition, despite significant differences in parent species [80,164,165,166]. The mechanisms discussed above explain the processing of different organic atmospheric precursors, which are found in both biomass burning [167] and anthropogenic emissions [168], and likely proceed through similar pathways in the atmosphere [60,61,62]. Oxy-aromatics can generate a rich product mixture that absorbs more light, forms better nuclei to form clouds, and partitions into clouds easily [60,61,62]. The absorptivity of the oxidation products grows in the visible region both in films and aqueous solutions, contributing to the “browning” of aerosols during the aging process [149]. The highly oxidized products contain carboxylic acid and hydroxyl moieties that justify the extremely hygroscopic nature of this class of aerosols from aromatic precursors [45,46]. Therefore, these kind of products are expected to easily partition into cloud waters and serve as precursors to the formation of humic-like materials [169,170]. Future efforts should explore some of these ideas further in the laboratory, by developing new experimental methods and instrumentation capable of investigating reactions at atmospheric interfaces.
Author Contributions
All authors conceived and wrote the paper at the University of Kentucky.
Funding
M.I.G. thanks research funding from the US. National Science Foundation under NSF CAREER award CHE-1255290 and under RII Track-2 FEC award No. 1539070.
Conflicts of Interest
The authors declare no conflict of interest. E.A.P.-L. is currently a Postdoctoral Research Associate in the School of Meteorology at the University of Oklahoma, Norman, OK, USA.
References
- Von Schneidemesser, E.; Monks, P.S.; Allan, J.D.; Bruhwiler, L.; Forster, P.; Fowler, D.; Lauer, A.; Morgan, W.T.; Paasonen, P.; Righi, M.; et al. Chemistry and the linkages between air quality and climate change. Chem. Rev. 2015, 115, 3856–3897. [Google Scholar] [CrossRef] [PubMed]
- Andreae, M.O. Aerosols before pollution. Science 2007, 315, 50–51. [Google Scholar] [CrossRef] [PubMed]
- Seinfeld, J.H.; Pandis, S.N. Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, 2nd ed.; Wiley: Hoboken, NJ, USA, 2006. [Google Scholar]
- Hallquist, M.; Wenger, J.C.; Baltensperger, U.; Rudich, Y.; Simpson, D.; Claeys, M.; Dommen, J.; Donahue, N.M.; George, C.; Goldstein, A.H.; et al. The formation, properties and impact of secondary organic aerosol: Current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. [Google Scholar] [CrossRef]
- Woodhouse, M.T.; Mann, G.W.; Carslaw, K.S.; Boucher, O. Sensitivity of cloud condensation nuclei to regional changes in dimethyl-sulphide emissions. Atmos. Chem. Phys. 2013, 13, 2723–2733. [Google Scholar] [CrossRef]
- Alexander, B.; Park, R.J.; Jacob, D.J.; Li, Q.B.; Yantosca, R.M.; Savarino, J.; Lee, C.C.W.; Thiemens, M.H. Sulfate formation in sea-salt aerosols: Constraints from oxygen isotopes. J. Geophys. Res. Atmos. 2005, 110. [Google Scholar] [CrossRef]
- Setyan, A.; Song, C.; Merkel, M.; Knighton, W.B.; Onasch, T.B.; Canagaratna, M.R.; Worsnop, D.R.; Wiedensohler, A.; Shilling, J.E.; Zhang, Q. Chemistry of new particle growth in mixed urban and biogenic emissions—Insights from CARES. Atmos. Chem. Phys. 2014, 14, 6477–6494. [Google Scholar] [CrossRef]
- Paasonen, P.; Asmi, A.; Petaja, T.; Kajos, M.K.; Aijala, M.; Junninen, H.; Holst, T.; Abbatt, J.P.D.; Arneth, A.; Birmili, W.; et al. Warming-induced increase in aerosol number concentration likely to moderate climate change. Nat. Geosci. 2013, 6, 438–442. [Google Scholar] [CrossRef]
- Wu, S.; Mickley, L.J.; Jacob, D.J.; Rind, D.; Streets, D.G. Effects of 2000–2050 changes in climate and emissions on global tropospheric ozone and the policy-relevant background surface ozone in the United States. J. Geophys. Res. Atmos. 2008, 113. [Google Scholar] [CrossRef]
- Ammann, M.; Klimont, Z.; Wagner, F. Regional and global emissions of air pollutants: Recent trends and future scenarios. Annu. Rev. Environ. Resour. 2013, 38, 31–55. [Google Scholar] [CrossRef]
- Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, C.; Palmer, P.I.; Geron, C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar] [CrossRef]
- Sanderson, M.G.; Jones, C.D.; Collins, W.J.; Johnson, C.E.; Derwent, R.G. Effect of climate change on isoprene emissions and surface ozone levels. Geophys. Res. Lett. 2003, 30. [Google Scholar] [CrossRef]
- Wiedinmyer, C.; Tie, X.; Guenther, A.; Neilson, R.; Granier, C. Future changes in biogenic isoprene emissions: How might they affect regional and global atmospheric chemistry? Earth Interact. 2006, 10, 1–19. [Google Scholar] [CrossRef]
- Lathière, J.; Hauglustaine, D.A.; Friend, A.D.; De Noblet-Ducoudré, N.; Viovy, N.; Folberth, G.A. Impact of climate variability and land use changes on global biogenic volatile organic compound emissions. Atmos. Chem. Phys. 2006, 6, 2129–2146. [Google Scholar] [CrossRef]
- Boucher, O.; Randall, D.; Artaxo, P.; Bretherton, C.; Feingold, G.; Forster, P.; Kerminen, V.-M.; Kondo, Y.; Liao, H.; Lohmann, U.; et al. Clouds and aerosols. In Climate Change 2013: The Physical Science Basis. Contribution of Working Group I to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; Stocker, T.F., Qin, D., Plattner, G.-K., Tignor, M., Allen, S.K., Boschung, J., Nauels, A., Xia, Y., Bex, V., Midgley, P.M., Eds.; Cambridge University Press: Cambridge, UK; New York, NY, USA, 2013; pp. 571–658. [Google Scholar]
- Wang, Y.; Shen, L.; Wu, S.; Mickley, L.; He, J.; Hao, J. Sensitivity of surface ozone over China to 2000–2050 global changes of climate and emissions. Atmos. Environ. 2013, 75, 374–382. [Google Scholar] [CrossRef]
- Doherty, R.M.; Wild, O.; Shindell, D.T.; Zeng, G.; MacKenzie, I.A.; Collins, W.J.; Fiore, A.M.; Stevenson, D.S.; Dentener, F.J.; Schultz, M.G.; et al. Impacts of climate change on surface ozone and intercontinental ozone pollution: A multi-model study. J. Geophys. Res. Atmos. 2013, 118, 3744–3763. [Google Scholar] [CrossRef]
- Naik, V.; Horowitz, L.W.; Fiore, A.M. Impact of preindustrial to present-day changes in short-lived pollutant emissions on atmospheric composition and climate forcing. J. Geophys. Res. Atmos. 2013, 118, 8086–8110. [Google Scholar] [CrossRef]
- Levy, H.; Horowitz, L.W.; Schwarzkopf, M.D.; Ming, Y.; Golaz, J.-C.; Naik, V.; Ramaswamy, V. The roles of aerosol direct and indirect effects in past and future climate change. J. Geophys. Res. Atmos. 2013, 118, 4521–4532. [Google Scholar] [CrossRef]
- Rotstayn, L.D.; Collier, M.A.; Chrastansky, A.; Jeffrey, S.J.; Luo, J.J. Projected effects of declining aerosols in RCP4.5: Unmasking global warming? Atmos. Chem. Phys. 2013, 13, 10883–10905. [Google Scholar] [CrossRef]
- Whitby, K.T. The physical characteristics of sulfur aerosols. Atmos. Environ. 1978, 12, 135–159. [Google Scholar] [CrossRef]
- Conte, M.; Donateo, A.; Dinoi, A.; Belosi, F.; Contini, D. Case study of particle number fluxes and size distributions during nucleation events in southeastern italy in the summer. Atmosphere 2015, 6, 942–959. [Google Scholar] [CrossRef]
- Hussein, T.; Martikainen, J.; Junninen, H.; Sogacheva, L.; Wagner, R.; Maso, M.D.; Riipinen, I.; Aalto, P.P.; Kulmala, M. Observation of regional new particle formation in the urban atmosphere. Tellus B 2008, 60, 509–521. [Google Scholar] [CrossRef]
- Aalto, P.; Hämeri, K.; Becker, E.; Weber, R.; Salm, J.; Mäkelä, J.M.; Hoell, C.; O’dowd, C.D.; Hansson, H.-C.; Väkevä, M.; et al. Physical characterization of aerosol particles during nucleation events. Tellus B 2001, 53, 344–358. [Google Scholar] [CrossRef]
- Williams, J.; de Reus, M.; Krejci, R.; Fischer, H.; Ström, J. Application of the variability-size relationship to atmospheric aerosol studies: Estimating aerosol lifetimes and ages. Atmos. Chem. Phys. 2002, 2, 133–145. [Google Scholar] [CrossRef]
- Petroff, A.; Zhang, L. Development and validation of a size-resolved particle dry deposition scheme for application in aerosol transport models. Geosci. Model Dev. 2010, 3, 753–769. [Google Scholar] [CrossRef]
- Pryor, S.; Binkowski, F. An analysis of the time scales associated with aerosol processes during dry deposition. Aerosol. Sci. Technol. 2004, 38, 1091–1098. [Google Scholar] [CrossRef]
- Jacobs, D.J. Introduction to Atmospheric Chemistry; Princeton University Press: Princeton, NJ, USA, 1999. [Google Scholar]
- Monks, P.S.; Archibald, A.T.; Colette, A.; Cooper, O.; Coyle, M.; Derwent, R.; Fowler, D.; Granier, C.; Law, K.S.; Mills, G.E.; et al. Tropospheric ozone and its precursors from the urban to the global scale from air quality to short-lived climate forcer. Atmos. Chem. Phys. 2015, 15, 8889–8973. [Google Scholar] [CrossRef]
- Brook, R.D.; Rajagopalan, S. Particulate matter, air pollution, and blood pressure. J. Am. Soc. Hypertens. 2009, 3, 332–350. [Google Scholar] [CrossRef] [PubMed]
- Pope, C.A.; Burnett, R.T.; Thun, M.J. Lung cancer, cardiopulmonary mortality, and long-term exposure to fine particulate air pollution. J. Am. Med. Assoc. 2002, 287, 1132–1141. [Google Scholar] [CrossRef]
- WHO. Review of Evidence on Health Aspects of Air Pollution (REVIHAAP) Project; WHO: Copenhagen, Denmark, 2013. [Google Scholar]
- Lim, S.S.; Vos, T.; Flaxman, A.D.; Danaei, G.; Shibuya, K.; Adair-Rohani, H.; Amann, M.; Anderson, H.R.; Andrews, K.G.; Aryee, M.; et al. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet 2012, 380, 2224–2260. [Google Scholar] [CrossRef]
- Tuomisto, J.T.; Wilson, A.; Evans, J.S.; Tainio, M. Uncertainty in mortality response to airborne fine particulate matter: Combining European air pollution experts. Reliab. Eng. Syst. Saf. 2008, 93, 732–744. [Google Scholar] [CrossRef]
- Lippmann, M.; Chen, L.C.; Gordon, T.; Ito, K.; Thurston, G.D. National Particle Component Toxicity (NPACT) Initiative: Integrated epidemiologic and toxicologic studies of the health effects of particulate matter components. Res. Rep. Health Eff. Inst. 2013, 177, 5–13. [Google Scholar]
- Lelieveld, J.; Evans, J.S.; Fnais, M.; Giannadaki, D.; Pozzer, A. The contribution of outdoor air pollution sources to premature mortality on a global scale. Nature 2015, 525, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Myhre, G.; Samset, B.H.; Schulz, M.; Balkanski, Y.; Bauer, S.; Berntsen, T.K.; Bian, H.; Bellouin, N.; Chin, M.; Diehl, T.; et al. Radiative forcing of the direct aerosol effect from AeroCom Phase II simulations. Atmos. Chem. Phys. 2013, 13, 1853–1877. [Google Scholar] [CrossRef]
- Schuyler, T.J.; Guzman, M.I. Unmanned aerial systems for monitoring trace tropospheric gases. Atmosphere 2017, 8, 206. [Google Scholar] [CrossRef]
- Petters, M.D.; Kreidenweis, S.M. A single parameter representation of hygroscopic growth and cloud condensation nucleus activity. Atmos. Chem. Phys. 2007, 7, 1961–1971. [Google Scholar] [CrossRef]
- Duplissy, J.; DeCarlo, P.F.; Dommen, J.; Alfarra, M.R.; Metzger, A.; Barmpadimos, I.; Prevot, A.S.H.; Weingartner, E.; Tritscher, T.; Gysel, M.; et al. Relating hygroscopicity and composition of organic aerosol particulate matter. Atmos. Chem. Phys. 2011, 11, 1155–1165. [Google Scholar] [CrossRef]
- Liu, J.; Scheuer, E.; Dibb, J.; Ziemba, L.D.; Thornhill, K.L.; Anderson, B.E.; Wisthaler, A.; Mikoviny, T.; Devi, J.J.; Bergin, M.; et al. Brown carbon in the continental troposphere. Geophys. Res. Lett. 2014, 41, 2191–2195. [Google Scholar] [CrossRef]
- Twomey, S. The influence of pollution on the shortwave albedo of clouds. J. Atmos. Sci. 1977, 34, 1149–1152. [Google Scholar] [CrossRef]
- Isaksen, I.S.A.; Granier, C.; Myhre, G.; Berntsen, T.K.; Dalsøren, S.B.; Gauss, M.; Klimont, Z.; Benestad, R.; Bousquet, P.; Collins, W.; et al. Atmospheric composition change: Climate-chemistry interactions. Atmos. Environ. 2009, 43, 5138–5192. [Google Scholar] [CrossRef]
- Albrecht, B.A. Aerosols, cloud microphysics, and fractional cloudiness. Science 1989, 245, 1227–1230. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, Z. Estimation of cloud condensation nuclei concentration from aerosol optical quantities: Influential factors and uncertainties. Atmos. Chem. Phys. 2014, 14, 471–483. [Google Scholar] [CrossRef]
- Kravitz, B.; Wang, H.; Rasch, P.J.; Morrison, H.; Solomon, A.B. Process-model simulations of cloud albedo enhancement by aerosols in the Arctic. Philos. Trans. R. Soc. A 2014, 372. [Google Scholar] [CrossRef] [PubMed]
- Usher, C.R.; Michel, A.E.; Grassian, V.H. Reactions on mineral dust. Chem. Rev. 2003, 103, 4883–4940. [Google Scholar] [CrossRef] [PubMed]
- Sokolik, I.N.; Winker, D.M.; Bergametti, G.; Gillette, D.A.; Carmichael, G.; Kaufman, Y.J.; Gomes, L.; Schuetz, L.; Penner, J.E. Introduction to special section: Outstanding problems in quantifying the radiative impacts of mineral dust. J. Geophys. Res. Atmos. 2001, 106, 18015–18027. [Google Scholar] [CrossRef]
- Ramanathan, V.; Carmichael, G. Global and regional climate changes due to black carbon. Nat. Geosci. 2008, 1, 221–227. [Google Scholar] [CrossRef]
- Flanner, M.G. Arctic climate sensitivity to local black carbon. J. Geophys. Res. Atmos. 2013, 118, 1840–1851. [Google Scholar] [CrossRef]
- AMAP. The Impact of Black Carbon on the Arctic Climate; Arctic Monitoring and Assessment Programme (AMAP): Oslo, Norway, 2011. [Google Scholar]
- Johnson, D.; Utembe, S.R.; Jenkin, M.E.; Derwent, R.G.; Hayman, G.D.; Alfarra, M.R.; Coe, H.; McFiggans, G. Simulating regional scale secondary organic aerosol formation during the TORCH 2003 campaign in the southern UK. Atmos. Chem. Phys. 2006, 6, 403–418. [Google Scholar] [CrossRef]
- Robinson, A.L.; Donahue, N.M.; Shrivastava, M.K.; Weitkamp, E.A.; Sage, A.M.; Grieshop, A.P.; Lane, T.E.; Pierce, J.R.; Pandis, S.N. Rethinking organic aerosols: Semivolatile emissions and photochemical aging. Science 2007, 315, 1259–1262. [Google Scholar] [CrossRef] [PubMed]
- Heald, C.L.; Jacob, D.J.; Park, R.J.; Russell, L.M.; Huebert, B.J.; Seinfeld, J.H.; Liao, H.; Weber, R.J. A large organic aerosol source in the free troposphere missing from current models. Geophys. Res. Lett. 2005, 32. [Google Scholar] [CrossRef]
- Kaiser, J.W.; Heil, A.; Andreae, M.O.; Benedetti, A.; Chubarova, N.; Jones, L.; Morcrette, J.J.; Razinger, M.; Schultz, M.G.; Suttie, M.; et al. Biomass burning emissions estimated with a global fire assimilation system based on observed fire radiative power. Biogeoscience 2012, 9, 527–554. [Google Scholar] [CrossRef]
- Tosca, M.G.; Randerson, J.T.; Zender, C.S. Global impact of smoke aerosols from landscape fires on climate and the Hadley circulation. Atmos. Chem. Phys. 2013, 13, 5227–5241. [Google Scholar] [CrossRef]
- Moise, T.; Flores, J.M.; Rudich, Y. Optical properties of secondary organic aerosols and their changes by chemical processes. Chem. Rev. 2015, 115, 4400–4439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, G.; Guo, S.; Zamora, M.L.; Ying, Q.; Lin, Y.; Wang, W.; Hu, M.; Wang, Y. Formation of Urban Fine Particulate Matter. Chem. Rev. 2015, 115, 3803–3855. [Google Scholar] [CrossRef] [PubMed]
- Pusede, S.E.; Steiner, A.L.; Cohen, R.C. Temperature and recent trends in the chemistry of continental surface ozone. Chem. Rev. 2015, 115, 3898–3918. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Camm, R.C.; Guzman, M.I. Catechol oxidation by ozone and hydroxyl radicals at the air–water interface. Environ. Sci. Technol. 2014, 48, 14352–14360. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Zhou, R.; Guzman, M.I. Heterogeneous oxidation of catechol. J. Phys. Chem. A 2015, 119, 10349–10359. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Guzman, M.I. Oxidation of substituted catechols at the air–water interface: Production of carboxylic acids, quinones, and polyphenols. Environ. Sci. Technol. 2017, 51, 4951–4959. [Google Scholar] [CrossRef] [PubMed]
- Rudich, Y.; Donahue, N.M.; Mentel, T.F. Aging of organic aerosol: Bridging the gap between laboratory and field studies. Ann. Rev. Phys. Chem. 2007, 58, 321–352. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.H.; Seinfeld, J.H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. [Google Scholar] [CrossRef]
- Updyke, K.M.; Nguyen, T.B.; Nizkorodov, S.A. Formation of brown carbon via reactions of ammonia with secondary organic aerosols from biogenic and anthropogenic precursors. Atmos. Environ. 2012, 63, 22–31. [Google Scholar] [CrossRef]
- Cappa, C.D.; Che, D.L.; Kessler, S.H.; Kroll, J.H.; Wilson, K.R. Variations in organic aerosol optical and hygroscopic properties upon heterogeneous OH oxidation. J. Geophys. Res. Atmos. 2011, 116. [Google Scholar] [CrossRef]
- Bond, T.C. Spectral dependence of visible light absorption by carbonaceous particles emitted from coal combustion. Geophys. Res. Lett. 2001, 28, 4075–4078. [Google Scholar] [CrossRef]
- Chung, C.E.; Ramanathan, V.; Decremer, D. Observationally constrained estimates of carbonaceous aerosol radiative forcing. Proc. Natl. Acad. Sci. USA 2012, 109, 11624–11629. [Google Scholar] [CrossRef] [PubMed]
- George, I.J.; Abbatt, J.P.D. Heterogeneous oxidation of atmospheric aerosol particles by gas-phase radicals. Nat. Chem. 2010, 2, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Kolb, C.E.; Cox, R.A.; Abbatt, J.P.D.; Ammann, M.; Davis, E.J.; Donaldson, D.J.; Garrett, B.C.; George, C.; Griffiths, P.T.; Hanson, D.R.; et al. An overview of current issues in the uptake of atmospheric trace gases by aerosols and clouds. Atmos. Chem. Phys. 2010, 10, 10561–10605. [Google Scholar] [CrossRef]
- Pöschl, U.; Rudich, Y.; Ammann, M. Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions—Part 1: General equations, parameters, and terminology. Atmos. Chem. Phys. 2007, 7, 5989–6023. [Google Scholar] [CrossRef]
- Rader, D.J.; Grasser, T.W.; Castaneda, J.N.; Trott, W.M.; Torczynski, J.R. Measurements of Thermal Accommodation Coefficients; Department of Energy: Livermore, CA, USA, 2005. [Google Scholar]
- Davidovits, P.; Worsnop, D.R.; Jayne, J.T.; Kolb, C.E.; Winkler, P.; Vrtala, A.; Wagner, P.E.; Kulmala, M.; Lehtinen, K.E.J.; Vesala, T.; et al. Mass accommodation coefficient of water vapor on liquid water. Geophys. Res. Lett. 2004, 31. [Google Scholar] [CrossRef]
- Vieceli, J.; Roeselová, M.; Potter, N.; Dang, L.X.; Garrett, B.C.; Tobias, D.J. Molecular dynamics simulations of atmospheric oxidants at the air−water interface: Solvation and accommodation of OH and O3. J. Phys. Chem. B 2005, 109, 15876–15892. [Google Scholar] [CrossRef] [PubMed]
- Crowley, J.N.; Ammann, M.; Cox, R.A.; Hynes, R.G.; Jenkin, M.E.; Mellouki, A.; Rossi, M.J.; Troe, J.; Wallington, T.J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume V—Heterogeneous reactions on solid substrates. Atmos. Chem. Phys. 2010, 10, 9059–9223. [Google Scholar] [CrossRef]
- Najera, J.J.; Percival, C.J.; Horn, A.B. Infrared spectroscopic studies of the heterogeneous reaction of ozone with dry maleic and fumaric acid aerosol particles. Phys. Chem. Chem. Phys. 2009, 11, 9093–9103. [Google Scholar] [CrossRef] [PubMed]
- Davidovits, P.; Kolb, C.E.; Williams, L.R.; Jayne, J.T.; Worsnop, D.R. Update 1 of: Mass Accommodation and Chemical Reactions at Gas−Liquid Interfaces. Chem. Rev. 2011, 111. [Google Scholar] [CrossRef]
- Ammann, M.; Pöschl, U. Kinetic model framework for aerosol and cloud surface chemistry and gas-particle interactions—Part 2: Exemplary practical applications and numerical simulations. Atmos. Chem. Phys. 2007, 7, 6025–6045. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J. Reactions at surfaces in the atmosphere: Integration of experiments and theory as necessary (but not necessarily sufficient) for predicting the physical chemistry of aerosols. Phys. Chem. Chem. Phys. 2009, 11, 7760–7779. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, K.; Tachibana, E.; Okuzawa, K.; Aggarwal, S.G.; Kanaya, Y.; Wang, Z.F. High abundances of water-soluble dicarboxylic acids, ketocarboxylic acids and α-dicarbonyls in the mountaintop aerosols over the North China Plain during wheat burning season. Atmos. Chem. Phys. 2013, 13, 8285–8302. [Google Scholar] [CrossRef]
- Mkoma, S.L.; Kawamura, K. Molecular composition of dicarboxylic acids, ketocarboxylic acids, α-dicarbonyls and fatty acids in atmospheric aerosols from Tanzania, East Africa during wet and dry seasons. Atmos. Chem. Phys. 2013, 13, 2235–2251. [Google Scholar] [CrossRef]
- Eugene, A.J.; Xia, S.-S.; Guzman, M.I. Aqueous photochemistry of glyoxylic acid. J. Phys. Chem. A 2016, 120, 3817–3826. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Guzman, M.I. Reactivity of ketyl and acetyl radicals from direct solar actinic photolysis of aqueous pyruvic acid. J. Phys. Chem. A 2017, 121, 2924–2935. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, R.; Arey, J. Atmospheric degradation of volatile organic compounds. Chem. Rev. 2003, 103, 4605–4638. [Google Scholar] [CrossRef] [PubMed]
- Pankow, J.F.; Asher, W.E. SIMPOL.1: A simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. Discuss. 2007, 7, 11839–11894. [Google Scholar] [CrossRef]
- Barsanti, K.C.; Pankow, J.F. Thermodynamics of the formation of atmospheric organic particulate matter by accretion reactions—Part 1: Aldehydes and ketones. Atmos. Environ. 2004, 38, 4371–4382. [Google Scholar] [CrossRef]
- Li, Y.J.; Cheong, G.Y.L.; Lau, A.P.S.; Chan, C.K. Acid-catalyzed condensed-phase reactions of limonene and terpineol and their impacts on gas-to-particle partitioning in the formation of organic aerosols. Environ. Sci. Technol. 2010, 44, 5483–5489. [Google Scholar] [CrossRef] [PubMed]
- Liggio, J.; Li, S.-M.; Brook, J.R.; Mihele, C. Direct polymerization of isoprene and a-pinene on acidic aerosols. Geophys. Res. Lett. 2007, 34. [Google Scholar] [CrossRef]
- Boyd, C.M.; Sanchez, J.; Xu, L.; Eugene, A.J.; Nah, T.; Tuet, W.Y.; Guzman, M.I.; Ng, N.L. Secondary organic aerosol formation from the β-pinene+NO3 system: Effect of humidity and peroxy radical fate. Atmos. Chem. Phys. 2015, 15, 7497–7522. [Google Scholar] [CrossRef]
- Williams, M.B.; Michelsen, R.R.H.; Axson, J.L.; Iraci, L.T. Uptake of acetone, acetaldehyde and ethanol in cold sulfuric acid solutions containing organic material: Carbon accretion mechanisms. Atmos. Environ. 2010, 44, 1145–1151. [Google Scholar] [CrossRef]
- Rincón, A.G.; Guzmán, M.I.; Hoffmann, M.R.; Colussi, A.J. Optical absorptivity versus molecular composition of model organic aerosol matter. J. Phys. Chem. A 2009, 113, 10512–10520. [Google Scholar] [CrossRef] [PubMed]
- Rincón, A.G.; Guzmán, M.I.; Hoffmann, M.R.; Colussi, A.J. Thermochromism of model organic aerosol matter. J. Phys. Chem. Lett. 2010, 1, 368–373. [Google Scholar] [CrossRef]
- Guzmán, M.I.; Colussi, A.J.; Hoffmann, M.R. Photoinduced oligomerization of aqueous pyruvic acid. J. Phys. Chem. A 2006, 110, 3619–3626. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Guzman, M.I. Reply to “Comment on ‘Reactivity of ketyl and acetyl radicals from direct solar actinic photolysis of aqueous pyruvic acid’”. J. Phys. Chem. A 2017, 121, 8741–8744. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Pillar, E.A.; Colussi, A.J.; Guzman, M.I. Enhanced acidity of acetic and pyruvic acids on the surface of water. Langmuir 2018, 34, 9307–9713. [Google Scholar] [CrossRef] [PubMed]
- Eugene, A.J.; Xia, S.-S.; Guzman, M.I. Negative production of acetoin in the photochemistry of aqueous pyruvic acid. Proc. Natl. Acad. Sci. USA 2013, 110, E4274–E4275. [Google Scholar] [CrossRef] [PubMed]
- Xia, S.-S.; Eugene, A.J.; Guzman, M.I. Cross photoreaction of glyoxylic and pyruvic acids in model aqueous aerosol. J. Phys. Chem. A 2018, 122, 6457–6466. [Google Scholar] [CrossRef] [PubMed]
- Canonica, S.; Kohn, T.; Mac, M.; Real, F.J.; Wirz, J.; von Gunten, U. Photosensitizer method to determine rate constants for the reaction of carbonate radical with organic compounds. Environ. Sci. Technol. 2005, 39, 9182–9188. [Google Scholar] [CrossRef] [PubMed]
- Vione, D.; Maurino, V.; Minero, C.; Pelizzetti, E.; Harrison, M.A.J.; Olariu, R.-I.; Arsene, C. Photochemical reactions in the tropospheric aqueous phase and on particulate matter. Chem. Soc. Rev. 2006, 35, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Colussi, A.; Hoffmann, M.R. Photogeneration of distant radical pairs in aqueous pyruvic acid glasses. J. Phys. Chem. A 2006, 110, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Hoffmann, M.R.; Colussi, A.J. Photolysis of pyruvic acid in ice: Possible relevance to CO and CO2 ice core record anomalies. J. Geophys. Res. Atmos. 2007, 112. [Google Scholar] [CrossRef]
- Gligorovski, S.; Strekowski, R.; Barbati, S.; Vione, D. Environmental implications of hydroxyl radicals (•OH). Chem. Rev. 2015, 115, 13051–13092. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.J.; MacDonald, S.M.; Shaw, M.D.; Kumar, R.; Saunders, R.W.; Parthipan, R.; Wilson, J.; Plane, J.M.C. Atmospheric iodine levels influenced by sea surface emissions of inorganic iodine. Nat. Geosci. 2013, 6, 108–111. [Google Scholar] [CrossRef]
- Kroll, J.H.; Smith, J.D.; Che, D.L.; Kessler, S.H.; Worsnop, D.R.; Wilson, K.R. Measurement of fragmentation and functionalization pathways in the heterogeneous oxidation of oxidized organic aerosol. Phys. Chem. Chem. Phys. 2009, 11, 8005–8014. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; He, K.; Duan, F.; Cheng, Y.; Ma, Y. Measurement of humic-like substances in aerosols: A review. Environ. Pollut. 2013, 181, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Sharpless, C.M.; Aeschbacher, M.; Page, S.E.; Wenk, J.; Sander, M.; McNeill, K. Photooxidation-induced changes in optical, electrochemical, and photochemical properties of humic substances. Environ. Sci. Technol. 2014, 48, 2688–2696. [Google Scholar] [CrossRef] [PubMed]
- Pillar, E.A.; Guzman, M.I.; Rodriguez, J.M. Conversion of iodide to hypoiodous acid and iodine in aqueous microdroplets exposed to ozone. Environ. Sci. Technol. 2013, 47, 10971–10979. [Google Scholar] [CrossRef] [PubMed]
- Wagner, P.E. A constant-angle mie scattering method (CAMS) for investigation of particle formation processes. J. Colloid Interface Sci. 1985, 105, 456–467. [Google Scholar] [CrossRef]
- Kupc, A.; Winkler, P.M.; Vrtala, A.; Wagner, P.E. Unusual temperature dependence of heterogeneous nucleation of water vapor on ag particles. Aerosol. Sci. Technol. 2013, 47, I–IV. [Google Scholar] [CrossRef]
- Vesala, T.; Kulmala, M.; Rudolf, R.; Vrtala, A.; Wagner, P.E. Models for condensational growth and evaporation of binary aerosol particles. J. Aerosol. Sci. 1997, 28, 565–598. [Google Scholar] [CrossRef]
- Quinlan, M.A.; Reihs, C.M.; Golden, D.M.; Tolbert, M.A. Heterogeneous reactions on model polar stratospheric cloud surfaces: Reaction of dinitrogen pentoxide on ice and nitric acid trihydrate. J. Phys. Chem. 1990, 94, 3255–3260. [Google Scholar] [CrossRef]
- Caloz, F.; Fenter, F.F.; Tabor, K.D.; Rossi, M.J. Paper I: Design and construction of a Knudsen-cell reactor for the study of heterogeneous reactions over the temperature range 130–750 K: Performances and limitations. Rev. Sci. Instrum. 1997, 68, 3172–3179. [Google Scholar] [CrossRef]
- Worsnop, D.R.; Zahniser, M.S.; Kolb, C.E.; Gardner, J.A.; Watson, L.R.; Van Doren, J.M.; Jayne, J.T.; Davidovits, P. The temperature dependence of mass accommodation of sulfur dioxide and hydrogen peroxide on aqueous surfaces. J. Phys. Chem. 1989, 93, 1159–1172. [Google Scholar] [CrossRef]
- Li, Y.Q.; Davidovits, P.; Kolb, C.E.; Worsnop, D.R. Mass and thermal accommodation coefficients of H2O (g) on liquid water as a function of temperature. J. Phys. Chem. A 2001, 105, 10627–10634. [Google Scholar] [CrossRef]
- Worsnop, D.R.; Shi, Q.; Jayne, J.T.; Kolb, C.E.; Swartz, E.; Davidovits, P. Gas-phase diffusion in droplet train measurements of uptake coefficients. J. Aerosol. Sci. 2001, 32, 877–891. [Google Scholar] [CrossRef]
- Ryder, O.S.; Campbell, N.R.; Morris, H.; Forestieri, S.; Ruppel, M.J.; Cappa, C.D.; Tivanski, A.; Prather, K.; Bertram, T.H. Role of organic coatings in regulating N2O5 reactive uptake to sea spray aerosol. J. Phys. Chem. A 2015, 119, 11683–11692. [Google Scholar] [CrossRef] [PubMed]
- Thornberry, T.; Abbatt, J.P.D. Heterogeneous reaction of ozone with liquid unsaturated fatty acids: Detailed kinetics and gas-phase product studies. Phys. Chem. Chem. Phys. 2004, 6, 84–93. [Google Scholar] [CrossRef]
- McCabe, J.; Abbatt, J.P.D. Heterogeneous loss of gas-phase ozone on n-hexane soot surfaces: Similar kinetics to loss on other chemically unsaturated solid surfaces. J. Phys. Chem. C 2009, 113, 2120–2127. [Google Scholar] [CrossRef]
- Arangio, A.M.; Slade, J.H.; Berkemeier, T.; Pöschl, U.; Knopf, D.A.; Shiraiwa, M. Multiphase chemical kinetics of OH radical uptake by molecular organic markers of biomass burning aerosols: Humidity and temperature dependence, surface reaction, and bulk diffusion. J. Phys. Chem. A 2015, 119, 4533–4544. [Google Scholar] [CrossRef] [PubMed]
- Garrett, B.C.; Schenter, G.K.; Morita, A. Molecular simulations of the transport of molecules across the liquid/vapor interface of water. Chem. Rev. 2006, 106, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Guascito, M.R.; Cesari, D.; Chirizzi, D.; Genga, A.; Contini, D. XPS surface chemical characterization of atmospheric particles of different sizes. Atmos. Environ. 2015, 116, 146–154. [Google Scholar] [CrossRef]
- Kirchner, U.; Vogt, R.; Natzeck, C.; Goschnick, J. Single particle MS, SNMS, SIMS, XPS, and FTIR spectroscopic analysis of soot particles during the AIDA campaign. J. Aerosol. Sci. 2003, 34, 1323–1346. [Google Scholar] [CrossRef]
- Qi, J.; Feng, L.; Li, X.; Zhang, M. An X-ray photoelectron spectroscopy study of elements on the surface of aerosol particles. J. Aerosol. Sci. 2006, 37, 218–227. [Google Scholar] [CrossRef]
- Faude, F.; Goschnick, J. XPS, SIMS and SNMS applied to a combined analysis of aerosol particles from a region of considerable air pollution in the upper Rhine valley. Fresen. J. Anal. Chem. 1997, 358, 67–72. [Google Scholar] [CrossRef]
- Washenfelder, R.A.; Attwood, A.R.; Brock, C.A.; Guo, H.; Xu, L.; Weber, R.J.; Ng, N.L.; Allen, H.M.; Ayres, B.R.; Baumann, K.; et al. Biomass burning dominates brown carbon absorption in the rural southeastern United States. Geophys. Res. Lett. 2015, 42, 653–664. [Google Scholar] [CrossRef]
- DeCarlo, P.F.; Kimmel, J.R.; Trimborn, A.M.; Northway, M.J.; Jayne, J.T.; Aiken, A.C.; Gonin, M.; Fuhrer, K.; Horvath, T.; Docherty, K.S.; et al. Field-deployable, high-resolution, time-of-flight aerosol mass spectrometer. Anal. Chem. 2006, 78, 8281–8289. [Google Scholar] [CrossRef] [PubMed]
- Canagaratna, M.R.; Jayne, J.T.; Jimenez, J.L.; Allan, J.D.; Alfarra, M.R.; Zhang, Q.; Onasch, T.B.; Drewnick, F.; Coe, H.; Middlebrook, A.; et al. Chemical and microphysical characterization of ambient aerosols with the aerodyne aerosol mass spectrometer. Mass Spectrom. Rev. 2007, 26, 185–222. [Google Scholar] [CrossRef] [PubMed]
- Pósfai, M.; Axisa, D.; Tompa, É.; Freney, E.; Bruintjes, R.; Buseck, P.R. Interactions of mineral dust with pollution and clouds: An individual-particle TEM study of atmospheric aerosol from Saudi Arabia. Atmos. Res. 2013, 122, 347–361. [Google Scholar] [CrossRef]
- O’Brien, R.E.; Wang, B.; Kelly, S.T.; Lundt, N.; You, Y.; Bertram, A.K.; Leone, S.R.; Laskin, A.; Gilles, M.K. Liquid–liquid phase separation in aerosol particles: Imaging at the nanometer scale. Environ. Sci. Technol. 2015, 49, 4995–5002. [Google Scholar] [CrossRef] [PubMed]
- Satsangi, P.G.; Yadav, S. Characterization of PM2.5 by X-ray diffraction and scanning electron microscopy—Energy dispersive spectrometer: Its relation with different pollution sources. Int. J. Environ. Sci. Technol. 2014, 11, 217–232. [Google Scholar] [CrossRef]
- Sakata, K.; Sakaguchi, A.; Tanimizu, M.; Takaku, Y.; Yokoyama, Y.; Takahashi, Y. Identification of sources of lead in the atmosphere by chemical speciation using X-ray absorption near-edge structure (XANES) spectroscopy. J. Environ. Sci. 2014, 26, 343–352. [Google Scholar] [CrossRef]
- Isaacman-VanWertz, G.; Massoli, P.; O’Brien, R.; Lim, C.; Franklin, J.P.; Moss, J.A.; Hunter, J.F.; Nowak, J.B.; Canagaratna, M.R.; Misztal, P.K.; et al. Chemical evolution of atmospheric organic carbon over multiple generations of oxidation. Nat. Chem. 2018, 10, 462–468. [Google Scholar] [CrossRef] [PubMed]
- Katrib, Y.; Biskos, G.; Buseck, P.R.; Davidovits, P.; Jayne, J.T.; Mochida, M.; Wise, M.E.; Worsnop, D.R.; Martin, S.T. Ozonolysis of mixed oleic-acid/stearic-acid particles: Reaction kinetics and chemical morphology. J. Phys. Chem. A 2005, 109, 10910–10919. [Google Scholar] [CrossRef] [PubMed]
- Guzman, M.I.; Athalye, R.R.; Rodriguez, J.M. Concentration effects and ion properties controlling the fractionation of halides during aerosol formation. J. Phys. Chem. A 2012, 116, 5428–5435. [Google Scholar] [CrossRef] [PubMed]
- Iribarne, J.V.; Thomson, B.A. On the evaporation of small ions from charged droplets. J. Chem. Phys. 1976, 64, 2287–2294. [Google Scholar] [CrossRef]
- Barnum, T.J.; Medeiros, N.; Hinrichs, R.Z. Condensed-phase versus gas-phase ozonolysis of catechol: A combined experimental and theoretical study. Atmos. Environ. 2012, 55, 98–106. [Google Scholar] [CrossRef]
- von Sonntag, C.; von Gunten, U. Chemistry of Ozone in Water and Wastewater Treatment; IWA Publishing: London, UK, 2012. [Google Scholar]
- Bertram, T.H.; Thornton, J.A.; Riedel, T.P. An experimental technique for the direct measurement of N2O5 reactivity on ambient particles. Atmos. Meas. Technol. 2009, 2, 231–242. [Google Scholar] [CrossRef]
- Morris, J.W.; Davidovits, P.; Jayne, J.T.; Jimenez, J.L.; Shi, Q.; Kolb, C.E.; Worsnop, D.R.; Barney, W.S.; Cass, G. Kinetics of submicron oleic acid aerosols with ozone: A novel aerosol mass spectrometric technique. Geophys. Res. Lett. 2002, 29, 71–74. [Google Scholar] [CrossRef]
- Sage, A.M.; Weitkamp, E.A.; Robinson, A.L.; Donahue, N.M. Reactivity of oleic acid in organic particles: Changes in oxidant uptake and reaction stoichiometry with particle oxidation. Phys. Chem. Chem. Phys. 2009, 11, 7951–7962. [Google Scholar] [CrossRef] [PubMed]
- Ziemann, P.J. Aerosol products, mechanisms, and kinetics of heterogeneous reactions of ozone with oleic acid in pure and mixed particles. Faraday Discuss. 2005, 130, 469–490. [Google Scholar] [CrossRef] [PubMed]
- Docherty, K.S.; Ziemann, P.J. Reaction of oleic acid particles with NO3 radicals: Products, mechanism, and implications for radical-initiated organic aerosol oxidation. J. Phys. Chem. A 2006, 110, 3567–3577. [Google Scholar] [CrossRef] [PubMed]
- Last, D.J.; Najera, J.J.; Percival, C.J.; Horn, A.B. A comparison of infrared spectroscopic methods for the study of heterogeneous reactions occurring on atmospheric aerosol proxies. Phys. Chem. Chem. Phys. 2009, 11, 8214–8225. [Google Scholar] [CrossRef] [PubMed]
- Leng, C.; Hiltner, J.; Pham, H.; Kelley, J.; Mach, M.; Zhang, Y.; Liu, Y. Kinetics study of heterogeneous reactions of ozone with erucic acid using an ATR-IR flow reactor. Phys. Chem. Chem. Phys. 2014, 16, 4350–4360. [Google Scholar] [CrossRef] [PubMed]
- Sillman, S. The relation between ozone, NOx and hydrocarbons in urban and polluted rural environments. Atmos. Environ. 1999, 33, 1821–1845. [Google Scholar] [CrossRef]
- Atkinson, R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 2000, 34, 2063–2101. [Google Scholar] [CrossRef]
- Pankow, J.F. An absorption model of the gas/aerosol partitioning involved in the formation of secondary organic aerosol. Atmos. Environ. 1994, 28, 189–193. [Google Scholar] [CrossRef]
- Chan, A.W.H.; Kroll, J.H.; Ng, N.L.; Seinfeld, J.H. Kinetic modeling of secondary organic aerosol formation: Effects of particle- and gas-phase reactions of semivolatile products. Atmos. Chem. Phys. 2007, 7, 4135–4147. [Google Scholar] [CrossRef]
- Andreae, M.O.; Gelencsér, A. Black carbon or brown carbon? The nature of light-absorbing carbonaceous aerosols. Atmos. Chem. Phys. 2006, 6, 3131–3148. [Google Scholar] [CrossRef]
- Enami, S.; Hoffmann, M.R.; Colussi, A.J. How phenol and α-tocopherol react with ambient ozone at gas/liquid interfaces. J. Phys. Chem. A 2009, 113, 7002–7010. [Google Scholar] [CrossRef] [PubMed]
- Enami, S.; Vecitis, C.D.; Cheng, J.; Hoffmann, M.R.; Colussi, A.J. Global inorganic source of atmospheric bromine. J. Phys. Chem. A 2007, 111, 8749–8752. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Zhang, J.; Cai, S.; Liao, Y.; Zhao, W.; Hu, C.; Gu, X.; Fang, L.; Zhang, W. Mass spectrometric study of aged benzene secondary organic aerosol in the presence of dry ammonium sulfate. J. Atmos. Chem. 2016, 73, 329–344. [Google Scholar] [CrossRef]
- Finlayson-Pitts, B.J. Introductory lecture: Atmospheric chemistry in the Anthropocene. Faraday Discuss. 2017, 200, 11–58. [Google Scholar] [CrossRef] [PubMed]
- Lavi, A.; Lin, P.; Bhaduri, B.; Carmieli, R.; Laskin, A.; Rudich, Y. Characterization of light-absorbing oligomers from reactions of phenolic compounds and Fe(III). ACS Earth Space Chem. 2017, 1, 637–646. [Google Scholar] [CrossRef]
- Sun, J.; Mei, Q.; Wei, B.; Huan, L.; Xie, J.; He, M. Mechanisms for ozone-initiated removal of biomass burning products from the atmosphere. Environ. Chem. 2018, 15, 83–91. [Google Scholar] [CrossRef]
- Magalhães, A.C.O.; Esteves da Silva, J.C.G.; Pinto da Silva, L. Density functional theory calculation of the absorption properties of brown carbon chromophores generated by catechol heterogeneous ozonolysis. ACS Earth Space Chem. 2017, 1, 353–360. [Google Scholar] [CrossRef]
- Sarwar, G.; Gantt, B.; Schwede, D.; Foley, K.; Mathur, R.; Saiz-Lopez, A. Impact of enhanced ozone deposition and halogen chemistry on tropospheric ozone over the northern hemisphere. Environ. Sci. Technol. 2015, 49, 9203–9211. [Google Scholar] [CrossRef] [PubMed]
- Holla, R.; Schmitt, S.; Frieß, U.; Pöhler, D.; Zingler, J.; Corsmeier, U.; Platt, U. Vertical distribution of BrO in the boundary layer at the Dead Sea. Environ. Chem. 2015, 12, 438–460. [Google Scholar] [CrossRef]
- Liu, L.; Zhang, X.; Li, Z.; Zhang, Y.; Ge, M. Gas-phase hydration of glyoxylic acid: Kinetics and atmospheric implications. Chemosphere 2017, 186, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Schnitzler, E.G.; Abbatt, J.P.D. Heterogeneous OH oxidation of secondary brown carbon aerosol. Atmos. Chem. Phys. Discuss. 2018, 2018, 1–29. [Google Scholar] [CrossRef]
- Saunders, R.W.; Kumar, R.; MacDonald, S.M.; Plane, J.M.C. Insights into the photochemical transformation of iodine in aqueous systems: Humic acid photosensitized reduction of iodate. Environ. Sci. Technol. 2012, 46, 11854–11861. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.D.; Estillore, A.D.; Morris, H.S.; Ray, K.K.; Alejandro, A.; Grassian, V.H.; Tivanski, A.V. Direct surface tension measurements of individual sub-micrometer particles using atomic force microscopy. J. Phys. Chem. A 2017, 121, 8296–8305. [Google Scholar] [CrossRef] [PubMed]
- Nissenson, P.; Wingen, L.M.; Hunt, S.W.; Finlayson-Pitts, B.J.; Dabdub, D. Rapid formation of molecular bromine from deliquesced NaBr aerosol in the presence of ozone and UV light. Atmos. Environ. 2014, 89, 491–506. [Google Scholar] [CrossRef]
- Desyaterik, Y.; Sun, Y.; Shen, X.; Lee, T.; Wang, X.; Wang, T.; Collett, J.L. Speciation of “brown” carbon in cloud water impacted by agricultural biomass burning in eastern China. J. Geophys. Res. Atmos. 2013, 118, 7389–7399. [Google Scholar] [CrossRef]
- Kanakidou, M.; Seinfeld, J.H.; Pandis, S.N.; Barnes, I.; Dentener, F.J.; Facchini, M.C.; Van Dingenen, R.; Ervens, B.; Nenes, A.; Nielsen, C.J.; et al. Organic aerosol and global climate modelling: A review. Atmos. Chem. Phys. 2005, 5, 1053–1123. [Google Scholar] [CrossRef]
- Hoque, M.; Kawamura, K.; Seki, O.; Hoshi, N. Spatial distributions of dicarboxylic acids, ω-oxoacids, pyruvic acid and α-dicarbonyls in the remote marine aerosols over the North Pacific. Mar. Chem. 2015, 172, 1–11. [Google Scholar] [CrossRef]
- Veres, P.; Roberts, J.M.; Burling, I.R.; Warneke, C.; de Gouw, J.; Yokelson, R.J. Measurements of gas-phase inorganic and organic acids from biomass fires by negative-ion proton-transfer chemical-ionization mass spectrometry. J. Geophys. Res. Atmos. 2010, 115. [Google Scholar] [CrossRef]
- Henze, D.K.; Seinfeld, J.H.; Ng, N.L.; Kroll, J.H.; Fu, T.M.; Jacob, D.J.; Heald, C.L. Global modeling of secondary organic aerosol formation from aromatic hydrocarbons: High- vs. low-yield pathways. Atmos. Chem. Phys. 2008, 8, 2405–2420. [Google Scholar] [CrossRef]
- Lin, P.; Engling, G.; Yu, J.Z. Humic-like substances in fresh emissions of rice straw burning and in ambient aerosols in the Pearl River Delta Region, China. Atmos. Chem. Phys. 2010, 10, 6487–6500. [Google Scholar] [CrossRef]
- Hoffer, A.; Gelencsér, A.; Guyon, P.; Kiss, G.; Schmid, O.; Frank, G.P.; Artaxo, P.; Andreae, M.O. Optical properties of humic-like substances (HULIS) in biomass-burning aerosols. Atmos. Chem. Phys. 2006, 6, 3563–3570. [Google Scholar] [CrossRef]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).