

Remediating Per- and Polyfluoroalkyl Substances (PFAS)-Contaminated Water by Foam Fractionation and Electrochemical Oxidation

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Foam Fractionation Experiments—Batch Conditions
2.3. Foam Fractionation Experiments—Tank Experiments
2.4. Water Sampling PFAS-Tainted Water
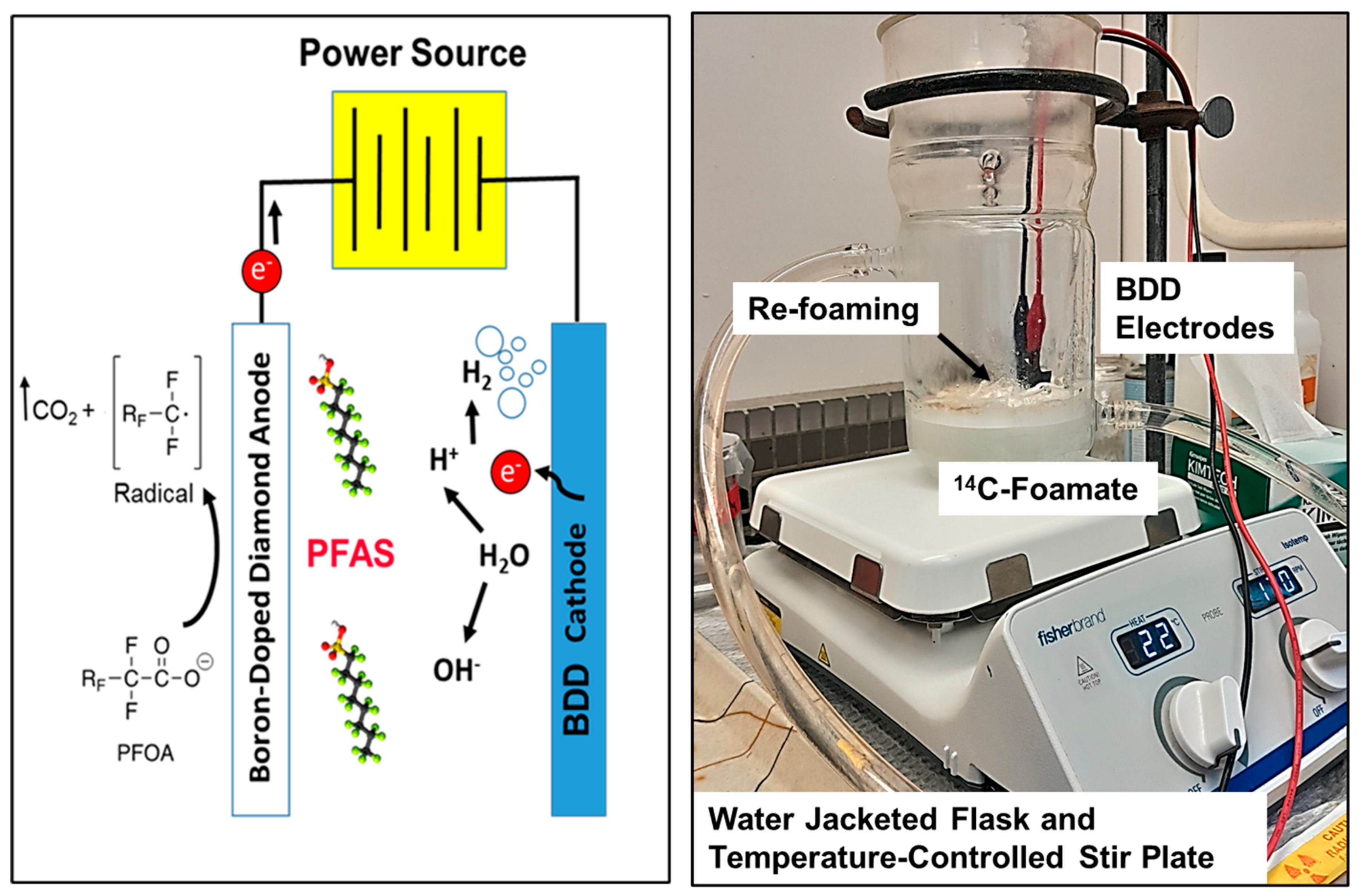
2.5. Electrochemical Oxidation
2.6. Statistical Analysis
3. Results and Discussion
3.1. Effects of Cosolvent Mass on PFAS Recovery and Enrichment

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Surfactant Mass | Foamate Volume | 14C Enrichment in Foam † | 14C Recovered in Foamate |
|---|---|---|---|
| g | mL | % | |
| 0.02 | 32 | 3.51 a ‡ | 56.99 a |
| 0.04 | 64 | 2.33 b | 75.75 b |
| 0.06 | 104 | 1.75 c | 92.35 c |
| 0.08 | 122 | 1.32 d | 81.80 d |
3.2. Effect of Flow Rate
| Flow Rate | Foamate Volume | 14C Enrichment in Foam † | 14C Recovered in Foamate |
|---|---|---|---|
| L/min | mL | % | |
| 4 | 94 | 1.78 a ‡ | 85.02 a |
| 6 | 118 | 1.49 b | 88.89 ab |
| 8 | 126 | 1.43 c | 91.13 bc |
3.3. Effect of pH
| pH | Foamate Volume | 14C Enrichment in Foam † | 14C Recovered in Foamate |
|---|---|---|---|
| mL | % | ||
| 3.6 | 124 | 1.40 a ‡ | 88.32 a |
| 6.4 | 112 | 1.64 b | 93.19 ab |
| 9.4 | 110 | 1.55 c | 86.42 bc |
3.4. Effect of Temperature
| Temperature | Foamate Volume | 14C Enrichment in Foam † | 14C Recovered in Foamate |
|---|---|---|---|
| °C | mL | % | |
| 16 | 124 | 1.38 c ‡ | 86.99 a |
| 20 | 122 | 1.52 a | 93.75 c |
| 23 | 140 | 1.30 d | 92.48 c |
| 26 | 122 | 1.45 b | 89.49 b |
3.5. Tank Experiments
| Water Source | Foamate Volume (mL) | 14C-Enrichment Factor † | 14C Recovered in Foamate (%) | Mass Balance | Time (min) |
|---|---|---|---|---|---|
| DI Water | 659 | 4.00 a ‡ | 75.50 b ‡ | 94 | 30 |
| Site B WWTP | 1206 | 2.60 b | 89.47 a | 91 | 30 |
| Site B WWTP | 1306 | 2.31 b | 86.19 a | 91 | 60 |
| Site A Drainage | 663 | 2.67 b | 50.78 c | 111 | 30 |
3.6. Electrochemical Oxidation of Foamate
4. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Suthersan, S.S.; Horst, J.; Ross, I.; Kalve, E.; Quinnan, J.; Houtz, E.; Burdick, J. Responding to emerging contaminant impacts: Situational management. Groundw. Monit. Remediat. 2016, 36, 22–32. [Google Scholar] [CrossRef]
- Key, B.L.; Howell, R.D.; Criddle, C.S. Fluorinated organics in the biosphere. Environ. Sci. Technol. 1997, 31, 2445–2454. [Google Scholar] [CrossRef]
- O’Hagan, D. Understanding organofluorine chemistry. An introduction to the C-F bond. Chem. Soc. Rev. 2008, 37, 308–318. [Google Scholar] [CrossRef] [PubMed]
- Conder, J.M.; Hoke, R.A.; Wolf, W.D.; Russell, M.H.; Buck, R.C. Are PFCAs Bioaccumulative? A critical review and comparison with regulatory criteria and persistent lipophilic compounds. Environ. Sci. Technol. 2008, 42, 995–1003. [Google Scholar] [CrossRef]
- Lau, C.; Anitole, K.; Hodes, C.; Lai, D.; Pfahles-Hutchens, A.; Seed, J. Perfluoroalkyl acids: A review of monitoring and toxicological findings. Toxicol. Sci. 2007, 99, 366–394. [Google Scholar] [CrossRef]
- Johansson, N.; Eriksson, P.; Viberg, H. Neonatal exposure to PFOS and PFOA in mice results in changes in proteins which are important for neuronal growth and synaptogenesis in the developing brain. Toxicol. Sci. 2009, 108, 412–418. [Google Scholar] [CrossRef]
- Liu, W.; Xu, L.; Li, X.; Jin, Y.H.; Sasaki, K.; Saito, N.; Sato, I.; Tsuda, S. Human nails analysis as biomarker of exposure to perfluoroalkyl compounds. Environ. Sci. Technol. 2011, 45, 8144–8150. [Google Scholar] [CrossRef]
- Mills, M.S.; Thurman, E.M.; Ertel, J.; Thorn, K.A. Chapter 11: Organic Geochemistry and sources of natural aquatic foams. In Humic and Fulvic Acids; Gaffney, J.S., Marley, N.A., Clark, S.B., Eds.; PFAS and Microplastic Enrichment in Surface Water Foams; American Chemical Society: Washington, DC, USA, 1996; pp. 151–192. [Google Scholar]
- Schwichtenberg, T.; Bogdan, D.; Carignan, C.C.; Reardon, P.; Rewerts, J.; Wanzek, T.; Field, J. PFAS and Dissolved Organic Carbon Enrichment in Surface Water Foams on a Northern U.S. Freshwater Lake. Environ. Sci. Technol. 2020, 54, 14455–14464. [Google Scholar] [CrossRef]
- Lee, Y.C.; Wang, P.Y.; Lo, S.L.; Huang, C.P. Recovery of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) from dilute water solution by foam flotation. Sep. Purif. Technol. 2017, 173, 280–285. [Google Scholar] [CrossRef]
- We, A.C.E.; Zamyadi, A.; Stickland, A.D.; Clarke, B.O.; Freguia, S. A review of foam fractionation for the removal of per- and polyfluoroalkyl substances (PFAS) from aqueous matrices. J. Hazard. Mater. 2024, 465, 133182. [Google Scholar] [CrossRef]
- Burns, D.J.; Stevenson, P.; Murphy, P.J.C. PFAS removal from groundwaters using Surface—Active Foam Fractionation. Remediat. J. 2021, 31, 19–33. [Google Scholar] [CrossRef]
- Buckley, T.; Karanam, K.; Xu, X.; Shukla, P.; Firouzi, M.; Rudolph, V. Effect of mono- and di-valent cations on PFAS removal from water using foam fractionation—A modelling and experimental study. Sep. Purif. Technol. 2022, 286, 120508. [Google Scholar] [CrossRef]
- Burghoff, B. Foam fractionation applications. J Biotechnol. 2012, 161, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, P.; Li, X. Foam Fractionation: Principles and Process Design, 1st ed.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- Carter, K.E.; Farrel, J. Oxidative destruction of perfluorooctane sulfate using boron-doped diamond film electrodes. Environ. Sci. Technol. 2009, 42, 6111–6115. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Farrell, J. Electrochemical oxidation of perfluorobutane sulfonate using boron-doped diamond film electrodes. J. Appl. Electrochem. 2009, 39, 1993–1999. [Google Scholar] [CrossRef]
- Matzek, L.W.; Tipton, M.J.; Farmer, A.T.; Steen, A.D.; Carter, K.E. Understanding electrochemically activated persulfate and its application to Ciprofloxacin abatement. Environ. Sci. Technol. 2018, 52, 5875–5883. [Google Scholar] [CrossRef]
- Schaefer, C.E.; Andaya, C.; Urtiaga, A.; McKenzie, E.R.; Higgins, C.P. Electrochemical treatment of perfluorooctanoic acid (PFOA) and perfluorooctane sulfonic acid (PFOS) in groundwater impacted by aqueous film forming foams (AFFFs). J. Hazard. Mater. 2015, 295, 170–175. [Google Scholar] [CrossRef]
- Schaefer, C.E.; Andaya, C.; Burant, A.; Condee, C.W.; Urtiagad, A.; Strathmann, T.J.; Higgins, C.P. Electrochemical treatment of perfluorooctanoic acid and perfluorooctane sulfonate: Insights into mechanisms and application to groundwater treatment. Chem. Eng. J. 2017, 317, 424–432. [Google Scholar] [CrossRef]
- Yanagida, A.; Webb, E.; Harris, C.E.; Christenson, M.; Comfort, S. Using Electrochemical Oxidation to Remove PFAS in Simulated Investigation-Derived Waste (IDW): Laboratory and Pilot-Scale Experiments. Water 2022, 14, 2708. [Google Scholar] [CrossRef]
- Kapałka, A.; Fóti, G.; Comninellis, C. Kinetic modelling of electrochemical mineralization of organic pollutants for wastewater treatment. J. Appl. Electrochem. 2008, 38, 7–16. [Google Scholar] [CrossRef]
- Steel, R.G.D.; Torrie, J.H. Principles and procedures of statistics. In A Biometrical Approach, 2nd ed.; McGraw-Hill Book Co.: New York, NY, USA, 1980; pp. 172–194. [Google Scholar]
- Hu, K.; Zhang, H.; Ouyang, M.; Kong, M.; Jiang, Q.; Wang, G.; Zhuang, L. Experimental and DFT studies on foam performances of lauryl ether sulfate-based anionic surface active ionic liquids. J. Mol. Liq. 2021, 342, 117519. [Google Scholar] [CrossRef]
- Meng, P.; Deng, S.; Maimaiti, A.; Wang, B.; Huang, J.; Wang, Y.; Cousins, I.T.; Yu, G. Efficient removal of perfluorooctane sulfonate from aqueous film-forming foam solution by aeration-foam collection. Chemosphere 2018, 203, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.-F.; Chien, W.-Y.; Liu, Y.-J.; Lee, Y.-C.; Lo, S.-L.; Hu, C.-Y. Perfluorooctanoic acid (PFOA) removal by flotation with cationic surfactants. Chemosphere 2021, 266, 128949. [Google Scholar] [CrossRef] [PubMed]
- Buckley, T.; Karanam, K.; Han, H.; Vo, H.N.P.; Shukla, P.; Firouzi, M.; Rudolph, V. Effect of different co-foaming agents on PFAS removal from the environment by foam fractionation. Water Res. 2023, 230, 119532. [Google Scholar] [CrossRef]
- Weisman, J. Chapter 15: Two-phase flow patterns. In Handbook of Fluids in Motion; Cheremisinoff, N.P., Gupta, R., Eds.; Ann Arbor Science Publ.: Ann Arbor, MI, USA, 1983; pp. 409–425. [Google Scholar]
- Boonyasuwat, S.; Chavadej, S.; Malakul, P.; Scamehorn, J.F. Anionic and cationic surfactant recovery from water using a multistage foam fractionator. Chem. Eng. J. 2003, 93, 241–252. [Google Scholar] [CrossRef]
- Dai, X.; Xie, Z.; Dorian, B.; Gray, S.; Zhang, J. Comparative study of PFAS treatment by UV, UV/ozone, and fractionations with air and ozonated air. Environ. Sci. Water Res. Technol. 2019, 5, 1897–1907. [Google Scholar] [CrossRef]
- Du, L.; Prokop, A.; Tanner, R.D. Effect of pH on the Startup of a Continuous Foam Fractionation Process Containing Ovalbumin. Sep. Sci. Technol. 2003, 38, 1093–1109. [Google Scholar] [CrossRef]
- Morrison, A.L.; Strezov, V.; Niven, R.K.; Taylor, M.P.; Wilson, S.P.; Wang, J.; Burns, D.J.; Murphy, P.J.C. Impact of salinity and temperature on removal of PFAS species from water by aeration in the absence of additional surfactants: A novel application of green chemistry using adsorptive bubble fractionation. Ind. Eng. Chem. Res. 2023, 62, 5635–5645. [Google Scholar] [CrossRef]
- Smith, S.J.; Lauria, M.; Ahrens, L.; McCleaf, P.; Hollman, P.; Bj¨alkefur Seroka, S.; Hamers, T.; Arp, H.P.H.; Wiberg, K. Electrochemical oxidation for treatment of pfas in contaminated water and fractionated foam—A pilot-scale study. ACS EST Water 2023, 3, 1201–1211. [Google Scholar] [CrossRef]
- Tasca, A.L.; Uwayezu, J.N.; Carabante, I.; Kumpiene, J. Electrochemical remediation of PFAS by Boron-Doped Diamond electrodes: A review. J. Environ. Chem. Eng. 2025, 13, 117044. [Google Scholar] [CrossRef]
- Radjenovic, J.; Duinslaeger, N.; Avvl, S.S.; Chaplin, B.P. Facing the challenge of poly- and perfluoroalkyl substances in water: Is electrochemical oxidation the answer? Environ. Sci. Technol. 2020, 54, 14815–14829. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Schaefer, C.E.; Bamer, J.T.; Lanza, H.A.; Wintle, D.; Maynard, K.G.; Murphy, P.; Anderson, R.H. Bench-scale testing of a novel soil PFAS treatment train for informed remedial planning and decision-making. Remediat. J. 2023, 33, 309–321. [Google Scholar] [CrossRef]
- Uwayezu, J.N.; Ren, Z.; Sonnenschein, S.; Leiviskä, T.; Lejon, T.; van Hees, P.; Karlsson, P.; Kumpiene, J.; Carabante, I. Combination of separation and degradation methods after PFAS soil washing. Sci. Total Environ. 2024, 907, 168137. [Google Scholar] [CrossRef]
- Lu, D.; Sha, S.; Luo, J.; Huang, Z.; Jackie, X.Z. Treatment train approaches for the remediation of per- and polyfluoroalkyl substances (PFAS): A critical review. J. Hazard. Mater. 2020, 386, 121963. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Comfort, S.; da Silva, A.A.; Powell, J.; Cain, R.; McGreer, A.; Dantas, R.F. Remediating Per- and Polyfluoroalkyl Substances (PFAS)-Contaminated Water by Foam Fractionation and Electrochemical Oxidation. Environments 2025, 12, 185. https://doi.org/10.3390/environments12060185
Comfort S, da Silva AA, Powell J, Cain R, McGreer A, Dantas RF. Remediating Per- and Polyfluoroalkyl Substances (PFAS)-Contaminated Water by Foam Fractionation and Electrochemical Oxidation. Environments. 2025; 12(6):185. https://doi.org/10.3390/environments12060185
Chicago/Turabian StyleComfort, Steve, Amanda Araújo da Silva, Jessica Powell, Rebecca Cain, Ashleigh McGreer, and Renato F. Dantas. 2025. "Remediating Per- and Polyfluoroalkyl Substances (PFAS)-Contaminated Water by Foam Fractionation and Electrochemical Oxidation" Environments 12, no. 6: 185. https://doi.org/10.3390/environments12060185
APA StyleComfort, S., da Silva, A. A., Powell, J., Cain, R., McGreer, A., & Dantas, R. F. (2025). Remediating Per- and Polyfluoroalkyl Substances (PFAS)-Contaminated Water by Foam Fractionation and Electrochemical Oxidation. Environments, 12(6), 185. https://doi.org/10.3390/environments12060185