
Copper Speciation in Wine Growing-Drain Waters: Mobilization, Transport, and Environmental Diffusion

Abstract
1. Introduction
2. Materials and Methods
2.1. Site and Samples
2.2. Chemicals
2.3. Instruments and Operating Conditions
- A UV-Vis spectrophotometer (Agilent Technologies 1100 series from Agilent, VWD G1314A, Tokyo, Japan) with a wavelength set at 254 nm to monitor organic matter. Beforehand, the proportionality of the response of the organic carbon to its concentration in the water suspensions considered was verified as described by Harguindeguy et al. [33]. Indeed, a constant molar extinction coefficient at the selected wavelength is a prerequisite for using UV-Vis as a concentration detector [19]. A UV-Vis DAD (1260 Infinity series, Agilent Technology, Tokyo, Japan) was used to provide organic matter spectra. The spectra were recorded between 200 and 700 nm, on samples filtered at 0.45 µm and fractionated by AF4;
- A multi-angle light scattering (MALS) detector (DAWN HELEOS, Wyatt Technology, Santa Barbara, CA, USA) to determine the gyration diameters online. The UV-Vis and MALS data were collected and processed with Astra 5.3.4.18 software (Wyatt Technology). The gyration radii and diameters were calculated using Zimm’s first-order fitting formalism;
- An atomic mass spectrometer (ICP-MS) (Agilent 7500ce; Agilent Technology, Tokyo, Japan) equipped with a Meinhard nebulizer, a refrigerated Scott chamber (2 °C), and a collision reaction cell (CRC) used in hydrogen mode. The AF4-ICP-MS coupling was carried out by a two-pump assembly (as precisely described elsewhere [37]). This assembly enabled both the fractionated colloidal particles to be carried to the ICP-MS, and the element standards to be introduced into the ICP-MS. According to the elemental composition previously determined in the considered samples (see Section 2.1 above), the monitored trace and major elements were 63Cu, 65Cu, 27Al, 54Fe, 56Fe, 39K, 55Mn, 23Na, 64Zn, and 66Zn.
2.4. Signal and Data Processing
2.4.1. Signal Filtering
2.4.2. UV-Vis Spectral Characterization
- The spectral slope was between 275 and 295 nm (S275-295), an indicator of the degree of aromaticity. It increases when the aromatic carbon content decreases;
- The spectral (slope) ratio, i.e., the ratio of the spectral slopes, was between 275 and 295 nm and between 350 and 400 nm (SR), an indicator of the molar mass. It increases when the molar mass decreases;
- The absorbance ratios were between 250 and 365 nm (AR250-365) and between 465 and 665 nm (AR465-665), and are indicators of the degree of aromaticity and therefore the degree of humification. These ratios correlate with the molar mass and can also be used to jointly evaluate the molar mass. They increase when aromaticity, humification, and molar mass decrease.
2.4.3. Deconvolution
2.4.4. Size Distribution
2.4.5. MALS as Concentration Detector
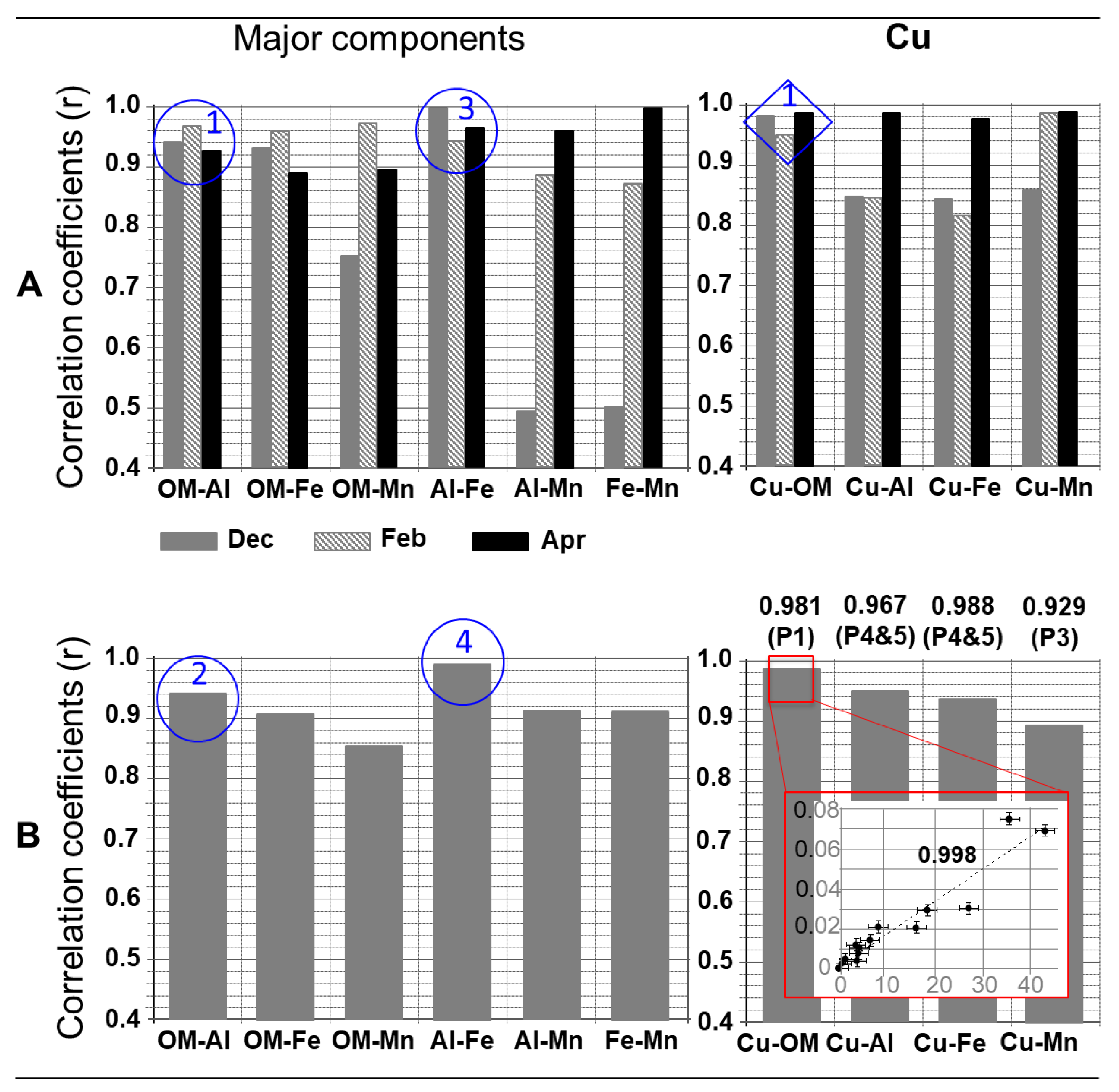
2.4.6. Correlation
- The signals of the fractograms of these components recorded by the different detectors (or the deduced distributions, which leads to similar results) for each of the samples;
- The concentrations of the components in the different colloidal populations identified for all the samples.
- A correlation coefficient close to 1 between the fractograms of 2 components, or between the concentrations of 2 components in all the colloidal populations of a given water sample, means that these 2 components are associated with each other throughout the colloidal continuum, or are associated in the same way in all populations. A significant but <<1 correlation coefficient means that these 2 components are associated on only part of the colloidal continuum, or are associated in the same way in some populations but not in all;
- A correlation coefficient close to 1 between the concentrations of 2 components in the same populations in the different water samples analyzed means that these 2 components are strongly associated in this population. A significant correlation coefficient, but << 1, means that the association is weaker.
3. Results and Discussion
3.1. Preliminary Study
3.2. Characterization of the Colloidal Phase
- P1, present in all three samples. Taking into account the response of the UV-Vis and ICP-MS detectors, P1 appeared to be mainly made up of organic matter. It may have contained some traces of manganese and iron, attributable to (hydr)oxides, given the soil composition. Note that despite very rapid elution, this population did not correspond to the AF4 dead volume peak (which was eluted before P1, lasting a few seconds, and which is usually not mentioned in size distributions [45]). P1 was an “early peak” (EP; also present in the soil leachate (Figure 3)) observed for small colloidal entities that were not very sensitive to the field applied in the Field Flow Fractionation methods, and are therefore poorly retained [55,56]. This organic material had an apparent molar mass of >10 kDa (AF4 cut-off threshold; equivalent to a diameter of approximately 2–3 nm [34]), and a gyration diameter varying between a few nanometers and 40 to 50 nm (peak at its base) and mainly between 10 and 20–25 nm (Figure 4, circle box on the right). Taking into account the results presented in Figure 3, this organic matter could either effectively correspond to humic acids having a molar mass of >10 kDa, or to humic supra-molecular structures consisting mainly of molecules with a molar mass of <10 kDa;
- P2, present in the samples from December and February. In very low concentrations, it contained only a few traces of manganese and iron, attributable to (hydr)oxides;
- P3, present in all three samples, and mainly in December and February, and P4, the main population of the colloidal phase (in terms of concentration, distribution range, and occurrence), present in all three samples. These two populations mainly contained particles rich in Al, attributable to the clays that were present significantly in the soil and humic acids. Note that P4 contributed significantly, even the majority (in December), to the presence of OM in waters;
- P5, present in the April sample only, with the same components as P4. The concentrations of OM and Al correlated strongly (r = 0.938). The ratio of concentrations between these two components was also of the same order of magnitude (approximately 1/10) between P4 and P5. This suggests that P5 contained aggregates mainly consisting of the particles initially present in P4. This was consistent with the size distributions of these two populations (Figure 4).
3.3. Copper Speciation
- Cooper, in the colloidal phase, was very preferentially complexed with the soil humic acids during its mobilization towards soil water, and the complexes formed were non-labile. The copper present in the dissolved phase (<10 kDa) could be in free and/or labile forms. However, the correlation between Cu and OM (Figure 5B, red box) on the dissolved-colloidal continuum suggests that the predominant form of dissolved Cu was the form complexed with humic acids;
- Copper was also associated with clays and (hydr)oxides, mainly via clay-humic complexes ((Cu-OM)-clays using the notation in Figure 5) and complexes between (hydr)oxides and clays, taking into account the previous remark.
3.4. Copper Behaviour and Fate
- The first significant rains on dry soil probably had a preferentially mechanical action. The fragmentation of the soil, the genesis of particles including inorganic-organic composite colloidal particles carrying Cu, and their migration through the soil in the water flow to the drain, were the predominant mobilization and transport processes;
- The rains, even heavy at the end of winter/early spring, on soil that remained damp, probably had a preferentially chemical and physical action. The leaching and weathering of the soil caused the release of dissolved and colloidal organic matter to which copper is complexed, and the genesis of colloidal particles of a smaller size, overall, in less mass concentration but in higher particle number concentrations than in December. This favored the aggregation phenomena reflected by P5 and a more balanced distribution of copper between dissolved and colloidal phases.
- The transport of copper from the soil, via drain water, to the aquatic system must be considered spatially and temporally. Spatially, because taking into account the distribution of copper between the dissolved and colloidal phases, the transport of copper on a significant scale, particularly to river waters, is possible [16]. Temporally, because knowing that the drain water flows several hours after a rainy episode, or even for several days and weeks during rainy periods, a significant copper quantity can be transported. This quantity can be estimated from, on the one hand, the measured rainfall and the copper concentrations determined in drain water and drained surfaces, and, on the other hand, the total stock of copper in the soil (65 mg kg−1 on average), considering that all the rain that fell infiltrated. Thus, during the rainy periods, around 3% per day on average of the copper stock could be transported outside the plots. At the maximum of the flow rates measured, up to 0.5% per hour of the copper stock could be transported. Given the alternation of dry periods / rainy episodes which promotes both leaching and weathering of soils, the quantities of copper transported in the long term are therefore indeed significant. This is in agreement with the observations of Bereswill et al. (2012). In their study, the copper contents in the surface sediments of watercourses were of the same order of magnitude as the concentrations in wine-growing soils studied [66]. This confirms that copper can be transported significantly, mainly in the colloidal form in our study, from vineyards to nearby river waters.
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lespes, G.; Zuliani, T.; Schaumlöffel, D. Need for revisiting the terminology about speciation. Environ. Sci. Pollut. Res. 2016, 23, 15767–15770. [Google Scholar] [CrossRef] [PubMed]
- Tessier, A.; Campbell, P.G.C.; Bisson, M. Sequential extraction procedure for the speciation of particulate trace metals. Anal. Chem. 1979, 51, 844–851. [Google Scholar] [CrossRef]
- Lespes, G. Nanoparticles in environment and health effect. In Metallomics: Analytical Techniques and Speciation Methods; Michalke, B., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Markt Schwaben, Germany, 2016; Volume 11, pp. 319–337. [Google Scholar]
- Lespes, G.; Faucher, S.; Slaveykova, V. Natural nanoparticles, anthropogenic nanoparticles, where is the frontier? Front. Environ. Sci. 2020, 8, 71. [Google Scholar] [CrossRef]
- Lead, J.R.; Wilkinson, K.J. Environmental colloids and particles: Current knowledge and future developments. In Environmental Colloids and Particles. Behaviour, Separation and Characterisation; Wilkinson, K.J., Lead, J.R., Eds.; Wiley and Sons: Chichester, UK, 2007; Volume 10, Chapter 1; pp. 1–14. [Google Scholar]
- Grolimund, D.; Barmettler, K.; Borkovec, M. Colloidal facilitated transport in natural porous media: Fundamental phenomena and modelling. In Colloidal Transport in Porous Media; Frimmel, F.H., von der Kammer, F., Flemming, H.C., Eds.; Springer: Berlin, Germany, 2007; pp. 3–27. [Google Scholar]
- Hochella, M.F., Jr.; Mogk, D.W.; Ranville, J.; Allen, I.C.; Luther, G.W.; Marr, L.C.; McGrail, B.P.; Murayama, M.; Qafoku, N.P.; Rosso, K.M.; et al. Natural, incidental, and engineered nanomaterials and their impacts on the Earth system. Science 2019, 363, eaau8299. [Google Scholar] [CrossRef] [PubMed]
- Lespes, G.; Gigault, J. Hyphenated analytical techniques for multidimensional characterisation of submicron particles: A review. Anal. Chim. Acta 2011, 692, 26–41. [Google Scholar] [CrossRef]
- Ferreira da Silva, B.; Pérez, S.; Gardinalli, P.; Singhal, R.K.; Mozeto, A.A.; Barcelo, D. Analytical chemistry of metallic nanoparticles in natural environments. Trends Anal. Chem. 2011, 30, 528–540. [Google Scholar]
- Zattoni, A.; Roda, B.; Borghi, F.; Marassi, V.; Reschiglian, P. Flow field-flow fractionation for the analysis of nanoparticles used in drug delivery. J. Pharm. Biomed. Anal. 2014, 87, 53–61. [Google Scholar] [CrossRef]
- Maria, E.; Crançon, P.; Le Coustumer, P.; Bridoux, M.; Lespes, G. Comparison of preconcentration methods of the colloidal phase of a uranium-containing soil suspension. Talanta 2020, 208, 120383. [Google Scholar] [CrossRef]
- Wahlund, K.-G.; Giddings, J.C. Properties of an asymmetrical flow field-flow fractionation channel having one permeable wall. Anal. Chem. 1987, 59, 1332–1339. [Google Scholar] [CrossRef]
- Schimpf, M.E.; Wahlund, K.-G. Asymmetrical flow field-flow fractionation as a method to study the behavior of humic acids in solution. J. Microcolumn Sep. 1997, 9, 535–543. [Google Scholar] [CrossRef]
- Baalousha, M.; Kammer, F.V.D.; Motelica-Heino, M.; Hilal, H.S.; Le Coustumer, P. Size fractionation and characterization of natural colloids by flow-field flow fractionation coupled to multi-angle laser light scattering. J. Chromatogr. A. 2006, 1104, 272–281. [Google Scholar] [CrossRef]
- Bouzas-Ramos, D.; García-Cortes, M.; Sanz-Medel, A.; Encinar, J.R.; Costa-Fernández, J.M. Assessment of the removal of side nanoparticulated populations generated during one-pot synthesis by asymmetric flow field-flow fractionation coupled to elemental mass spectrometry. J. Chromatogr. A 2017, 1519, 156–161. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lespes, G.; de Carsalade du pont, V. Field flow fractionation for nanoparticle characterization. J. Sep. Sci. 2022, 45, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Schurtenberger, P.; Newman, M.E. Characterization of biological and environmental particles using static and dynamic light scattering. In Environmental Particles; Buffle, J., van Leeuwen, H.P., Eds.; Lewis Boca Raton: London, UK, 1993; Volume 2, Chapter 2; pp. 37–115. [Google Scholar]
- Lespes, G.; Huclier, S.; Battu, S.; Rolland-Sabaté, A. Field flow Fractionation (FFF): Practical and experimental aspects. In Particle Separation Techniques: Fundamentals, Instrumentation, and Selected Applications; Handbook in Separation Science; Contado, C., Ed.; Elsevier: Amsterdam, The Netherlands, 2022; Chapter 19; pp. 621–657. [Google Scholar]
- Hassellov, M.; Readman, J.W.; Ranville, J.F.; Tiede, K. Nanoparticle analysis and characterization methodologies in environmental risk assessment of engineered nanoparticles. Ecotoxicology 2008, 17, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Van der Horst, C.; Silwana, B.; Iwuoha, E.; Somerset, V. Spectroscopic and voltammetric analysis of platinum group metals in road dust and roadside soil. Environment 2018, 5, 120. [Google Scholar] [CrossRef]
- Faucher, S.; Le Coustumer, P.; Lespes, G. Nanoanalytics: History, concepts and specificities. Environ. Sci. Pollut. Res. 2019, 26, 5267–5281. [Google Scholar] [CrossRef]
- El Hadri, H.; Chery, P.; Jalabert, S.; Lee, A.; Potin-Gautier, M.; Lespes, G. Assessment of diffuse contamination of agricultural soil by copper in Aquitaine region by using French national databases. Sci. Total Environ. 2012, 441, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Flores-Vélez, L.M.; Ducaroir, J.; Jaunet, A.M.; Robert, M. Study of the distribution of copper in an acid sandy vineyard soil by three different methods. Eur. J. Soil Sci. 1996, 47, 523–532. [Google Scholar] [CrossRef]
- Besnard, E.; Chenu, C.; Robert, M. Influence of organic amendments on copper distribution among particle-size and density fractions in Champagne vineyard soils. Environ. Pollut. 2001, 112, 329–337. [Google Scholar] [CrossRef]
- Mc Kenzie, R.M. The adsorption of lead and other heavy metals on oxides of manganese and iron. Aust. J. Soil Res. 1980, 18, 61–73. [Google Scholar] [CrossRef]
- Mc Bride, M.B. Forms and distribution of copper in solid and solution phases of soil. In Copper in Soils and Plants; Loneragan, J.F., Robson, A.D., Graham, R.D., Eds.; Academic Press: Cambridge, MA, USA, 1981; pp. 25–45. [Google Scholar]
- Sauve, S.; Mc Bride, M.B.; Norvell, W.A.; Hendershot, W.H. Copper solubility and speciation of in situ contaminated soils: Effects of copper level; pH and organic matter. Wat. Air Soil Poll. 1997, 100, 133–149. [Google Scholar] [CrossRef]
- Fernandez-Calvino, D.; Novoa-Munoz, J.C.; Lopez-Pezperiago, E.; Arias-Estevez, M. Changes in copper content and distribution in young old and abandoned vineyard acid soil due to land use changes. Land Degrad. Dev. 2008, 19, 165–177. [Google Scholar] [CrossRef]
- Ma, Y.B.; Lombi, E.; Oliver, I.W.; Nolan, A.L.; McLaughlin, M.J. Long-term aging of copper added to soils. Environ. Sci. Technol. 2006, 40, 6310–6317. [Google Scholar] [CrossRef] [PubMed]
- Anatole-Monnier, L. Effets de la Contamination Cuprique des Sols Viticoles sur la Sensibilité de la Vigne à un Cortège de Bio-agresseurs. Ph.D. Thesis, University of Bordeaux, Bordeaux, France, 2014; 199p. [Google Scholar]
- Komárek, M.; Cadková, E.; Chrastný, V.; Bordas, F.; Bollinger, J.-C. Contamination of vineyard soils with fungicides: A review of environmental and toxicological aspects. Environ. Int. 2010, 36, 138–151. [Google Scholar] [CrossRef] [PubMed]
- El Hadri, H. Pollution Diffuse des Sols par des Contaminants Minéraux et Impacts Environnementaux Liés aux Pratiques Agricoles. Ph.D. Thesis, University of Pau and Pays de l’Adour, Pau, France, 2012; 213p. [Google Scholar]
- Harguindeguy, S.; Crançon, P.; Potin-Gautier, M.; Pointurier, F.; Lespes, G. Colloidal mobilization from soil and transport of uranium in (sub)-surface waters. Environ. Sci. Pollut. Res. 2019, 26, 5294–5304. [Google Scholar] [CrossRef]
- de Carsalade du pont, V.; Faucher, S.; Lespes, G. Nanoparticles in waters and soil. In Field Flow Fractionation: Principles and Applications; Guegen, C., Baalousha, M., Williams, K.R., Eds.; Wiley-VCH: Weinheim, Germany, 2024; in press. [Google Scholar]
- Dubascoux, S.; v d Kammer, F.; Le Hecho, I.; Potin-Gautier, M.; Lespes, G. Optimisation of asymmetrical Field Flow Fractionation for environmental nanoparticles separation. J. Chromatogr. A 2008, 1206, 160–165. [Google Scholar] [CrossRef]
- El Hadri, H.; Gigault, J.; Chery, P.; Potin-Gautier, M.; Lespes, G. Optimization of flow-field flow fractionation for the characterization of natural colloids. Anal. Bioanal. Chem. 2014, 406, 1639–1649. [Google Scholar] [CrossRef] [PubMed]
- Dubascoux, S.; Le Hecho, I.; Potin-Gautier, M.; Lespes, G. On line and off-line quantification of trace elements associated to colloids by As-Fl-FFF and ICP-MS. Talanta 2008, 77, 60–65. [Google Scholar] [CrossRef] [PubMed]
- von der Kammer, F. Characterization of Environmental Colloids Applying Field-Flow Fractionation—Multi Detection Analysis with Emphasis on Light Scattering Techniques. Ph.D. Thesis, Technical University of Hamburg-Harburg, Hamburg, Germany, 2004; 234p. [Google Scholar]
- Masson, M.; Guigues, N.; Arhror, M.; Raveau, S.; Brosse, C.; Forquet, N. Caractérisation de la Matière Organique d’Eaux Résiduaires et d’Eaux de Surface par les Sondes Spectrophotométriques UV-Visible; Research Report IRSTEA; Agence Française de la Biodiversité: Vincennes, France, 2019; 77p, hal-02609331. [Google Scholar]
- Enev, V.; Pospisilova, L.; Klucakova, M.; Liptaj, T.; Doskocil, L. Spectral characterization of selected humic substances. Soil Water Res. 2014, 9, 9–17. [Google Scholar] [CrossRef]
- Polak, J.; Bartoszek, M.; Sułkowski, W.W. Comparison of some spectroscopic and physico-chemical properties of humic acids extracted from sewage sludge and bottom sediments. J. Mol. Struc. 2009, 924–926, 309–312. [Google Scholar] [CrossRef]
- Peacock, M.; Evans, C.D.; Fenner, N.; Freeman, C.; Gough, R.; Jones, T.G.; Lebron, I. UV-visible absorbance spectroscopy as a proxy for peatland dissolved organic carbon (DOC) quantity and quality: Considerations on wavelength and absorbance degradation. Environ. Sci. Process Impacts 2014, 16, 1445. [Google Scholar] [CrossRef] [PubMed]
- Helms, J.R.; Stubbins, A.; Ritchie, J.D.; Minor, E.C.; Kieber, D.J.; Mopper, K. Absorption spectral slopes and slope ratios as indicators of molecular weight, source, and photobleaching of chromophoric dissolved organic matter. Limnol. Oceanogr. 2008, 53, 955–969. [Google Scholar] [CrossRef]
- Chen, H.; Zheng, B.; Song, Y.; Qin, Y. Correlation between molecular absorption spectral slope ratios and fluorescence humification indices in characterizing CDOM. Aquat. Sci. 2011, 73, 103–112. [Google Scholar] [CrossRef]
- Schimpf, M.E.; Caldwell, K.; Giddings, J.C. (Eds.) Field-Flow Fractionation Handbook; Wiley and Sons: New York, NY, USA, 2000. [Google Scholar]
- Andersson, M.; Wittgren, B.; Wahlund, K.-G. Accuracy in Multiangle Light Scattering Measurements for Molar Mass and Radius Estimations. Model Calculations and Experiments. Anal. Chem. 2003, 75, 4279–4291. [Google Scholar] [CrossRef]
- Teraoka, I. Polymer Solutions: An Introduction to Physical Properties; Wiley and Sons: New York, NY, USA, 2002. [Google Scholar]
- Zimm, B.H. The Scattering of Light and the Radial Distribution Function of High Polymer Solutions. J. Chem. Phys. 1948, 16, 1093–1099. [Google Scholar] [CrossRef]
- Bancon-Montigny, C.; Lespes, G.; Potin-Gautier, M. Organotin survey in Adour-Garonne Basin. Water Res. 2004, 38, 933–946. [Google Scholar] [CrossRef] [PubMed]
- Masson, M. Synthèse Bibliographique sur la Faisabilité des Sondes Spectrophotométriques pour la Caractérisation In Situ de la Matière Organique; Report Aquaref; HAL: Lyon, France, 2015; 20p. [Google Scholar]
- Faucher, S.; Ivaneev, A.I.; Fedotov, P.S.; Lespes, G. Characterization of volcanic ash nanoparticles and study of their fate in aqueous medium by asymmetric flow field-flow fractionation—Multidetection. Environ. Sci. Pollut. Res. 2021, 28, 31850–31860. [Google Scholar] [CrossRef] [PubMed]
- Suraj, A.P.; Khalid, A.M.D.; Rajesh, K.; Asgar, A.; Yasmin, S. Humic acid from Shilajit: A physico-chemical and spectroscopic characterization. J. Serb. Chem. Soc. 2010, 75, 413–422. [Google Scholar]
- McKnight, D.M.; Boyer, E.W.; Westerhoff, P.K.; Doran, P.T.; Kulbe, T.; Andersen, D.T. Spectrofluorometric characterization of dissolved organic matter for indication of precursor organic material and aromaticity. Limnol. Oceanogr. 2001, 46, 38–48. [Google Scholar] [CrossRef]
- Ukalska-Jaruga, A.; Bejger, R.; Debaene, G.; Smreczak, B. Characterization of Soil Organic Matter Individual Fractions (Fulvic Acids, Humic Acids, and Humins) by Spectroscopic and Electrochemical Techniques in Agricultural Soils. Agronomy 2021, 11, 1067. [Google Scholar] [CrossRef]
- Gale, B.K.; Srinivas, M. Cyclical electrical fiel flow fractionation. Electrophoresis 2005, 26, 1623–1632. [Google Scholar] [CrossRef] [PubMed]
- Srinivas, M.; Sant, H.J.; Gale, B.K. Optimisation of cyclical electrical field flow fractionation. Electrophoresis 2010, 31, 3372–3379. [Google Scholar] [CrossRef] [PubMed]
- Adediran, S.A.; Kramer, J.R. Copper adsorption on clay, iron-manganese oxide and organic fractions along a salinity gradient. Appl. Geochem. 1987, 2, 213–216. [Google Scholar] [CrossRef]
- Mcbride, M.B. Reactions controlling heavy metal solubility in soils. Adv. Soil Sci. 1989, 10, 1–56. [Google Scholar]
- Bravin, M.N. Processus Rhizosphèriques Déterminant la Biodisponibilité du Cuivre pour le Blé dur Cultivé en Sols à Antécédant Viticole. Ph.D. Thesis, Montpellier SupAgro, Montpellier, France, 2008; 237p. [Google Scholar]
- Duchaufour, P. Introduction à la Science du Sol. In Sol, Végétation, Environnement, 6th ed.; de l’Abrégé de Pédologie; Dunod: Paris, France, 2001. [Google Scholar]
- Hizal, J.; Apak, R. Modeling of copper(II) and lead(II) adsorption on kaolinite- based clay minerals individually and in the presence of humic acid. J. Colloid Interface Sci. 2006, 295, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sposito, G. The Chemistry of Soils; Oxford University Press: Oxford, UK, 2008. [Google Scholar]
- Bradl, H.B. Adsorption of heavy metal ions on soils and soils constituents. J. Colloid Interface Sci. 2004, 277, 1–18. [Google Scholar] [CrossRef]
- Hodgson, J.F.; Lindsay, W.L.; Trierweiler, J.F. Micronutrient Cation Complexing in Soil Solution: II. Complexing Of Zinc and Copper in Displaced Solution from Calcareous Soils. Soil Sci. Soc. Am. J. 1966, 30, 723–726. [Google Scholar] [CrossRef]
- Sauvé, S.; Dumestre, A.; Mcbride, M.; Hendershot, W. Derivation of soil quality criteria using predicted chemical speciation of Pb2+ and Cu2+. Environ. Toxicol. Chem. 1998, 17, 1481–1489. [Google Scholar] [CrossRef]
- Bereswill, R.; Golla, B.; Streloke, M.; Schulz, R. Entry and toxicity of organic pesticides and copper in vineyard streams: Erosion rills jeopardise the efficiency of riparian buffer strips. Agric. Ecosyst. Environ. 2012, 146, 81–92. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | Sample | Gyration Diameter (nm) | Concentration (mg L−1) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Range (Baseline) | Peak Top | Total Colloidal | Main Components | Cu | |||||
| OM | Al | Fe | Mn | ||||||
| P1 | December | 3–60 | 15 ± 1 | 4.49 ± 0.05 | 4.2 ± 0.4 | – | – | 0.048 ± 0.003 | 0.010 ± 0.001 |
| February | 8–65 | 20 ± 2 | 1.69 ± 0.02 | 1.44 ± 0.04 | – | – | 0.008 ± 0.001 | 0.005 ± 0.001 | |
| April | 9–40 | 15 ± 1 | 6.18 ± 0.5 | 6.4 ± 0.5 | – | 0.008 ± 0.001 | 0.0014 ± 0.0003 | 0.014 ± 0.001 | |
| P2 | December | 20–230 | 100 ± 5 | 0.22 ± 0.01 | – | – | 0.088 ± 0.006 | 0.020 ± 0.001 | − |
| February | 90–200 | 110 ± 5 | 0.101 ± 0.009 | – | – | 0.030 ± 0.002 | − | − | |
| April | – | – | – | – | – | − | − | − | |
| P3 | December | 70–200 | 140 ± 5 | 138 ± 14 | 4.1 ± 0.4 | 20 ± 2 | 0.11 ± 0.01 | 0.24 ± 0.02 | 0.008 ± 0.001 |
| February | 60–200 | 125 ± 5 | 134 ± 11 | 3.7 ± 0.5 | 17 ± 1 | 0.13 ± 0.01 | 0.010 ± 0.001 | 0.004 ± 0.001 | |
| April | 1–200 | 100 ± 4 | 0.70 ± 0.05 | 0.55 ± 0.06 | − | 0.074 ± 0.008 | 0.013 ± 0.001 | 0.002 ± 0.001 | |
| P4 | December | 70–420 | 220 ± 7 | 4258 ± 350 | 42 ± 3 | 622 ± 44 | 2.6 ± 0.1 | 0.13 ± 0.01 | 0.069 ± 0.006 |
| February | 60–360 | 195 ± 6 | 824 ± 75 | 18 ± 2 | 105 ± 7 | 0.56 ± 0.05 | 0.044 ± 0.003 | 0.029 ± 0.003 | |
| April | 15–380 | 170 ± 6 | 917 ± 85 | 16 ± 2 | 154 ± 11 | 0.52 ± 0.04 | 0.063 ± 0.005 | 0.021 ± 0.002 | |
| P5 | December | – | – | − | – | – | – | – | – |
| February | – | – | – | – | – | – | – | – | |
| April | 100–660 | 300 ± 13 | 1276 ± 110 | 27 ± 2 | 214 ± 15 | 0.55 ± 0.04 | 0.068 ± 0.008 | 0.030 ± 0.003 | |
| TOTAL concentration (mg L−1) | December | / | / | 4400 ± 400 | 15 ± 1 | 641 ± 45 | 2.8 ± 0.3 | 0.44 ± 0.02 | 0.087 ± 0.008 |
| February | 980 ± 090 | 58 ± 4 | 122 ± 11 | 0.72 ± 0.08 | 0.061 ± 0.005 | 0.038 ± 0.004 | |||
| April | 2200 ± 200 | 33 ± 2 | 367 ± 49 | 1.15 ± 0.02 | 0.15 ± 0.01 | 0.067 ± 0.006 | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
De Carsalade du Pont, V.; Ben Azzouz, A.; El Hadri, H.; Chéry, P.; Lespes, G. Copper Speciation in Wine Growing-Drain Waters: Mobilization, Transport, and Environmental Diffusion. Environments 2024, 11, 19. https://doi.org/10.3390/environments11010019
De Carsalade du Pont V, Ben Azzouz A, El Hadri H, Chéry P, Lespes G. Copper Speciation in Wine Growing-Drain Waters: Mobilization, Transport, and Environmental Diffusion. Environments. 2024; 11(1):19. https://doi.org/10.3390/environments11010019
Chicago/Turabian StyleDe Carsalade du Pont, Valentin, Amani Ben Azzouz, Hind El Hadri, Philippe Chéry, and Gaëtane Lespes. 2024. "Copper Speciation in Wine Growing-Drain Waters: Mobilization, Transport, and Environmental Diffusion" Environments 11, no. 1: 19. https://doi.org/10.3390/environments11010019
APA StyleDe Carsalade du Pont, V., Ben Azzouz, A., El Hadri, H., Chéry, P., & Lespes, G. (2024). Copper Speciation in Wine Growing-Drain Waters: Mobilization, Transport, and Environmental Diffusion. Environments, 11(1), 19. https://doi.org/10.3390/environments11010019