C-Phycocyanin and Phycocyanobilin as Remyelination Therapies for Enhancing Recovery in Multiple Sclerosis and Ischemic Stroke: A Preclinical Perspective

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. C-Phycocyanin and Phycocyanobilin
3. Mechanisms of Remyelination: An Overview
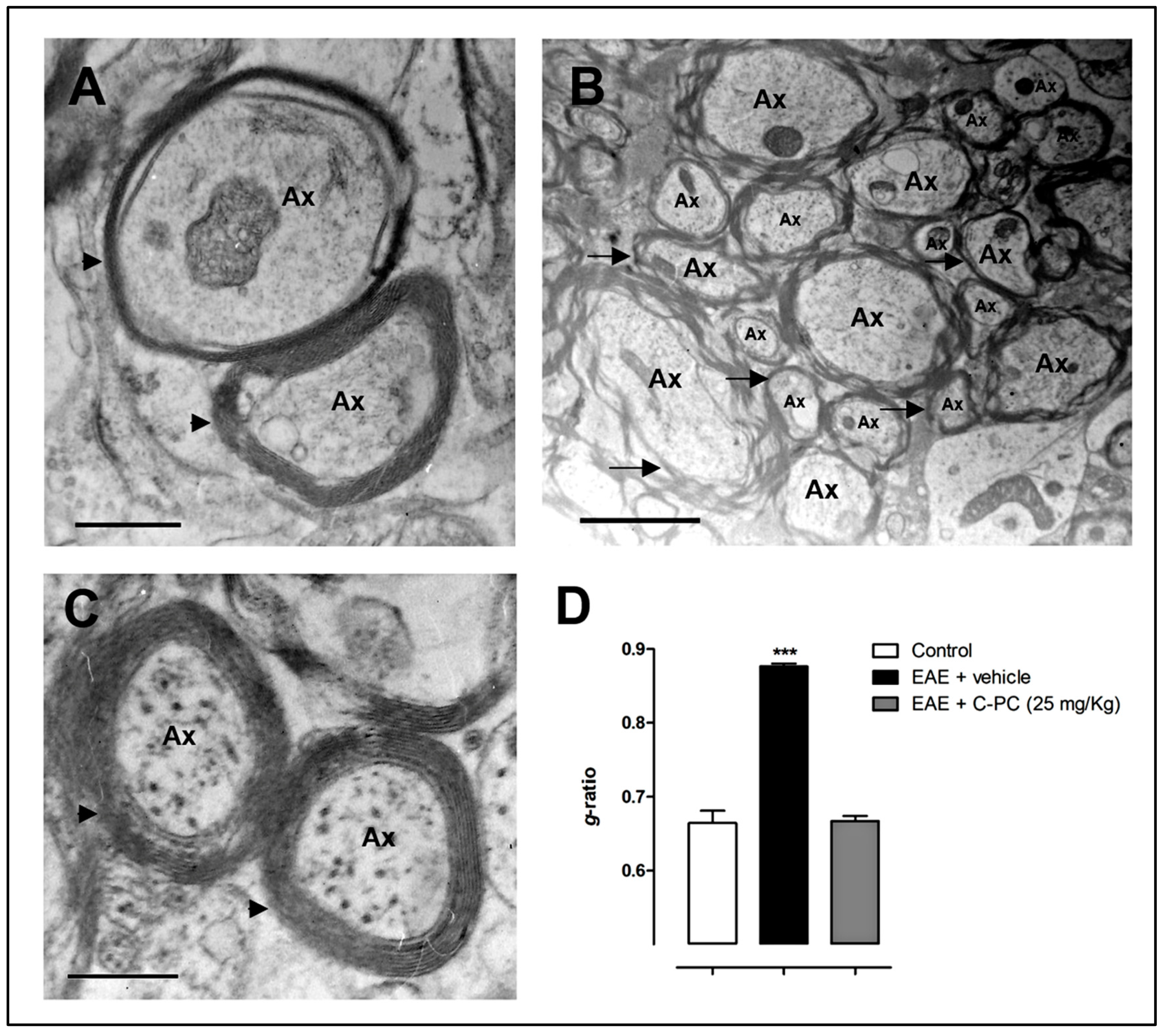
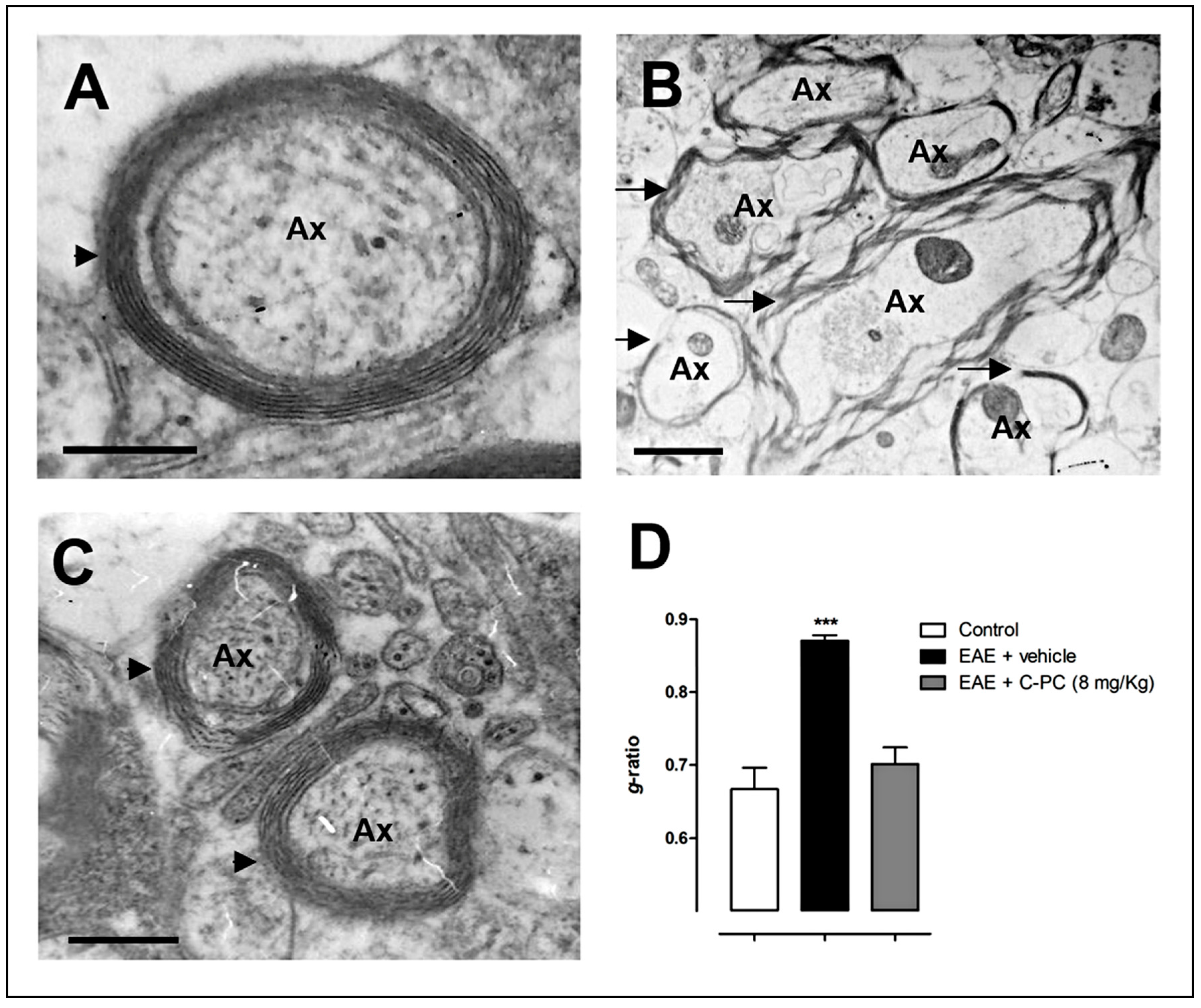
4. C-PC Remyelinating Actions in MS Models
5. PCB Remyelinating Effects in Animal Models of Focal and Global Cerebral Ischemia
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280. [Google Scholar] [CrossRef] [PubMed]
- Snaidero, N.; Simons, M. Myelination at a glance. J. Cell Sci. 2014, 127, 2999–3004. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Göttle, P.; Hartung, H.P.; Küry, P. Pushing forward: Remyelination as the new frontier in CNS diseases. Trends Neurosci. 2016, 39, 246–263. [Google Scholar] [CrossRef] [PubMed]
- Jones, T.A. Motor compensation and its effects on neural reorganization after stroke. Nat. Rev. Neurosci. 2017, 18, 267–280. [Google Scholar] [CrossRef] [PubMed]
- Mark, V.W. Stroke and Behavior. Neurol. Clin. 2016, 34, 205–234. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Zhuang, J.; Li, J.; Ooi, E.; Bloom, J.; Poon, C.; Lax, D.; Rosenbaum, D.M.; Barone, F.C. Long-term post-stroke changes include myelin loss, specific deficits in sensory and motor behaviors and complex cognitive impairment detected using active place avoidance. PLoS ONE 2013, 8, e57503. [Google Scholar] [CrossRef] [PubMed]
- Ho, P.W.; Reutens, D.C.; Phan, T.G.; Wright, P.M.; Markus, R.; Indra, I.; Young, D.; Donnan, G.A. Is white matter involved in patients entered into typical trials of neuroprotection? Stroke 2005, 36, 2742–2744. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.E.; Tittgemeyer, M.; Imperati, D.; Diekhoff, S.; Ameli, M.; Fink, G.R.; Grefkes, C. Degeneration of corpus callosum and recovery of motor function after stroke: A multimodal magnetic resonance imaging study. Hum. Brain Mapp. 2012, 33, 2941–2956. [Google Scholar] [CrossRef] [PubMed]
- Nylander, A.; Hafler, D.A. Multiple sclerosis. J. Clin. Investig. 2012, 122, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Whitwell, J. Setting new standards in multiple sclerosis care and research. Lancet Neurol. 2012, 11, 835. [Google Scholar] [CrossRef]
- Gelfand, J.M. Multiple sclerosis: Diagnosis, differential diagnosis and clinical presentation. Handb. Clin. Neurol. 2014, 122, 269–290. [Google Scholar] [CrossRef] [PubMed]
- Ziemssen, T. Symptom management in patients with multiple sclerosis. J. Neurol. Sci. 2011, 311, S48–S52. [Google Scholar] [CrossRef]
- Simmons, R.D.; Tribe, K.L.; McDonald, E.A. Living with multiple sclerosis: Longitudinal changes in employment and the importance of symptom management. J. Neurol. 2010, 257, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Westad, A.; Venugopal, A.; Snyder, E. The multiple sclerosis market. Nat. Rev. Drug Discov. 2017, 16, 675–676. [Google Scholar] [CrossRef] [PubMed]
- Montalban, X.; Hauser, S.L.; Kappos, L.; Arnold, D.L.; Bar-Or, A.; Comi, G.; de Seze, J.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Hauser, S.L.; Bar-Or, A.; Comi, G.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Lublin, F.; Montalban, X.; Rammohan, K.W.; Selmaj, K.; et al. Ocrelizumab versus Interferon Beta-1a in Relapsing Multiple Sclerosis. N. Engl. J. Med. 2017, 376, 221–234. [Google Scholar] [CrossRef] [PubMed]
- FDA News Release. FDA Approves New Drug to Treat Multiple Sclerosis. First Drug Approved for Primary Progressive MS. 29 March 2017. Available online: https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm549325.htm (accessed on 8 November 2017).
- Dendrou, C.A.; Fugger, L. Immunomodulation in multiple sclerosis: Promises and pitfalls. Curr. Opin. Immunol. 2017, 49, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Kremer, D.; Küry, P.; Dutta, R. Promoting remyelination in multiple sclerosis: Current drugs and future prospects. Mult. Scler. 2015, 21, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Romay, C.; Armesto, J.; Remirez, D.; González, R.; Ledón, N.; García, I. Antioxidant and anti-inflammatory properties of C-phycocyanin from blue-green algae. Inflamm. Res. 1998, 47, 36–41. [Google Scholar] [CrossRef] [PubMed]
- McCarty, M.F. Clinical potential of Spirulin as a source of Phycocyanobilin. J. Med. Food. 2007, 10, 566–570. [Google Scholar] [CrossRef] [PubMed]
- Fu, E.; Friedman, L.; Siegelman, H.W. Mass-spectral identification and purification of phycoerythrobilin and phycocyanobilin. Biochem. J. 1979, 179, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mukougawa, K.; Kanamoto, H.; Kobayashi, T.; Yokota, A.; Kohchi, T. Metabolic engineering to produce phytochromes with phytochromobilin, phycocyanobilin, or phycoerythrobilin chromophore in Escherichia coli. FEBS Lett. 2006, 580, 1333–1338. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Rojas, B.; Hernández-Juárez, J.; Pedraza-Chaverri, J. Nutraceutical properties of phycocyanin. J. Funct. Food 2014, 11, 375–392. [Google Scholar] [CrossRef]
- Remirez, D.; Ledón, N.; González, R. Role of histamine in the inhibitory effects of phycocyanin in experimental models of allergic inflammatory response. Mediat. Inflamm. 2002, 11, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Leung, P.O.; Lee, H.H.; Kung, Y.C.; Tsai, M.F.; Chou, T.C. Therapeutic effect of C-phycocyanin extracted from blue green algae in a rat model of acute lung injury induced by lipopolysaccharide. Evid. Based Complement. Altern. Med. 2013, 916590. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J. Why does remyelination fail in multiple sclerosis? Nat. Rev. Neurosci. 2002, 3, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, T.; Miron, V.; Cui, Q.; Wegner, C.; Antel, J.; Brück, W. Differentiation block of oligodendroglial progenitor cells as a cause for remyelination failure in chronic multiple sclerosis. Brain 2008, 131, 1749–1758. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.J.; Ffrench-Constant, C. Remyelination in the CNS: From biology to therapy. Nat. Rev. Neurosci. 2008, 9, 839–855. [Google Scholar] [CrossRef] [PubMed]
- McMurran, C.E.; Jones, C.A.; Fitzgerald, D.C.; Franklin, R.J.M. CNS remyelination and the innate immune system. Front. Cell Dev. Biol. 2016, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Zawadzka, M.; Rivers, L.E.; Fancy, S.P.; Zhao, C.; Tripathi, R.; Jamen, F.; Young, K.; Goncharevich, A.; Pohl, H.; Rizzi, M.; et al. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell 2010, 6, 578–590. [Google Scholar] [CrossRef] [PubMed]
- Irvine, K.A.; Blakemore, W.F. Remyelination protects axons from demyelination-associated axon degeneration. Brain 2008, 131, 1464–1477. [Google Scholar] [CrossRef] [PubMed]
- Manrique-Hoyos, N.; Jürgens, T.; Grønborg, M.; Kreutzfeldt, M.; Schedensack, M.; Kuhlmann, T.; Schrick, C.; Brück, W.; Urlaub, H.; Simons, M.; et al. Late motor decline after accomplished remyelination: Impact for progressive multiple sclerosis. Ann. Neurol. 2012, 71, 227–244. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, J.J.; Messier, C. From precursors to myelinating oligodendrocytes: Contribution of intrinsic and extrinsic factors to white matter plasticity in the adult brain. Neuroscience 2014, 269, 343–366. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.; Tourtellotte, W.W.; Rudick, R.; Trapp, B.D. Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N. Engl. J. Med. 2002, 346, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, K.A.; Nanescu, S.E.; Psachoulia, K.; Huang, J.K. Oligodendrocyte regeneration: Its significance in myelin replacement and neuroprotection in multiple sclerosis. Neuropharmacology 2016, 110 Pt B, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Arai, K.; Lo, E.H. An oligovascular niche: Cerebral endothelial cells promote the survival and proliferation of oligodendrocyte precursor cells. J. Neurosci. 2009, 29, 4351–4355. [Google Scholar] [CrossRef] [PubMed]
- Domingues, H.S.; Portugal, C.C.; Socodato, R.; Relvas, J.B. Oligodendrocyte, astrocyte and microglia crosstalk in myelin development, damage and repair. Front. Cell Dev. Biol. 2016, 4, 71. [Google Scholar] [CrossRef] [PubMed]
- Plemel, J.R.; Liu, W.Q.; Yong, V.W. Remyelination therapies: A new direction and challenge in multiple sclerosis. Nat. Rev. Drug Discov. 2017, 16, 617–634. [Google Scholar] [CrossRef] [PubMed]
- Itoh, K.; Maki, T.; Lok, J.; Arai, K. Mechanisms of cell-cell interaction in oligodendrogenesis and remyelination after stroke. Brain Res. 2015, 1623, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Denic, A.; Johnson, A.J.; Bieber, A.J.; Warrington, A.E.; Rodriguez, M.; Pirko, I. The relevance of animal models in multiple sclerosis research. Pathophysiology 2011, 18, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Pentón-Rol, G.; Martínez-Sánchez, G.; Cervantes-Llanos, M.; Lagumersindez-Denis, N.; Acosta-Medina, E.F.; Falcón-Cama, V.; Alonso-Ramírez, R.; Valenzuela-Silva, C.; Rodríguez-Jiménez, E.; Llópiz-Arzuaga, A.; et al. C-Phycocyanin ameliorates experimental autoimmune encephalomyelitis and induces regulatory T cells. Int. Immunopharmacol. 2011, 11, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.A.; Akirav, E.M. Remyelination in multiple sclerosis: Cellular mechanisms and novel therapeutic approaches. J. Neurosci. Res. 2015, 93, 687–696. [Google Scholar] [CrossRef] [PubMed]
- Haider, L.; Zrzavy, T.; Hametner, S.; Hoftberger, R.; Bagnato, F.; Grabner, G.; Trattnig, S.; Pfeifenbring, S.; Brück, W.; Lassmann, H. The topography of demyelination and neurodegeneration in the multiple sclerosis brain. Brain 2016, 139 Pt 3, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Lagumersindez-Denis, N.; Wrzos, C.; Mack, M.; Winkler, A.; van der Meer, F.; Reinert, M.C.; Hollasch, H.; Flach, A.; Brühl, H.; Cullen, E.; et al. Differential contribution of immune effector mechanisms to cortical demyelination in multiple sclerosis. Acta Neuropathol. 2017, 134, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Santis, A.G.; López-Cabrera, M.; Hamann, J.; Strauss, M.; Sánchez-Madrid, F. Structure of the gene coding for the human early lymphocyte activation antigen CD69: A C-type lectin receptor evolutionarily related with the gene families of natural killer cell-specific receptors. Eur. J. Immunol. 1994, 24, 1692–1697. [Google Scholar] [CrossRef] [PubMed]
- Koutrolos, M.; Berer, K.; Kawakami, N.; Wekerle, H.; Krishnamoorthy, G. Treg cells mediate recovery from EAE by controlling effector T cell proliferation and motility in the CNS. Acta Neuropathol. Commun. 2014, 2, 163. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Longhi, R.; Lissoni, F.; Trabattoni, D.; Mendozzi, L.; Caputo, D.; Clerici, M. CD4+CD25+FoxP3+PD1− regulatory T cells in acute and stable relapsing-remitting multiple sclerosis and their modulation by therapy. FASEB J. 2008, 22, 3500–3508. [Google Scholar] [CrossRef] [PubMed]
- Dombrowski, Y.; O’Hagan, T.; Dittmer, M.; Penalva, R.; Mayoral, S.R.; Bankhead, P.; Fleville, S.; Eleftheriadis, G.; Zhao, C.; Naughton, M.; et al. Regulatory T cells promote myelin regeneration in the central nervous system. Nat. Neurosci. 2017, 20, 674–680. [Google Scholar] [CrossRef] [PubMed]
- Simmons, D.L.; Botting, R.M.; Hla, T. Cyclooxygenase isozymes: The biology of prostaglandin synthesis and inhibition. Pharmacol. Rev. 2004, 56, 387–437. [Google Scholar] [CrossRef] [PubMed]
- Yagami, T.; Koma, H.; Yamamoto, Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the Central Nervous System. Mol. Neurobiol. 2016, 53, 4754–4771. [Google Scholar] [CrossRef] [PubMed]
- Carlson, N.G.; Hill, K.E.; Tsunoda, I.; Fujinami, R.S.; Rose, J.W. The pathologic role for COX-2 in apoptotic oligodendrocytes in virus induced demyelinating disease: Implications for multiple sclerosis. J. Neuroimmunol. 2006, 174, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Shiow, L.R.; Favrais, G.; Schirmer, L.; Schang, A.L.; Cipriani, S.; Andres, C.; Wright, J.N.; Nobuta, H.; Fleiss, B.; Gressens, P.; et al. Reactive astrocyte COX2-PGE2 production inhibits oligodendrocyte maturation in neonatal white matter injury. Glia 2017, 65, 2024–2037. [Google Scholar] [CrossRef] [PubMed]
- Carlson, N.G.; Bellamkonda, S.; Schmidt, L.; Redd, J.; Huecksteadt, T.; Weber, L.M.; Davis, E.; Wood, B.; Maruyama, T.; Rose, J.W. The role of the prostaglandin E2 receptors in vulnerability of oligodendrocyte precursor cells to death. J. Neuroinflamm. 2015, 12, 101. [Google Scholar] [CrossRef] [PubMed]
- Reddy, C.M.; Bhat, V.B.; Kiranmai, G.; Reddy, M.N.; Reddanna, P.; Madyastha, K.M. Selective inhibition of cyclooxygenase-2 by C-phycocyanin, a biliprotein from Spirulina platensis. Biochem. Biophys. Res. Commun. 2000, 277, 599–603. [Google Scholar] [CrossRef] [PubMed]
- Mardini, I.A.; FitzGerald, G.A. Selective inhibitors of cyclooxygenase-2: A growing class of anti-inflammatory drugs. Mol. Interv. 2001, 1, 30–38. [Google Scholar] [PubMed]
- Stichtenoth, D.O.; Frölich, J.C. The second generation of COX-2 inhibitors: What advantages do the newest offer? Drugs 2003, 63, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Jensen, G.S.; Drapeau, C.; Lenninger, M.; Benson, K.F. Clinical safety of a high dose of Phycocyanin-enriched aqueous extract from Arthrospira (Spirulina) platensis: Results from a randomized, double-blind, placebo-controlled study with a focus on anticoagulant activity and platelet activation. J. Med. Food 2016, 19, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Ramdial, K.; Franco, M.C.; Estevez, A.G. Cellular mechanisms of peroxynitrite-induced neuronal death. Brain Res. Bull. 2017, 133, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.Y.; Ljunggren-Rose, A.; Chandramohan, N.; Whetsell, W.O., Jr.; Sriram, S. In Vitro and in vivo induction and activation of nNOS by LPS in oligodendrocytes. J. Neuroimmunol. 2010, 229, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Marik, C.; Felts, P.A.; Bauer, J.; Lassmann, H.; Smith, K.J. Lesion genesis in a subset of patients with multiple sclerosis: A role for innate immunity? Brain 2007, 130 Pt 11, 2800–2815. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Palma, L.; Pehar, M.; Cassina, P.; Peluffo, H.; Castellanos, R.; Anesetti, G.; Beckman, J.S.; Barbeito, L. Involvement of nitric oxide on kainate-induced toxicity in oligodendrocyte precursors. Neurotox. Res. 2003, 5, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Lepka, K.; Volbracht, K.; Bill, E.; Schneider, R.; Rios, N.; Hildebrandt, T.; Ingwersen, J.; Prozorovski, T.; Lillig, C.H.; van Horssen, J.; et al. Iron-sulfur glutaredoxin 2 protects oligodendrocytes against damage induced by nitric oxide release from activated microglia. Glia 2017, 65, 1521–1534. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.B.; Madyastha, K.M. Scavenging of peroxynitrite by phycocyanin and phycocyanobilin from Spirulina platensis: Protection against oxidative damage to DNA. Biochem. Biophys. Res. Commun. 2001, 285, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Cherng, S.C.; Cheng, S.N.; Tarn, A.; Chou, T.C. Anti-inflammatory activity of C-phycocyanin in lipopolysaccharide-stimulated RAW 264.7 macrophages. Life Sci. 2007, 81, 1431–1435. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.C.; Liu, K.S.; Yang, T.J.; Hwang, J.H.; Chan, Y.C.; Lee, I.T. Spirulina and C-phycocyanin reduce cytotoxicity and inflammation-related genes expression of microglial cells. Nutr. Neurosci. 2012, 15, 252–256. [Google Scholar] [CrossRef] [PubMed]
- Pentón-Rol, G.; Lagumersindez-Denis, N.; Muzio, L.; Bergami, A.; Furlan, R.; Fernández-Massó, J.R.; Nazábal-Gálvez, M.; Llópiz-Arzuaga, A.; Herrera-Rolo, T.; Véliz-Rodríguez, T.; et al. Comparative neuroregenerative effects of C-Phycocyanin and IFN-Beta in a model of Multiple Sclerosis in mice. J. Neuroimmune Pharmacol. 2016, 11, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Schultz, V.; van der Meer, F.; Wrzos, C.; Scheidt, U.; Bahn, E.; Stadelmann, C.; Brück, W.; Junker, A. Acutely damaged axons are remyelinated in multiple sclerosis and experimental models of demyelination. Glia 2017, 65, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Ligon, K.L.; Fancy, S.P.; Franklin, R.J.; Rowitch, D.H. Olig gene function in CNS development and disease. Glia 2006, 54, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Fancy, S.P.; Zhao, C.; Franklin, R.J. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol. Cell. Neurosci. 2004, 27, 247–254. [Google Scholar] [CrossRef] [PubMed]
- Southwood, C.; He, C.; Garbern, J.; Kamholz, J.; Arroyo, E.; Gow, A. CNS myelin paranodes require Nkx6-2 homeoprotein transcriptional activity for normal structure. J. Neurosci. 2004, 24, 11215–11225. [Google Scholar] [CrossRef] [PubMed]
- Frank, M. MAL, a proteolipid in glycosphingolipid enriched domains: Functional implications in myelin and beyond. Prog. Neurobiol. 2000, 60, 531–544. [Google Scholar] [CrossRef]
- Schaeren-Wiemers, N.; Bonnet, A.; Erb, M.; Erne, B.; Bartsch, U.; Kern, F.; Mantei, N.; Sherman, D.; Suter, U. The raft-associated protein MAL is required for maintenance of proper axon-glia interactions in the central nervous system. J. Cell Biol. 2004, 166, 731–742. [Google Scholar] [CrossRef] [PubMed]
- Minic, S.L.; Stanic-Vucinic, D.; Mihailovic, J.; Krstic, M.; Nikolic, M.R.; Cirkovic-Velickovic, T. Digestion by pepsin releases biologically active chromopeptides from C-phycocyanin, a blue-colored biliprotein of microalga Spirulina. J. Proteom. 2016, 147, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Lo, E.H. A new penumbra: Transitioning from injury into repair after stroke. Nat. Med. 2008, 14, 497–500. [Google Scholar] [CrossRef] [PubMed]
- Farkas, E.; Luiten, P.G.; Bari, F. Permanent, bilateral common carotid artery occlusion in the rat: A model for chronic cerebral hypoperfusion-related neurodegenerative diseases. Brain Res. Rev. 2007, 54, 162–180. [Google Scholar] [CrossRef] [PubMed]
- Marín-Prida, J.; Pavón-Fuentes, N.; Llópiz-Arzuaga, A.; Fernández-Massó, J.R.; Delgado-Roche, L.; Mendoza-Marí, Y.; Santana, S.P.; Cruz-Ramírez, A.; Valenzuela-Silva, C.; Nazábal-Gálvez, M.; et al. Phycocyanobilin promotes PC12 cell survival and modulates immune and inflammatory genes and oxidative stress markers in acute cerebral hypoperfusion in rats. Toxicol. Appl. Pharmacol. 2013, 272, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Liesz, A.; Suri-Payer, E.; Veltkamp, C.; Doerr, H.; Sommer, C.; Rivest, S.; Giese, T.; Veltkamp, R. Regulatory T cells are key cerebroprotective immunomodulators in acute experimental stroke. Nat. Med. 2009, 15, 192–199. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Akiyoshi, K.; Vandenbark, A.A.; Hurn, P.D.; Offner, H. CD4+ FoxP3+ regulatory T-cells in cerebral ischemic stroke. Metab. Brain Dis. 2011, 26, 87–90. [Google Scholar] [CrossRef] [PubMed]
- O’Garra, A.; Vieira, P. Regulatory T cells and mechanisms of immune system control. Nat. Med. 2004, 10, 801–805. [Google Scholar] [CrossRef] [PubMed]
- Dobolyi, A.; Vincze, C.; Pál, G.; Lovas, G. The neuroprotective functions of transforming growth factor Beta proteins. Int. J. Mol. Sci. 2012, 13, 8219–8258. [Google Scholar] [CrossRef] [PubMed]
- Palazuelos, J.; Klingener, M.; Aguirre, A. TGFβ signaling regulates the timing of CNS myelination by modulating oligodendrocyte progenitor cell cycle exit through SMAD3/4/FoxO1/Sp1. J. Neurosci. 2014, 34, 7917–7930. [Google Scholar] [CrossRef] [PubMed]
- Kolbinger, F.; Huppertz, C.; Mir, A.; Padova, F.D. IL-17A and Multiple Sclerosis: Signaling pathways, producing cells and target cells in the Central Nervous System. Curr. Drug Targets 2016, 17, 1882–1893. [Google Scholar] [CrossRef] [PubMed]
- Kang, Z.; Liu, L.; Spangler, R.; Spear, C.; Wang, C.; Gulen, M.F.; Veenstra, M.; Ouyang, W.; Ransohoff, R.M.; Li, X. IL-17-induced Act1-mediated signaling is critical for cuprizone-induced demyelination. J. Neurosci. 2012, 32, 8284–8292. [Google Scholar] [CrossRef] [PubMed]
- Baxi, E.G.; DeBruin, J.; Tosi, D.M.; Grishkan, I.V.; Smith, M.D.; Kirby, L.A.; Strasburger, H.J.; Fairchild, A.N.; Calabresi, P.A.; Gocke, A.R. Transfer of myelin-reactive th17 cells impairs endogenous remyelination in the central nervous system of cuprizone-fed mice. J. Neurosci. 2015, 35, 8626–8639. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhang, J.; Wang, L.; Chen, Y.; Wan, Y.; He, Y.; Jiang, L.; Ma, J.; Liao, R.; Zhang, X.; et al. Interleukin-1β impedes oligodendrocyte progenitor cell recruitment and white matter repair following chronic cerebral hypoperfusion. Brain Behav. Immun. 2017, 60, 93–105. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Giammarco, M.L.; De Nuccio, C.; Ajmone-Cat, M.A.; Visentin, S.; De Simone, R.; Minghetti, L. Docosahexaenoic acid promotes oligodendrocyte differentiation via PPAR-γ signalling and prevents tumor necrosis factor-α-dependent maturational arrest. Biochim. Biophys. Acta 2017, 1862, 1013–1023. [Google Scholar] [CrossRef] [PubMed]
- Wakita, H.; Tomimoto, H.; Akiguchi, I.; Matsuo, A.; Lin, J.X.; Ihara, M.; McGeer, P.L. Axonal damage and demyelination in the white matter after chronic cerebral hypoperfusion in the rat. Brain Res. 2002, 924, 63–70. [Google Scholar] [CrossRef]
- Bartolucci, M.; Ravera, S.; Garbarino, G.; Ramoino, P.; Ferrando, S.; Calzia, D.; Candiani, S.; Morelli, A.; Panfoli, I. Functional expression of electron transport chain and FoF1-ATP synthase in optic nerve myelin sheath. Neurochem. Res. 2015, 40, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, S.; Castelvetri, L.C.; Simons, M. Metabolism and functions of lipids in myelin. Biochim. Biophys. Acta 2015, 1851, 999–1005. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, S.; Benvenuti, F.; Scoglio, S.; Canestrari, F. Oxygen radical absorbance capacity of phycocyanin and phycocyanobilin from the food supplement Aphanizomenon flos-aquae. J. Med. Food 2010, 13, 223–227. [Google Scholar] [CrossRef] [PubMed]
- Adibhatla, R.M.; Hatcher, J.F. Lipid oxidation and peroxidation in CNS health and disease: From molecular mechanisms to therapeutic opportunities. Antioxid. Redox Signal. 2010, 12, 125–169. [Google Scholar] [CrossRef] [PubMed]
- Windle, V.; Szymanska, A.; Granter-Button, S.; White, C.; Buist, R.; Peeling, J.; Corbett, D. An analysis of four different methods of producing focal cerebral ischemia with endothelin-1 in the rat. Exp. Neurol. 2006, 201, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Huang, Y.; Zhang, R.; Cai, T.; Cai, Y. Medical application of Spirulina platensis derived C-Phycocyanin. Evid. Based Complement. Altern. Med. 2016, 2016, 7803846. [Google Scholar] [CrossRef]
- Pentón-Rol, G.; Marín-Prida, J.; Pardo-Andreu, G.; Martínez-Sánchez, G.; Acosta-Medina, E.F.; Valdivia-Acosta, A.; Lagumersindez-Denis, N.; Rodríguez-Jiménez, E.; Llópiz-Arzuaga, A.; López-Saura, P.A.; et al. C-Phycocyanin is neuroprotective against global cerebral ischemia/reperfusion injury in gerbils. Brain Res. Bull. 2011, 86, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Marín-Prida, J. Neuroprotective Effects and Molecular Mechanisms Associated to C-Phycocyanin and Phycocyanobilin in Experimental Models of Cerebral Ischemia. Ph.D. Thesis, University of Medical Sciences of Havana, Havana, Cuba, 2015. Open Access Repository of Ph.D. Thesis of the Virtual Library of Health, Cuba. Available online: http://tesis.repo.sld.cu/901/ (accessed on 12 May 2015). (In Spanish).
- Bermejo-Bescós, P.; Piñero-Estrada, E.; Villar del Fresno, A.M. Neuroprotection by Spirulina platensis protean extract and phycocyanin against iron-induced toxicity in SH-SY5Y neuroblastoma cells. Toxicol. In Vitro 2008, 22, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Marín-Prida, J.; Pentón-Rol, G.; Rodrigues, F.P.; Alberici, L.C.; Stringhetta, K.; Leopoldino, A.M.; Naal, Z.; Polizello, A.C.; Llópiz-Arzuaga, A.; Rosa, M.N.; et al. C-Phycocyanin protects SH-SY5Y cells from oxidative injury, rat retina from transient ischemia and rat brain mitochondria from Ca2+/phosphate-induced impairment. Brain Res. Bull. 2012, 89, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Rimbau, V.; Camins, A.; Pubill, D.; Sureda, F.X.; Romay, C.; González, R.; Jiménez, A.; Escubedo, E.; Camarasa, J.; Pallàs, M. C-phycocyanin protects cerebellar granule cells from low potassium/serum deprivation-induced apoptosis. Naunyn Schmiedebergs Arch. Pharmacol. 2001, 364, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Rimbau, V.; Camins, A.; Romay, C.; González, R.; Pallàs, M. Protective effects of C-phycocyanin against kainic acid-induced neuronal damage in rat hippocampus. Neurosci. Lett. 1999, 276, 75–78. [Google Scholar] [CrossRef]
- Hwang, J.H.; Chen, J.C.; Chan, Y.C. Effects of C-phycocyanin and Spirulina on salicylate-induced tinnitus, expression of NMDA receptor and inflammatory genes. PLoS ONE 2013, 8, e58215. [Google Scholar] [CrossRef] [PubMed]
- Min, S.K.; Park, J.S.; Luo, L.; Kwon, Y.S.; Lee, H.C.; Shim, H.J.; Kim, I.D.; Lee, J.K.; Shin, H.S. Assessment of C-phycocyanin effect on astrocytes-mediated neuroprotection against oxidative brain injury using 2D and 3D astrocyte tissue model. Sci. Rep. 2015, 5, 14418. [Google Scholar] [CrossRef] [PubMed]
- Pires, A.; Fortuna, A.; Alves, G.; Falcão, A. Intranasal drug delivery: How, why and what for? J. Pharm. Pharm. Sci. 2009, 12, 288–311. [Google Scholar] [CrossRef] [PubMed]
- Tymianski, M. Neuroprotective therapies: Preclinical reproducibility is only part of the problem. Sci. Transl. Med. 2015, 7, 299fs32. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Xiong, L.J.; Tong, Y.; Mao, M. The neuroprotective roles of BDNF in hypoxic ischemic brain injury. Biomed. Rep. 2013, 1, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Howe, C.L.; Mobley, W.C. Nerve growth factor signaling, neuroprotection and neural repair. Annu. Rev. Neurosci. 2001, 24, 1217–1281. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Maki, T.; Shindo, A.; Liang, A.C.; Maeda, M.; Egawa, N.; Itoh, K.; Lo, E.K.; Lok, J.; Ihara, M.; et al. Astrocytes promote oligodendrogenesis after white matter damage via Brain-Derived Neurotrophic Factor. J. Neurosci. 2015, 35, 14002–14008. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pentón-Rol, G.; Marín-Prida, J.; Falcón-Cama, V. C-Phycocyanin and Phycocyanobilin as Remyelination Therapies for Enhancing Recovery in Multiple Sclerosis and Ischemic Stroke: A Preclinical Perspective. Behav. Sci. 2018, 8, 15. https://doi.org/10.3390/bs8010015
Pentón-Rol G, Marín-Prida J, Falcón-Cama V. C-Phycocyanin and Phycocyanobilin as Remyelination Therapies for Enhancing Recovery in Multiple Sclerosis and Ischemic Stroke: A Preclinical Perspective. Behavioral Sciences. 2018; 8(1):15. https://doi.org/10.3390/bs8010015
Chicago/Turabian StylePentón-Rol, Giselle, Javier Marín-Prida, and Viviana Falcón-Cama. 2018. "C-Phycocyanin and Phycocyanobilin as Remyelination Therapies for Enhancing Recovery in Multiple Sclerosis and Ischemic Stroke: A Preclinical Perspective" Behavioral Sciences 8, no. 1: 15. https://doi.org/10.3390/bs8010015
APA StylePentón-Rol, G., Marín-Prida, J., & Falcón-Cama, V. (2018). C-Phycocyanin and Phycocyanobilin as Remyelination Therapies for Enhancing Recovery in Multiple Sclerosis and Ischemic Stroke: A Preclinical Perspective. Behavioral Sciences, 8(1), 15. https://doi.org/10.3390/bs8010015