
Peripheral Inflammatory Markers Contributing to Comorbidities in Autism

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Subjects
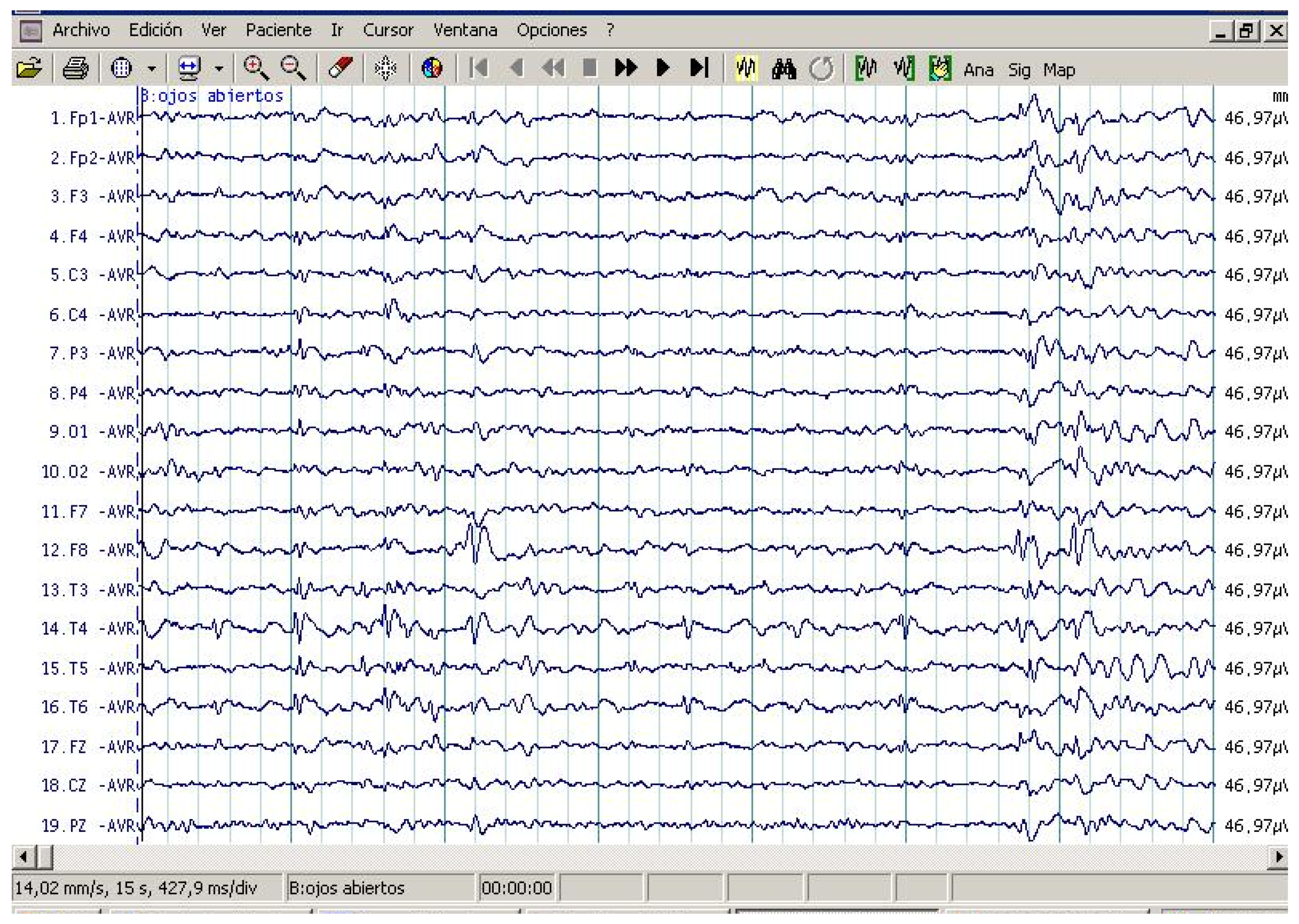
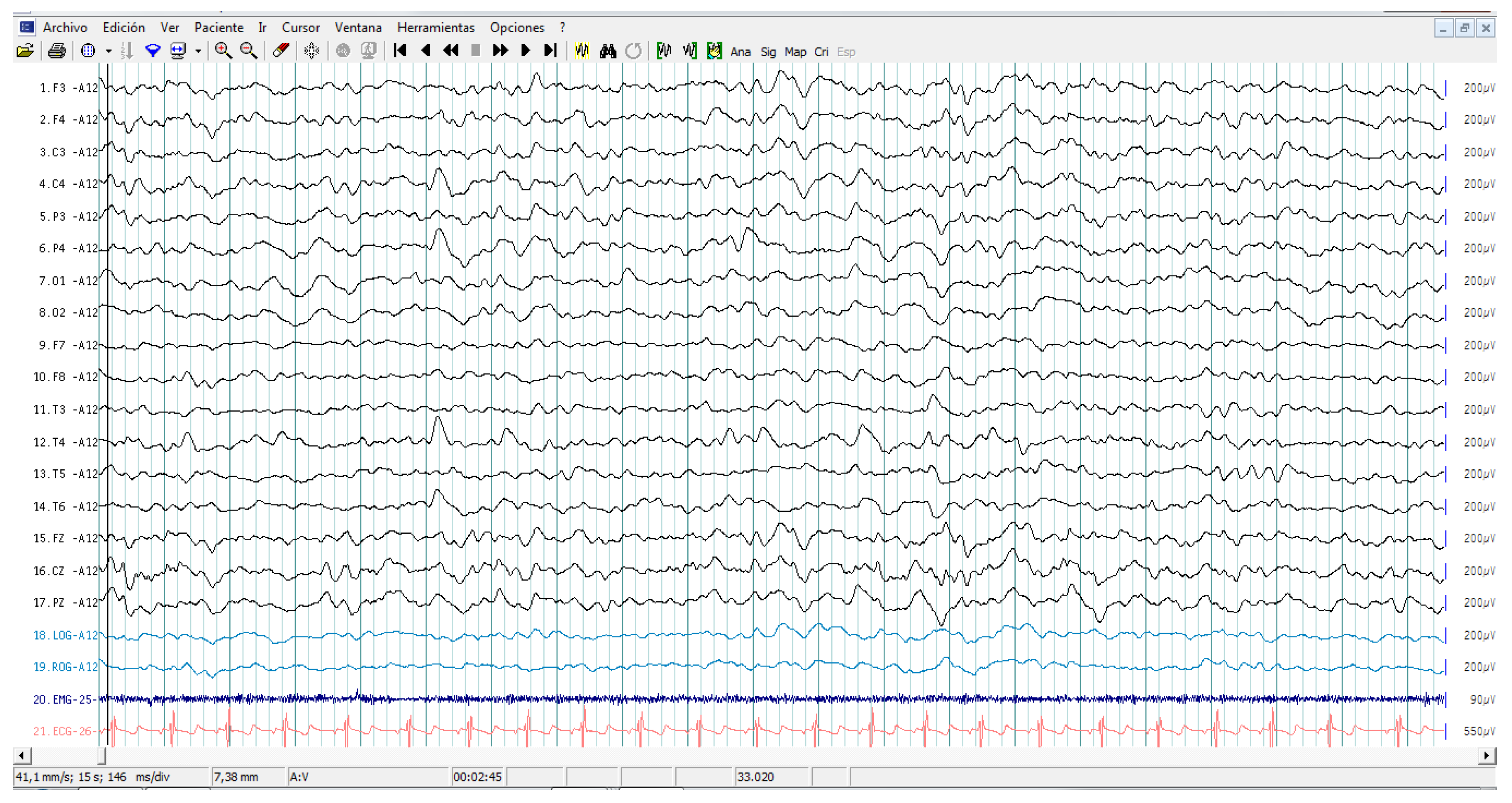
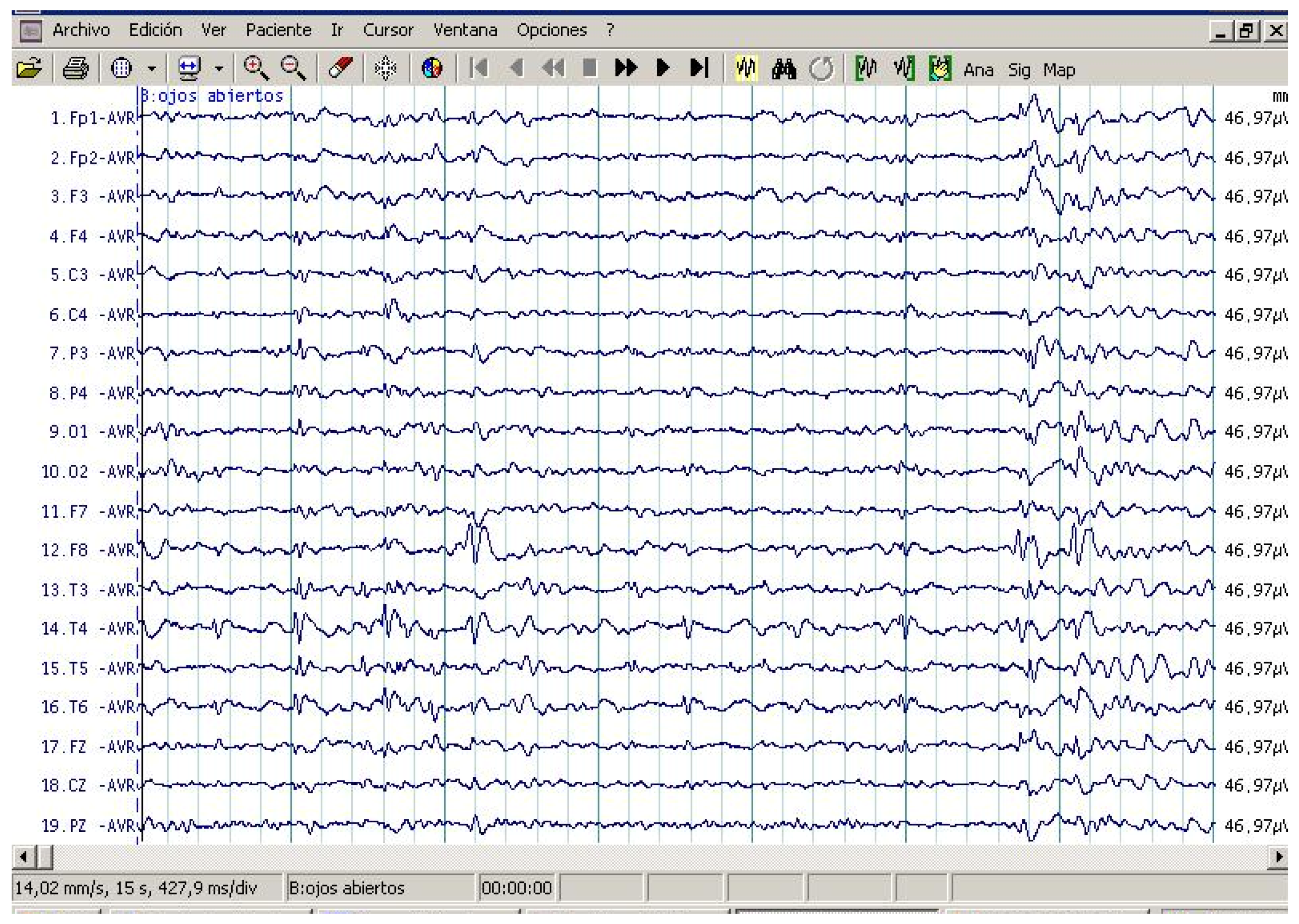
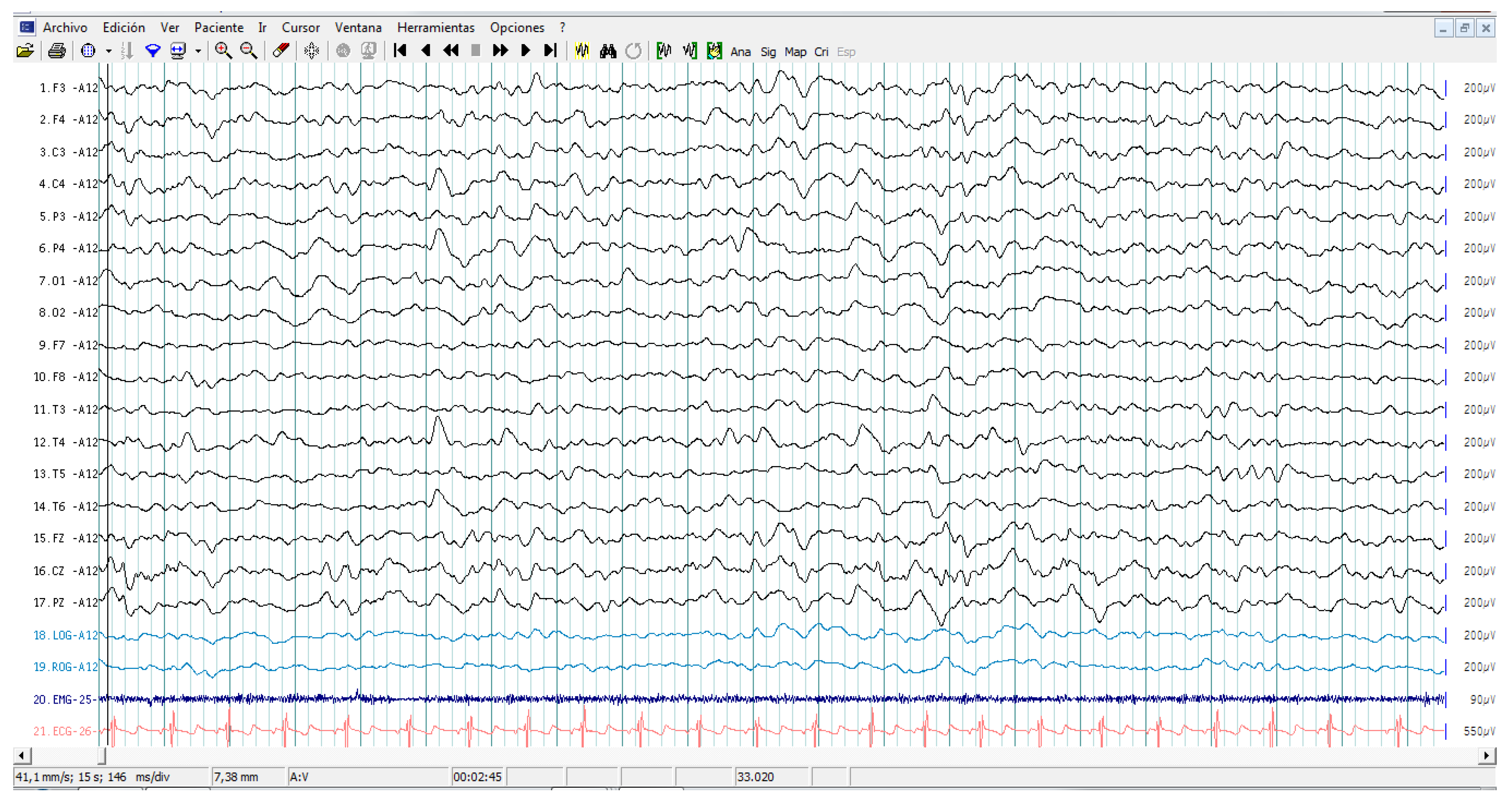
2.2. EEG Assessments
2.3. Cytokine Profile Measurement
2.3.1. Samples Collection
2.3.2. Cytokine Analysis
2.4. Data Analysis
3. Results
3.1. Clinical and Demography Description of the Group
3.2. EEG Assessment Records in Autistic Patients
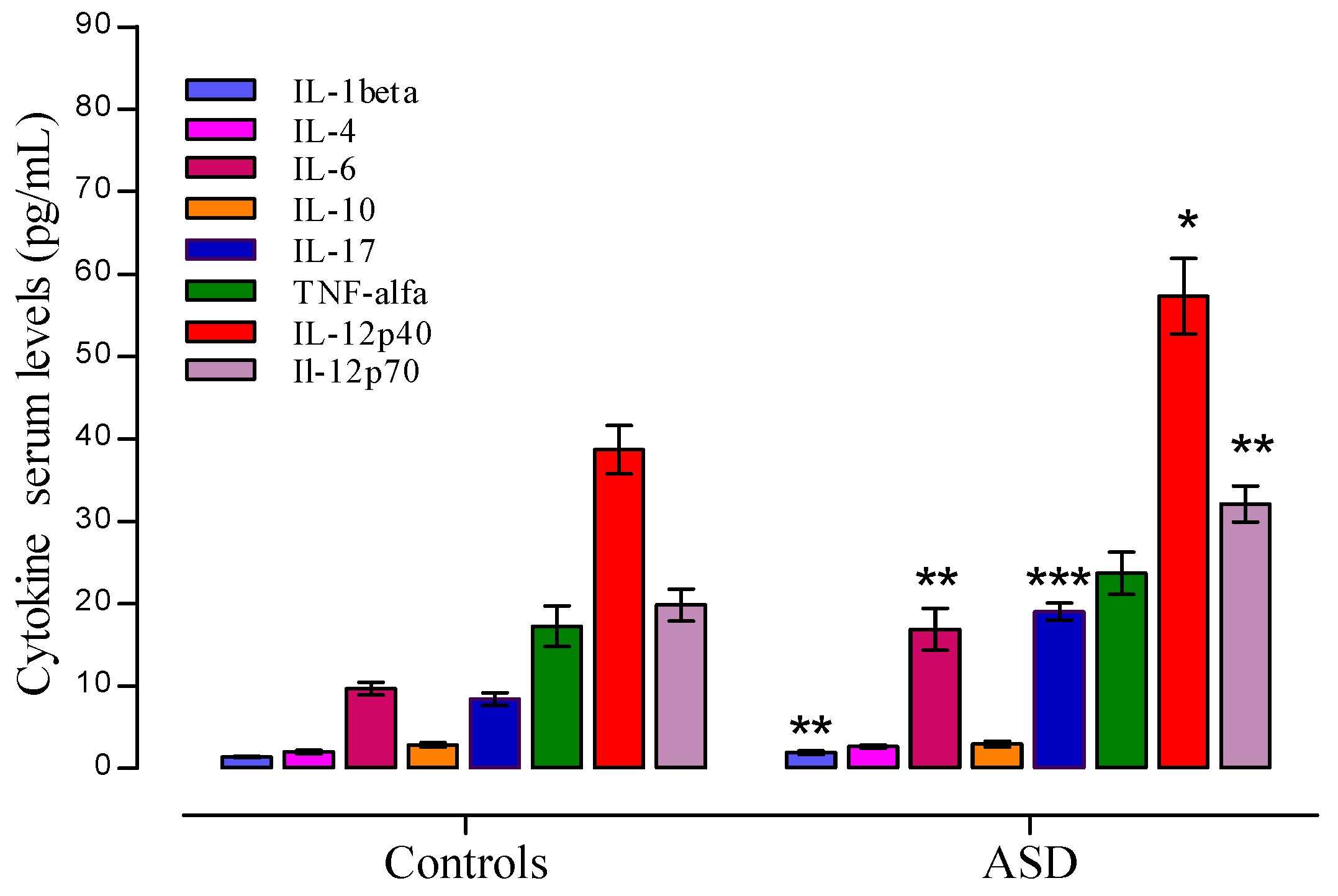
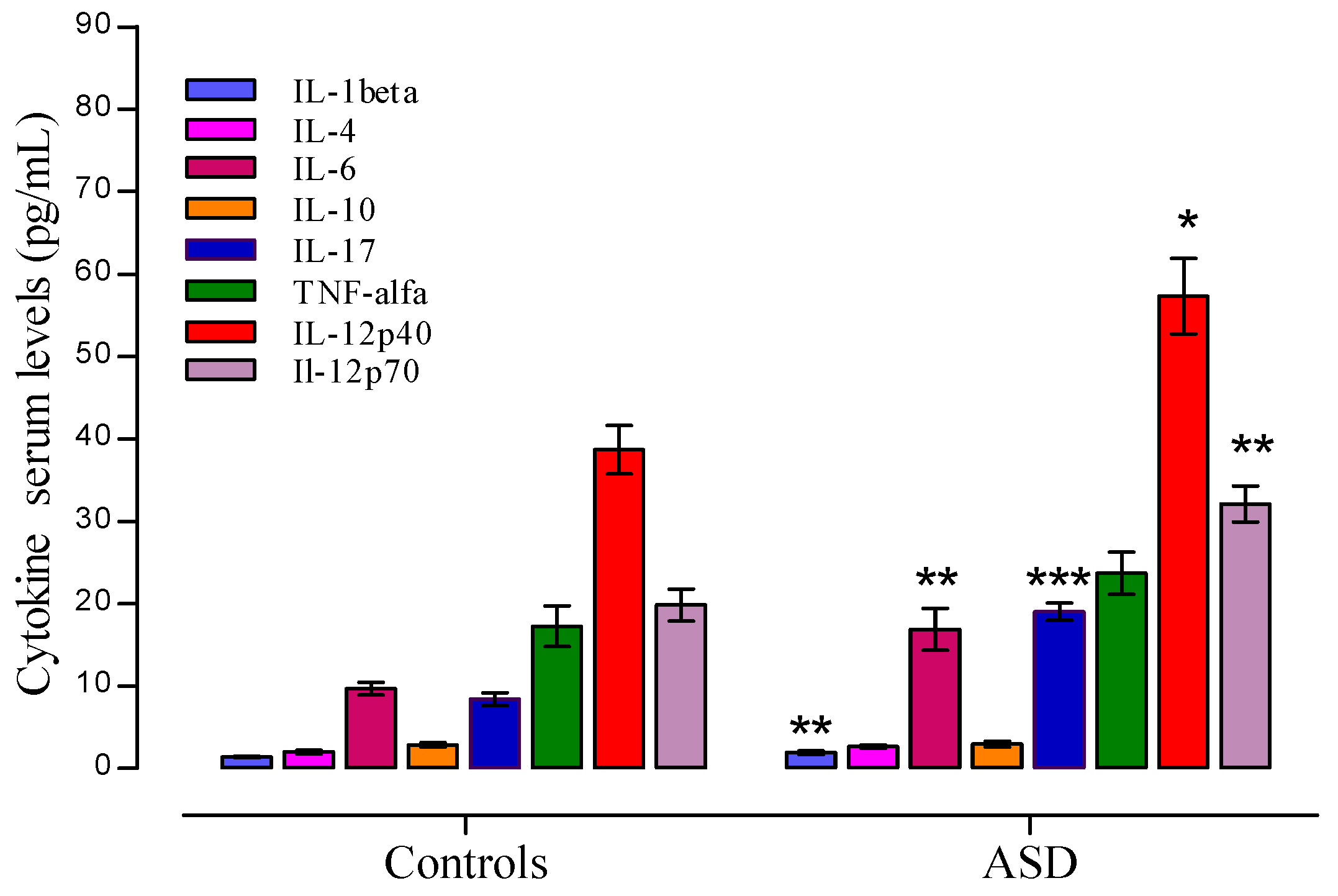
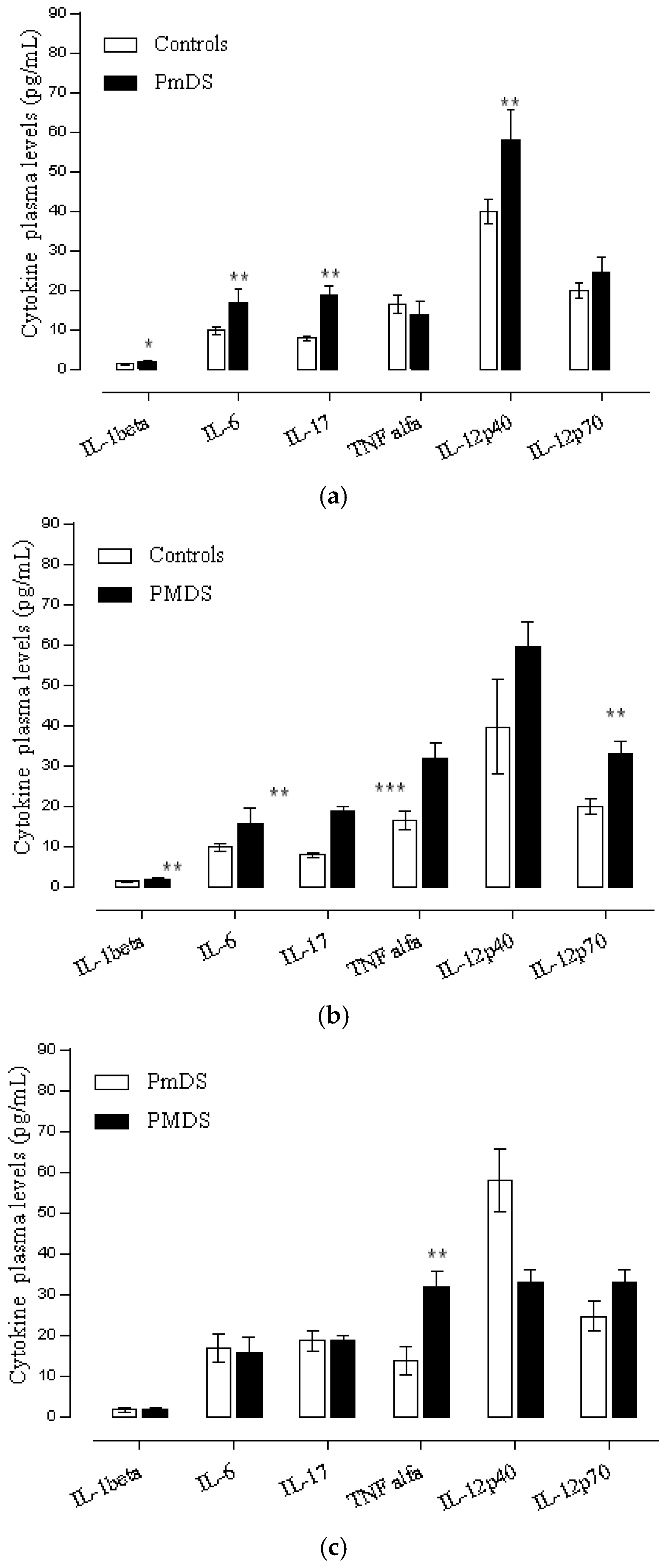
3.3. Cytokine Profile Level in ASD
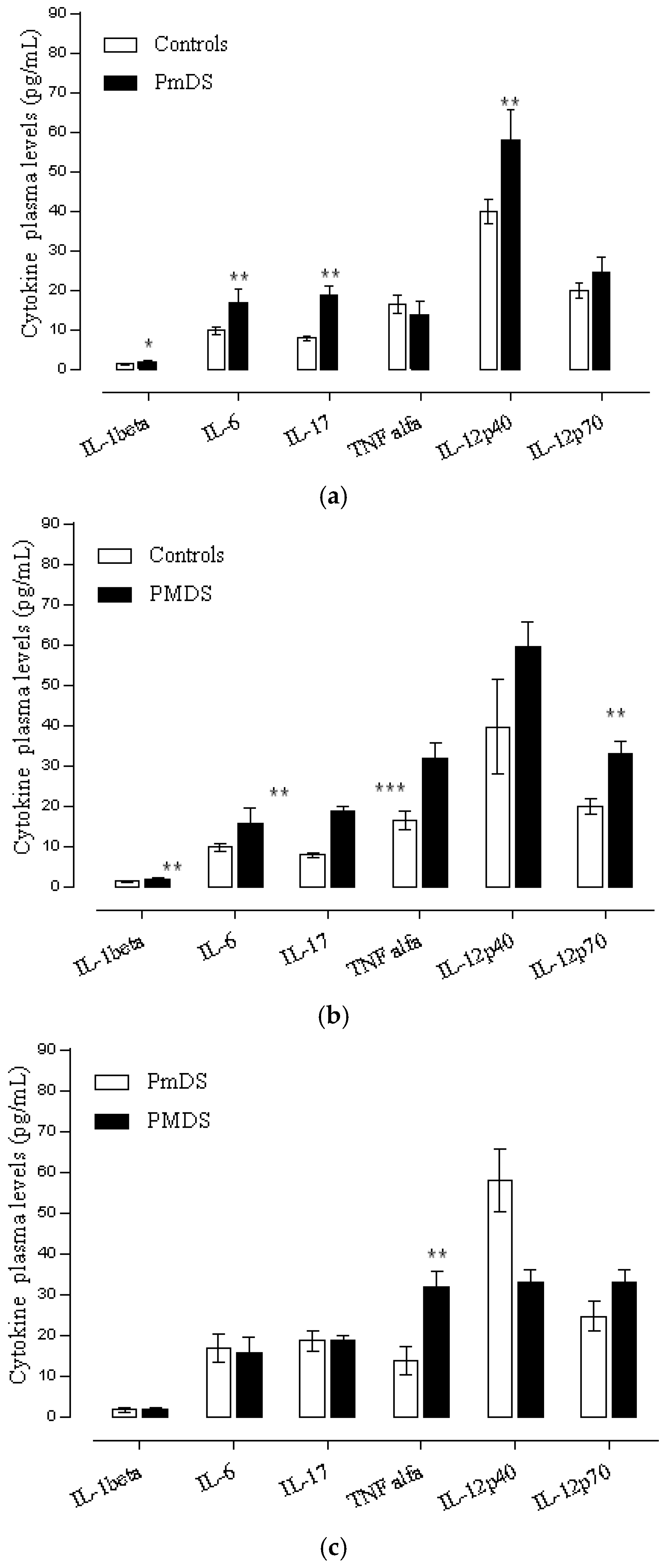
3.4. Cytokine Profile and Behavioral Clinical Outcomes in Autistic Group
Cytokine Profile and Disease Severity in Autistic Patients
3.5. Cytokine Profile and EEG Findings in Autistic Patients
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lintas, C.; Persico, A.M. Autistic phenotypes and genetic testing: State-of-the-art for the clinical geneticist. J. Med. Genet. 2009, 46, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.; Haapanen, L.; Boyce, R.; Duncanson, P.; Braunschweig, D.; Delwiche, L.; Hansen, R.; Hertz-Picciotto, I.; Ashwood, P.; van de Water, J. Autoantibodies to cerebellum in children with autism associate with behavior. Brain Behav. Immun. 2011, 25, 514–523. [Google Scholar] [CrossRef] [PubMed]
- Garay, P.A.; McAllister, A.K. Novel roles for immune molecules in neural development: Implications for neurodevelopmental disorders. Front. Synaptic. Neurosci. 2010, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Aktas, O.; Smorodchenko, A.; Brocke, S.; Infante-Duarte, C.; Schulze, T.U.; Vogt, J.; Prozorovski, T.; Meier, S.; Osmanova, V.; Pohl, E.; et al. Neuronal damage in autoimmune neuroinflammation mediated by the death ligand TRAIL. Neuron 2005, 46, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Arima, Y.; Harada, M.; Kamimura, D.; Park, J.H.; Kawano, F.; Yull, F.E.; Kawamoto, T.; Iwakura, Y.; Betz, U.A.; Marquez, G.; et al. Regional neural activation defines a gateway for autoreactive T cells to cross the blood-brain barrier. Cell 2012, 148, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Odoardi, F.; Sie, C.; Streyl, K.; Ulaganathan, V.K.; Schlager, C.; Lodygin, D.; Heckelsmiller, K.; Nietfeld, W.; Ellwart, J.; Klinkert, W.E.; et al. T cells become licensed in the lung to enter the central nervous system. Nature 2012, 488, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Aube, B.; Levesque, S.A.; Pare, A.; Chamma, E.; Kebir, H.; Gorina, R.; Lecuyer, M.A.; Alvarez, J.I.; de Koninck, Y.; Engelhardt, B.; et al. Neutrophils mediate blood-spinal cord barrier disruption in demyelinating neuroinflammatory diseases. J. Immunol. 2014, 193, 2438–2454. [Google Scholar] [CrossRef] [PubMed]
- Mostafa, G.A.; Al-Ayadhi, L.Y. The relationship between the increased frequency of serum antineuronal antibodies and the severity of autism in children. Eur. J. Paediatr. Neurol. 2012, 16, 464–468. [Google Scholar] [CrossRef] [PubMed]
- Careaga, M.; Ashwood, P. Autism spectrum disorders: From immunity to behavior. Methods Mol. Biol. 2012, 934, 219–240. [Google Scholar] [PubMed]
- Enstrom, A.M.; Onore, C.E.; van de Water, J.A.; Ashwood, P. Differential monocyte responses to TLR ligands in children with autism spectrum disorders. Brain Behav. Immun. 2010, 24, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.K.; Fortino, V.R.; Pelaez, D.; Cheung, H.S. Progress of mesenchymal stem cell therapy for neural and retinal diseases. World J. Stem Cells 2014, 6, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Hirtz, D.; Mamounas, L.A. NINDS epilepsy and autism spectrum disorders workshop report. Neurology 2013, 81, 1630–1636. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R. Autism and social cognition in epilepsy: Implications for comprehensive epilepsy care. Curr. Opin. Neurol. 2013, 26, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Cuccaro, M. Epilepsy and autism: Neurodevelopmental perspective. Curr. Neurol. Neurosci. Rep. 2011, 11, 428–434. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Alessandri, M.; Cuccaro, M. Autism spectrum disorders and epilepsy: Moving towards a comprehensive approach to treatment. Brain Dev. 2010, 32, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Moshe, S.L.; Rapin, I. Convulsing toward the pathophysiology of autism. Brain Dev. 2009, 31, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Khandaker, G.M.; Stochl, J.; Zammit, S.; Lewis, G.; Jones, P.B. A population-based longitudinal study of childhood neurodevelopmental disorders, IQ and subsequent risk of psychotic experiences in adolescence. Psychol. Med. 2014, 44, 3229–3238. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Rapin, I. Epilepsy in autism. Lancet Neurol. 2002, 1, 352–358. [Google Scholar] [CrossRef]
- Autism and Developmental Disabilities Monitoring Network Surveillance Year 2008 Principal Investigators; Centers for Disease Control and Prevention. Prevalence of autism spectrum disorders—Autism and Developmental Disabilities Monitoring Network, 14 sites, United States, 2008. MMWR Surveill. Summ. 2012, 61, 1–19. [Google Scholar]
- McKnight, W.; Shafer, E.; Wray, C. New autism spectrum criteria proposed: Some who qualify for diagnosis might not after 2013. Pediatr. Ann. 2012, 41, 89. [Google Scholar] [CrossRef]
- Battle, D.E. Diagnostic and Statistical Manual of Mental Disorders (DSM). Codas 2013, 25, 191–192. [Google Scholar] [CrossRef] [PubMed]
- Beighley, J.S.; Matson, J.L.; Rieske, R.D.; Jang, J.; Cervantes, P.E.; Goldin, R.L. Comparing challenging behavior in children diagnosed with autism spectrum disorders according to the DSM-IV-TR and the proposed DSM-5. Dev. Neurorehabil. 2013, 16, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Lopez, C.; Narbona, J. Clinical usefulness of IDEA and CARS: Concordance with DSM-IV-TR in children and adolescents with suspicion of PDD. An. Pediatr. (Barc.) 2014, 80, 71–76. [Google Scholar] [PubMed]
- George, B.; Padmam, M.S.; Nair, M.K.; Leena, M.L.; Prasanna, G.L.; Russell, P.S. CDC Kerala 11: Diagnosis of autism among children between 2 and 6 y—Comparison of CARS against DSM-IV-TR. Indian J. Pediatr. 2014, 81 (Suppl. 2), S125–S128. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.; van de Water, J. The immune system’s role in the biology of autism. Curr. Opin. Neurol. 2010, 23, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, P.A.; Dingman, A.L.; Palmer, T.D. Placental TNF-alpha signaling in illness-induced complications of pregnancy. Am. J. Pathol. 2011, 178, 2802–2810. [Google Scholar] [CrossRef] [PubMed]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [PubMed]
- Ramos, P.S.; Sajuthi, S.; Langefeld, C.D.; Walker, S.J. Immune function genes CD99L2, JARID2 and TPO show association with autism spectrum disorder. Mol. Autism 2012, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Mondal, K.; Ramachandran, D.; Patel, V.C.; Hagen, K.R.; Bose, P.; Cutler, D.J.; Zwick, M.E. Excess variants in AFF2 detected by massively parallel sequencing of males with autism spectrum disorder. Hum. Mol. Genet. 2012, 21, 4356–4364. [Google Scholar] [CrossRef] [PubMed]
- Skafidas, E.; Testa, R.; Zantomio, D.; Chana, G.; Everall, I.P.; Pantelis, C. Predicting the diagnosis of autism spectrum disorder using gene pathway analysis. Mol. Psychiatry 2014, 19, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [PubMed]
- De Theije, C.G.; Wu, J.; da Silva, S.L.; Kamphuis, P.J.; Garssen, J.; Korte, S.M.; Kraneveld, A.D. Pathways underlying the gut-to-brain connection in autism spectrum disorders as future targets for disease management. Eur. J. Pharmacol. 2011, 668 (Suppl. 1), S70–S80. [Google Scholar] [CrossRef] [PubMed]
- De Theije, C.G.; Koelink, P.J.; Korte-Bouws, G.A.; Lopes da, S.S.; Korte, S.M.; Olivier, B.; Garssen, J.; Kraneveld, A.D. Intestinal inflammation in a murine model of autism spectrum disorders. Brain Behav. Immun. 2014, 37, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Garbett, K.; Ebert, P.J.; Mitchell, A.; Lintas, C.; Manzi, B.; Mirnics, K.; Persico, A.M. Immune transcriptome alterations in the temporal cortex of subjects with autism. Neurobiol. Dis. 2008, 30, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Manzardo, A.M.; Henkhaus, R.; Dhillon, S.; Butler, M.G. Plasma cytokine levels in children with autistic disorder and unrelated siblings. Int. J. Dev. Neurosci. 2012, 30, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Onore, C.; van de Water, J.; Ashwood, P. Decreased levels of EGF in plasma of children with autism spectrum disorder. Autism Res. Treat. 2012, 2012, 205362. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.K.; Warren, R.P.; Odell, J.D.; Cole, P. Changes of soluble interleukin-2, interleukin-2 receptor, T8 antigen, and interleukin-1 in the serum of autistic children. Clin. Immunol. Immunopathol. 1991, 61, 448–455. [Google Scholar] [CrossRef]
- Al-Ayadhi, L.Y.; Mostafa, G.A. A lack of association between elevated serum levels of S100B protein and autoimmunity in autistic children. J. Neuroinflamm. 2012, 9, 54. [Google Scholar] [CrossRef] [PubMed]
- Abrahams, B.S.; Geschwind, D.H. Advances in autism genetics: On the threshold of a new neurobiology. Nat. Rev. Genet. 2008, 9, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, A.A.; Deuster, P.A.; Francis, J.L.; Bonsall, R.W.; Tracy, R.P.; Kop, W.J. Neurohormonal and inflammatory hyper-responsiveness to acute mental stress in depression. Biol. Psychol. 2010, 84, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Petitto, J.M.; Huang, Z.; Meola, D.; Ha, G.K.; Dauer, D. Interleukin-2 and the septohippocampal system: Intrinsic actions and autoimmune processes relevant to neuropsychiatric disorders. Methods Mol. Biol. 2012, 829, 433–443. [Google Scholar] [PubMed]
- Pucak, M.L.; Carroll, K.A.; Kerr, D.A.; Kaplin, A.I. Neuropsychiatric manifestations of depression in multiple sclerosis: Neuroinflammatory, neuroendocrine, and neurotrophic mechanisms in the pathogenesis of immune-mediated depression. Dialogues Clin. Neurosci. 2007, 9, 125–139. [Google Scholar] [PubMed]
- Vezzani, A.; Friedman, A. Brain inflammation as a biomarker in epilepsy. Biomark. Med. 2011, 5, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Li, X.; Zhong, Y. Inflammatory Cytokines: Potential Biomarkers of Immunologic Dysfunction in Autism Spectrum Disorders. Mediat. Inflamm. 2015, 2015, 531518. [Google Scholar] [CrossRef] [PubMed]
- Goines, P.E.; Ashwood, P. Cytokine dysregulation in autism spectrum disorders (ASD): Possible role of the environment. Neurotoxicol. Teratol. 2013, 36, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Enstrom, A.M.; van de Water, J.A.; Ashwood, P. Autoimmunity in autism. Curr. Opin. Investig. Drugs 2009, 10, 463–473. [Google Scholar] [PubMed]
- Vojdani, A. The Role of Th17 in Neuroimmune Disorders: A Target for CAM Therapy. Part III. Evid.-Based Complement. Altern. Med. 2011, 2011, 548086. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Agramonte, M.A.; Gonzalez-Quevedo, A.; Goncalvez, C.A. Glial and Axonal Pathology in Multiple Sclerosis. In Immunology to Target Cancer Inflammation and Infections; Kanwar, J.R., Ed.; InTech: Rijeka, Croatia, 2012; pp. 103–131. [Google Scholar]
- Banaschewski, T.; Poustka, L.; Holtmann, M. Autism and ADHD across the life span. Differential diagnoses or comorbidity? Nervenarzt 2011, 82, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Bonus, B.; Assion, H.J.; Deister, A. Coincidence of epilepsy and Asperger syndrome. Case report and review. Nervenarzt 1997, 68, 759–764. [Google Scholar] [CrossRef] [PubMed]
- Chiang, H.L.; Gau, S.S. Comorbid psychiatric conditions as mediators to predict later social adjustment in youths with autism spectrum disorder. J. Child Psychol. Psychiatry 2015. [Google Scholar] [CrossRef] [PubMed]
- Areta, J.E. Two main mistakes in classificatory and clinical function of DSM-IV, in the case of differential diagnosis between Asperger and autistic disorder. Vertex 2009, 20, 174–183. [Google Scholar] [PubMed]
- Clarke, A.R.; Barry, R.J.; Dupuy, F.E.; Heckel, L.D.; McCarthy, R.; Selikowitz, M.; Johnstone, S.J. Behavioural differences between EEG-defined subgroups of children with Attention-Deficit/Hyperactivity Disorder. Clin. Neurophysiol. 2011, 122, 1333–1341. [Google Scholar] [CrossRef] [PubMed]
- Ballaban-Gil, K.; Tuchman, R. Epilepsy and epileptiform EEG: Association with autism and language disorders. Ment. Retard. Dev. Disabil. Res. Rev. 2000, 6, 300–308. [Google Scholar] [CrossRef]
- Musso, T.; Espinoza-Delgado, I.; Pulkki, K.; Gusella, G.L.; Longo, D.L.; Varesio, L. IL-2 induces IL-6 production in human monocytes. J. Immunol. 1992, 148, 795–800. [Google Scholar] [PubMed]
- Casanova, M.F.; van Kooten, I.A.; Switala, A.E.; van Engeland, E.H.; Heinsen, H.; Steinbusch, H.W.; Hof, P.R.; Trippe, J.; Stone, J.; Schmitz, C. Minicolumnar abnormalities in autism. Acta Neuropathol. 2006, 112, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Casanova, M.F.; Sokhadze, E.; Opris, I.; Wang, Y.; Li, X. Autism spectrum disorders: Linking neuropathological findings to treatment with transcranial magnetic stimulation. Acta Paediatr. 2015, 104, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.R.; Hou, H.; Obregon, D.F.; Tian, J.; Zhu, Y.; Zou, Q.; Nikolic, W.V.; Bengtson, M.; Mori, T.; Murphy, T.; et al. Aberrant T-lymphocyte development and function in mice overexpressing human soluble amyloid precursor protein-alpha: Implications for autism. FASEB J. 2012, 26, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.; Luthert, P.; Dean, A.; Harding, B.; Janota, I.; Montgomery, M.; Rutter, M.; Lantos, P. A clinicopathological study of autism. Brain 1998, 121, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sokhadze, E.M.; El-Baz, A.S.; Li, X.; Sears, L.; Casanova, M.F.; Tasman, A. Relative Power of Specific EEG Bands and Their Ratios during Neurofeedback Training in Children with Autism Spectrum Disorder. Front. Hum. Neurosci. 2016, 9, 723. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R. Autism and epilepsy: What has regression got to do with it? Epilepsy Curr. 2006, 6, 107–111. [Google Scholar] [CrossRef] [PubMed]
- Napolioni, V.; Ober-Reynolds, B.; Szelinger, S.; Corneveaux, J.J.; Pawlowski, T.; Ober-Reynolds, S.; Kirwan, J.; Persico, A.M.; Melmed, R.D.; Craig, D.W.; et al. Plasma cytokine profiling in sibling pairs discordant for autism spectrum disorder. J. Neuroinflamm. 2013, 10, 38. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; Marventano, I.; Guerini, F.R.; Mancuso, R.; Ceresa, L.; Zanzottera, M.; Rusconi, B.; Maggioni, E.; Tinelli, C.; Clerici, M. An autistic endophenotype results in complex immune dysfunction in healthy siblings of autistic children. Biol. Psychiatry 2009, 66, 978–984. [Google Scholar] [CrossRef] [PubMed]
- Tuchman, R.; Cuccaro, M.; Alessandri, M. Autism and epilepsy: Historical perspective. Brain Dev. 2010, 32, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Robinson-Agramonte, M.A.; Gonzalez-Fraguela, M.E.; Bergado-Rosado, J. Chapter 7 Autism, Development and Neural Plasticity. In Translational Approaches to Autism Spectrum Disorder; Robinson-Agramonte, M.A., Ed.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 119–135. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | Age (Years) | Sex | CARS Score | Severity |
|---|---|---|---|---|
| 1 | 9 | M | 26 | Mild |
| 2 | 7.8 | M | 35.5 | Moderate |
| 3 | 7.8 | M | 36.5 | Moderate |
| 4 | 5 | M | 33 | Moderate |
| 5 | 6.1 | M | 36 | Moderate |
| 6 | 4 | F | 46 | Severe |
| 7 | 6.6 | F | 33.5 | Moderate |
| 8 | 4 | M | 36 | Moderate |
| 9 | 9.1 | M | 28 | Mild |
| 10 | 8 | M | 37 | Moderate |
| 11 | 5.2 | F | 36.5 | Moderate |
| 12 | 3 | M | 35.5 | Moderate |
| 13 | 3 | F | 27 | Mild |
| 14 | 7.2 | M | 34.5 | Moderate |
| 15 | 9.1 | M | 35 | Moderate |
| 16 | 5 | F | 30 | Mild |
| 17 | 5 | M | 31 | Mild |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Inga Jácome, M.C.; Morales Chacòn, L.M.; Vera Cuesta, H.; Maragoto Rizo, C.; Whilby Santiesteban, M.; Ramos Hernandez, L.; Noris García, E.; González Fraguela, M.E.; Fernandez Verdecia, C.I.; Vegas Hurtado, Y.; et al. Peripheral Inflammatory Markers Contributing to Comorbidities in Autism. Behav. Sci. 2016, 6, 29. https://doi.org/10.3390/bs6040029
Inga Jácome MC, Morales Chacòn LM, Vera Cuesta H, Maragoto Rizo C, Whilby Santiesteban M, Ramos Hernandez L, Noris García E, González Fraguela ME, Fernandez Verdecia CI, Vegas Hurtado Y, et al. Peripheral Inflammatory Markers Contributing to Comorbidities in Autism. Behavioral Sciences. 2016; 6(4):29. https://doi.org/10.3390/bs6040029
Chicago/Turabian StyleInga Jácome, Martha Cecilia, Lilia Maria Morales Chacòn, Hector Vera Cuesta, Carlos Maragoto Rizo, Mabel Whilby Santiesteban, Lesyanis Ramos Hernandez, Elena Noris García, Maria Elena González Fraguela, Caridad Ivette Fernandez Verdecia, Yamilé Vegas Hurtado, and et al. 2016. "Peripheral Inflammatory Markers Contributing to Comorbidities in Autism" Behavioral Sciences 6, no. 4: 29. https://doi.org/10.3390/bs6040029
APA StyleInga Jácome, M. C., Morales Chacòn, L. M., Vera Cuesta, H., Maragoto Rizo, C., Whilby Santiesteban, M., Ramos Hernandez, L., Noris García, E., González Fraguela, M. E., Fernandez Verdecia, C. I., Vegas Hurtado, Y., Siniscalco, D., Gonçalves, C. A., & Robinson-Agramonte, M. D. l. A. (2016). Peripheral Inflammatory Markers Contributing to Comorbidities in Autism. Behavioral Sciences, 6(4), 29. https://doi.org/10.3390/bs6040029