
Burn Injury: Mechanisms of Keratinocyte Cell Death

Abstract
1. Introduction
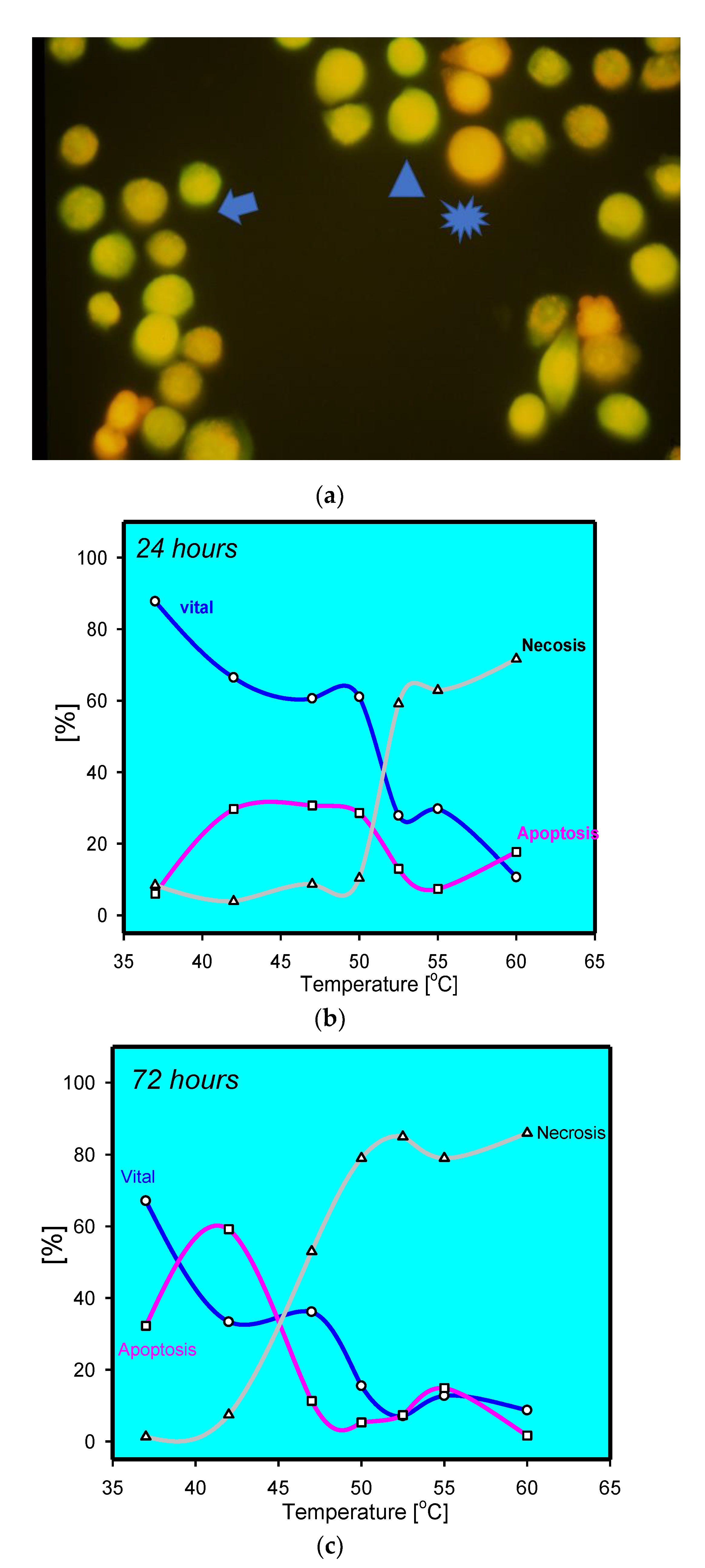
2. Accidental Cell Death (ACD)
3. Apoptosis of Keratinocytes In Vitro
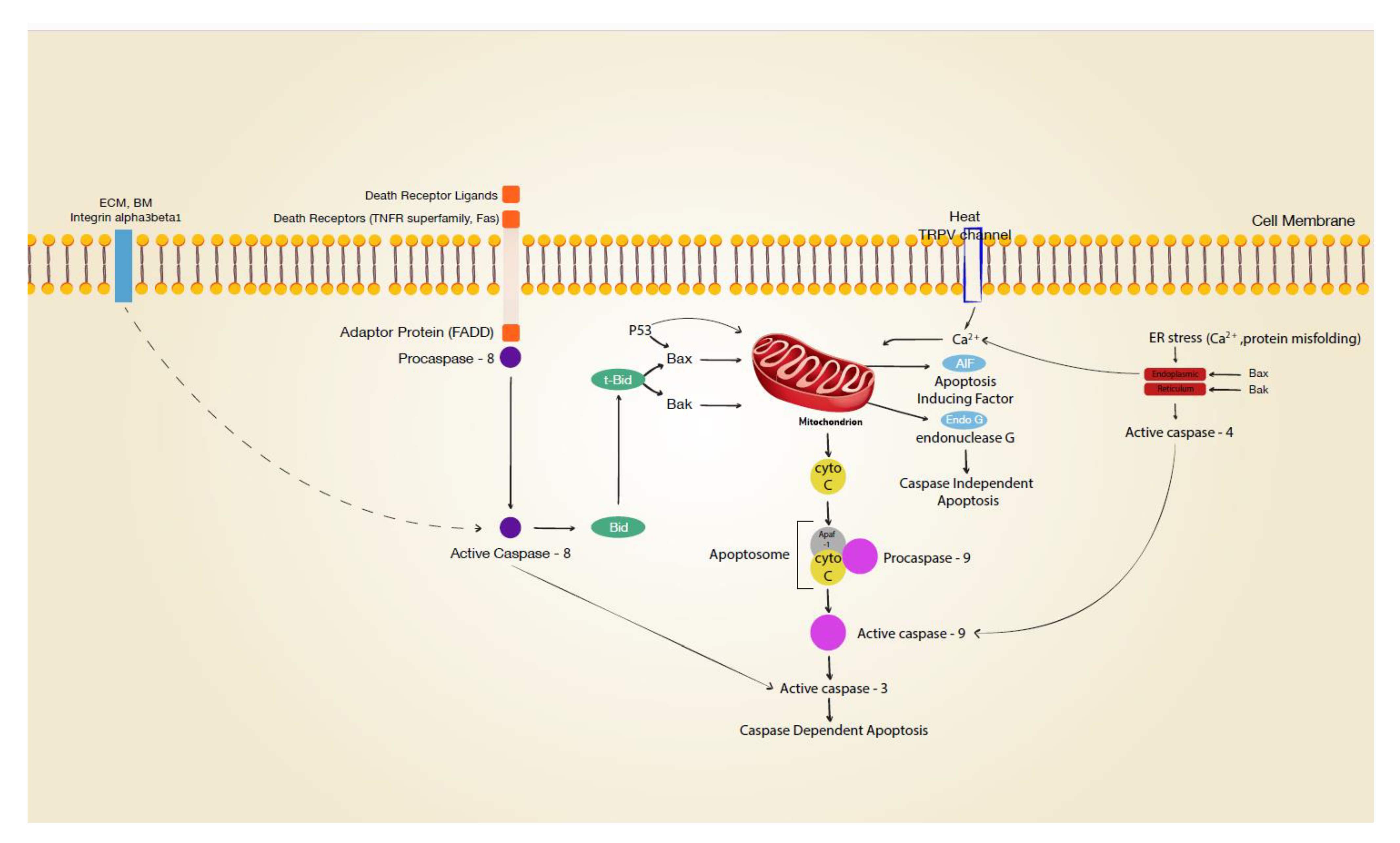
3.1. Intrinsic Apoptosis
3.2. Death-Receptor-Linked Apoptosis
3.3. Apoptosis of the Skin
3.4. Inhibition of Keratinocyte Proliferation
4. Therapeutic Outlook
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Moritz, A.R.; Henriques, F.C., Jr. Studies of termal injury: II. The relative importance of time and surface temperature in the causation of cutaneous burns. Am. J. Pathol. 1947, 23, 695–720. [Google Scholar]
- Jackson, D.M. The diagnosis of the depth of burning. BJS 2005, 40, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef] [PubMed]
- Gravante, G.; Palmieri, M.B.; Esposito, G.; Delogu, D.; Santeusanio, G.; Filingeri, V.; Montone, A. Apoptotic Death in Deep Partial Thickness Burns vs. Normal Skin of Burned Patients. J. Surg. Res. 2007, 141, 141–145. [Google Scholar] [CrossRef]
- McNamara, A.R.; Zamba, K.D.; Sokolich, J.C.; Jaskille, A.D.; Light, T.D.; Griffin, M.A.; Meyerholz, D.K. Apoptosis is Differentially Regulated by Burn Severity and Dermal Location. J. Surg. Res. 2010, 162, 258–263. [Google Scholar] [CrossRef] [PubMed]
- Lanier, S.T.; McClain, S.A.; Lin, F.; Singer, A.J.; Clark, R.A.F. Spatiotemporal progression of cell death in the zone of ischemia surrounding burns. Wound Repair Regen. 2011, 19, 622–632. [Google Scholar] [CrossRef]
- Mertz, I. Morphologic and Immunohistochemical Analysis in Proliferation and Apoptosis after Thermic Stress in Skin. Medical Degree, University of Tuebingen, Tuebingen, Germany, 2011. Available online: http://hdl.handle.net/10900/45846 (accessed on 2 May 2021).
- Lippens, S.; Hoste, E.; Vandenabeele, P.; Agostinis, P.; Declercq, W. Cell death in the skin. Apoptosis 2009, 14, 549–569. [Google Scholar] [CrossRef]
- Gandarillas, A. Epidermal differentiation, apoptosis, and senescence: Common pathways? Exp. Gerontol. 2000, 35, 53–62. [Google Scholar] [CrossRef]
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340. [Google Scholar] [CrossRef]
- Gilhar, A.; Ullmann, Y.; Karry, R.; Shalaginov, R.; Assy, B.; Serafimovich, S.; Kalish, R. Ageing of human epidermis: The role of apoptosis, Fas and telomerase. Br. J. Dermatol. 2004, 150, 56–63. [Google Scholar] [CrossRef]
- Chaturvedi, V.; Qin, J.Z.; Stennett, L.; Choubey, D.; Nickoloff, B.J. Resistance to UV-induced apoptosis in human keratinocytes during accelerated senescence is associated with functional inactivation of p53. J. Cell Physiol. 2004, 198, 100–109. [Google Scholar] [CrossRef]
- Takigawa, M.; Ofuji, S. Early changes in human epidermis following thermal burn: An electron microscopic study. Acta Derm. Venereol. 1977, 57, 187–193. [Google Scholar]
- Matylevitch, N.P.; Schuschereba, S.T.; Mata, J.R.; Gilligan, G.R.; Lawlor, D.F.; Goodwin, C.W.; Bowman, P.D. Apoptosis and accidental cell death in cultured human keratinocytes after thermal injury. Am. J. Pathol. 1998, 153, 567–577. [Google Scholar] [CrossRef]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef]
- Rennekampff, O.H.; Sauter, M.; Schaller, H.E.; Rodemann, H.P. Cooling Has No Benefit on Sequential Cell Death after Thermal Injury In Vitro. J. Burn. Care Rehabil. 2003, 24, S77. [Google Scholar] [CrossRef]
- Green, D.R.; Green, J.C. Mitochondria and apoptosis. Science 1998, 281, 1309–1312. [Google Scholar] [CrossRef]
- Yasuhara, S.; Asai, A.; Sahani, N.D.; Martyn, J.A.J. Mitochondria, endoplasmic reticulum, and alternative pathways of cell death in critical illness. Crit. Care Med. 2007, 35, S488–S495. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Caterina, M.J. TRPV channels as thermosensory receptors in epithelial cells. Pflügers Arch. 2005, 451, 160–167. [Google Scholar] [CrossRef] [PubMed]
- Radtke, C.; Sinis, N.; Sauter, M.; Jahn, S.; Kraushaar, U.; Guenther, E.; Rodemann, H.P.; Rennekampff, H.-O. TRPV Channel Expression in Human Skin and Possible Role in Thermally Induced Cell Death. J. Burn. Care Res. 2011, 32, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Moriello, A.S.; De Petrocellis, L. Assay of TRPV1 Receptor Signaling. Methods Mol. Biol. 2016, 1412, 65–76. [Google Scholar] [CrossRef]
- Yi, G.; Li, L.; Luo, M.; He, X.; Zou, Z.; Gu, Z.; Su, L. Heat stress induces intestinal injury through lysosome- and mitochondria-dependent pathway in vivo and in vitro. Oncotarget 2017, 8, 40741–40755. [Google Scholar] [CrossRef]
- Chinnathambi, S.; Tomanek-Chalkley, A.; Bickenbach, J.R. HSP70 and EndoG Modulate Cell Death by Heat in Human Skin Keratinocytes in vitro. Cells Tissues Organs 2008, 187, 131–140. [Google Scholar] [CrossRef]
- Fatokun, A.A.; Dawson, V.L.; Dawson, T.M. Parthanatos: Mitochondrial-linked mechanisms and therapeutic opportunities. Br. J. Pharmacol. 2014, 171, 2000–2016. [Google Scholar] [CrossRef]
- Banerjee, J.; Lodhi, N.; Nguyen, B.-N. The Role of Poly(ADP-Ribose) Polymerase-1 in Cutaneous Wound Healing. Adv. Wound Care 2019, 8, 634–643. [Google Scholar] [CrossRef] [PubMed]
- Gallo, R.L.; E Kochevar, I.; Granstein, R.D. Ultraviolet radiation induces a change in cell membrane potential in vitro: A possible signal for ultraviolet radiation induced alteration in cell activity. Photochem. Photobiol. 1989, 49, 655–662. [Google Scholar] [CrossRef]
- Holmes, C.J.; Plichta, J.; Gamelli, R.L.; Radek, K.A. Burn Injury Alters Epidermal Cholinergic Mediators and Increases HMGB1 and Caspase 3 in Autologous Donor Skin and Burn Margin. Shock 2017, 47, 175–183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomková, H.; Fujimoto, W.; Arata, J. Expression of the bcl-2 family of genes in the course of keratinocyte differentiation. Eur. J. Dermatol. 1999, 9, 191–196. [Google Scholar] [PubMed]
- Katiyar, S.K.; Roy, A.M.; Baliga, M.S. Silymarin induces apoptosis primarily through a p53-dependent pathway involving Bcl-2/Bax, cytochrome c release, and caspase activation. Mol. Cancer Ther. 2005, 4, 207–216. [Google Scholar] [PubMed]
- Wolff, S.; Erster, S.; Palacios, G.; Moll, U.M. p53’s mitochondrial translocation and MOMP action is independent of Puma and Bax and severely disrupts mitochondrial membrane integrity. Cell Res. 2008, 18, 733–744. [Google Scholar] [CrossRef]
- Erster, S.; Moll, U.M. Stress-induced p53 runs a transcription-independent death program. Biochem. Biophys. Res. Commun. 2005, 331, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Gu, Z.T.; Wang, H.; Li, L.; Liu, Y.S.; Deng, X.B.; Huo, S.F.; Yuan, F.F.; Liu, Z.F.; Tong, H.S.; Su, L. Heat stress induces apoptosis through transcription-independent p53-mediated mitochondrial pathways in human umbilical vein endothelial cell. Sci. Rep. 2014, 4, 4469. [Google Scholar] [CrossRef]
- Gu, Z.T.; Li, L.; Wu, F.; Zhao, P.; Yang, H.; Liu, Y.S.; Geng, Y.; Zhao, M.; Su, L. Heat stress induced apoptosis is triggered by transcription-independent p53, Ca2+ dyshomeostasis and the subsequent Bax mitochondrial translocation. Sci. Rep. 2015, 5, 11497. [Google Scholar] [CrossRef]
- Sauter, M. Differenzierter Zellltod von Keratinozyten. Medical Degree, University of Tuebingen, Tuebingen, Germany, 2009. [Google Scholar]
- Hikim, A.P.S.; Lue, Y.; Yamamoto, C.M.; Vera, Y.; Rodriguez, S.; Yen, P.H.; Soeng, K.; Wang, C.; Swerdloff, R.S. Key Apoptotic Pathways for Heat-Induced Programmed Germ Cell Death in the Testis. Endocrinology 2003, 144, 3167–3175. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Oakes, S.A.; Opferman, J.T.; Cheng, E.H.; Sorcinelli, M.D.; Pozzan, T.; Korsmeyer, S.J. BAX and BAK Regulation of Endoplasmic Reticulum Ca2+: A Control Point for Apoptosis. Science 2003, 300, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Shellman, Y.G.; Howe, W.R.; Miller, L.A.; Goldstein, N.B.; Pacheco, T.R.; Mahajan, R.L.; LaRue, S.M.; Norris, D.A. Hyperthermia induces endoplasmic reticulum-mediated apoptosis in melanoma and non-melanoma skin cancer cells. J. Investig. Dermatol. 2008, 128, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Zaidi, S.F.; Rehman, R.; Kondo, T. Hyperthermia and protein homeostasis: Cytoprotection and cell death. J. Therm. Biol. 2020, 91, 102615. [Google Scholar] [CrossRef] [PubMed]
- Rao, R.V.; Ellerby, H.M.; Bredesen, D.E.; Rao, R.V.; Ellerby, H.M.; Bredesen, D.E. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004, 11, 372–380. [Google Scholar] [CrossRef] [PubMed]
- Bahar, E.; Kim, H.; Yoon, H. ER Stress-Mediated Signaling: Action Potential and Ca2+ as Key Players. Int. J. Mol. Sci. 2016, 17, 1558. [Google Scholar] [CrossRef]
- Obeng, E.A.; Boise, L. Caspase-12 and Caspase-4 Are Not Required for Caspase-dependent Endoplasmic Reticulum Stress-induced Apoptosis. J. Biol. Chem. 2005, 280, 29578–29587. [Google Scholar] [CrossRef]
- Lam, M.; Lawrence, D.A.; Ashkenazi, A.; Walter, P. Confirming a critical role for death receptor 5 and caspase-8 in apoptosis induction by endoplasmic reticulum stress. Cell Death Differ. 2018, 25, 1530–1531. [Google Scholar] [CrossRef]
- Ashkenazi, A.; Dixit, V.M. Death receptors: Signaling and Modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- De Araujo, E.; Dessirier, V.; Laprée, G.; Valeyrie-Allanore, L.; Ortonne, N.; Stathopoulos, E.N.; Bagot, M.; Bensussan, A.; Mockenhaupt, M.; Roujeau, J.-C.; et al. Death ligand TRAIL, secreted by CD1a+ and CD14+ cells in blister fluids, is involved in killing keratinocytes in toxic epidermal necrolysis. Exp. Dermatol. 2011, 20, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Rennekampff, H.O.; Hansbrough, J.F.; Kiessig, V.; Doré, C.; Sticherling, M.; Schröder, J.M. Bioactive inteleukin-8 is expressed in wounds and enhances wound healing. J. Surg. Res. 2000, 93, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Nassif, A.; Moslehi, H.; Le Gouvello, S.; Bagot, M.; Lyonnet, L.; Michel, L.; Boumsell, L.; Bensussan, A.; Roujeau, J.C. Evaluation of the potential role of cytokines in toxic epidermal necrolysis. J. Investig. Dermatol. 2004, 123, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Viard-Leveugle, I.; Bullani, R.R.; Meda, P.; Micheau, O.; Limat, A.; Saurat, J.H.; Tschopp, J.; French, L.E. Intracellular localization of keratinocyte Fas ligand explains lack of cytolytic activity under physiological conditions. J. Biol. Chem. 2003, 278, 16183–16188. [Google Scholar] [CrossRef]
- van Ruissen, F.; Jansen, B.J.; Cerneus, S.; Cloin, W.; Bergers, M.; van Erp, P.E.; Schalkwijk, J. Tumor necrosis factor related apoptosis inducing ligand triggers apoptosis in dividing but not in differentiating human epidermal keratinocytes. J. Investig. Dermatol. 2003, 121, 1433–1439. [Google Scholar] [CrossRef]
- Marconi, A.; Atzei, P.; Panza, C.; Fila, C.; Tiberio, R.; Truzzi, F.; Wachter, T.; Leverkus, M.; Pincelli, C. FLICE/caspase-8 activation triggers anoikis induced by beta1-integrin blockade in human keratinocytes. J. Cell Sci. 2004, 117, 5815–5823. [Google Scholar] [CrossRef]
- Jadhav, S.S.; Meeks, C.J.; Mordwinkin, N.M.; Espinoza, T.B.; Louie, S.G.; Dizerega, G.S.; Rodgers, K.E. Effect of combined radiation injury on cell death and inflammation in skin. Apoptosis 2015, 20, 892–906. [Google Scholar] [CrossRef]
- Tenenhaus, M.; Bhavsar, D. (UCSD: San Diego, CA, USA). Division of Plastic Surgery. Personal communication, 2010. [Google Scholar]
- Reddy, A.S.; Abraham, A.; McClain, S.A.; Clark, R.A.; Ralen, P.; Sandoval, S.; Singer, A.J. The Role of Necroptosis in Burn Injury Progression in a Rat Comb Burn Model. Acad. Emerg. Med. 2015, 22, 1181–1186. [Google Scholar] [CrossRef]
- Linkermann, A.; Green, D.R. Necroptosis. N. Engl. J. Med. 2014, 370, 455–465. [Google Scholar] [CrossRef]
- Liu, L.; Tang, Z.; Zeng, Y.; Liu, Y.; Zhou, L.; Yang, S.; Wang, D. Role of necroptosis in infection-related, immune-mediated, and autoimmune skin diseases. J. Dermatol. 2021. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, M.-B.; Luo, H.-Y.; Shi, C.-H.; Xu, Y.-M. Necroptosis in neurodegenerative diseases: A potential therapeutic target. Cell Death Dis. 2017, 8, e2905. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, G.; Yao, M.; Fang, Y. Rip 1-dependent endothelial necroptosis participates in ischemia-reperfusion injury of mouse flap. J. Dermatol. Sci. 2020, 97, 30–40. [Google Scholar] [CrossRef]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta Bioenerg. 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Bretland, A.J.; Lawry, J.; Sharrard, R.M. A study of death by anoikis in cultured epithelial cells. Cell Prolif. 2001, 34, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell–matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Ruoslahti, E. Integrins and anoikis. Curr. Opin. Cell Biol. 1997, 9, 701–706. [Google Scholar] [CrossRef]
- Manohar, A.; Shome, S.G.; Lamar, J.; Stirling, L.; Iyer, V.; Pumiglia, K.; DiPersio, C.M. Alpha 3 beta 1 integrin promotes keratinocyte cell survival through activation of a MEK/ERK signaling pathway. J. Cell Sci. 2004, 117, 4043–4054. [Google Scholar] [CrossRef] [PubMed]
- Lotti, R.; Marconi, A.; Truzzi, F.; Dallaglio, K.; Gemelli, C.; Borroni, R.G.; Palazzo, E.; Pincelli, C. A previously unreported function of β(1)B integrin isoform in caspase-8-dependent integrin-mediated keratinocyte death. J. Investig. Dermatol. 2010, 130, 2569–2577. [Google Scholar] [CrossRef] [PubMed]
- Rousselle, P.; Aumailley, M. Kalinin is more efficient than laminin in promoting adhesion of primary keratinocytes and some other epithelial cells and has a different requirement for integrin receptors. J. Cell Biol. 1994, 125, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zeglinski, M.; Turner, C.; Raithatha, S.A.; Wu, Z.; Russo, V.; Oram, C.; Hiroyasu, S.; Nabai, L.; Zhao, H.; et al. Topical small molecule granzyme B inhibitor improves remodeling in a murine model of impaired burn wound healing. Exp. Mol. Med. 2018, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Li, W.H.; Lee, Y.M.; Kim, J.Y.; Kang, S.; Kim, S.; Kim, K.H.; Park, C.-H.; Chung, J.H. Transient Receptor Potential Vanilloid-1 Mediates Heat-Shock-Induced Matrix Metalloproteinase-1 Expression in Human Epidermal Keratinocytes. J. Investig. Dermatol. 2007, 127, 2328–2335. [Google Scholar] [CrossRef] [PubMed]
- Schmauss, D.; Rezaeian, F.; Finck, T.; Machens, H.-G.; Wettstein, R.; Harder, Y. Treatment of Secondary Burn Wound Progression in Contact Burns—A Systematic Review of Experimental Approaches. J. Burn. Care Res. 2015, 36, e176–e189. [Google Scholar] [CrossRef]
- Rosique, M.J.; Rosique, R.G.; Faria, F.M.; Oliveira, C.C.; Farina, J.A.; Évora, P.R. Methylene blue reduces progression of burn and increases skin survival in an experimental rat model. Burns 2017, 43, 1702–1708. [Google Scholar] [CrossRef] [PubMed]
- Fang, Q.; Guo, S.; Zhou, H.; Han, R.; Wu, P.; Han, C. Astaxanthin protects against early burn-wound progression in rats by attenuating oxidative stress-induced inflammation and mitochondria-related apoptosis. Sci. Rep. 2017, 7, srep41440. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Y.; Ren, K.-K.; Zhang, W.-J.; Xiao, L.; Wu, H.-Y.; Liu, Q.-Y.; Ding, T.; Zhang, X.-C.; Nie, W.-J.; Ke, Y.; et al. Human amniotic mesenchymal stem cells and their paracrine factors promote wound healing by inhibiting heat stress-induced skin cell apoptosis and enhancing their proliferation through activating PI3K/AKT signaling pathway. Stem Cell Res. Ther. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Giles, N.; Rea, S.; Beer, T.; Wood, F.M.; Fear, M.W. A peptide inhibitor of c-Jun promotes wound healing in a mouse full-thickness burn model. Wound Repair Regen. 2008, 16, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Li, L.; Hu, Q.; Ma, L.; Liu, L.; Chu, W.; Zhang, H. Rapamycin reduces burn wound progression by enhancing autophagy in deep second-degree burn in rats. Wound Repair Regen. 2013, 21, 852–859. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, A.; O’Brien, K.; Chen, M.; Wong, A.; Garner, W.; Woodley, D.T.; Li, W. Dual therapeutic functions of F-5 fragment in burn wounds: Preventing wound progression and promoting wound healing in pigs. Mol. Ther. Methods Clin. Dev. 2016, 3, 16041. [Google Scholar] [CrossRef]
- Uraloğlu, M.; Ural, A.; Efe, G.; Yuluğ, E.; Livaoğlu, M.; Karaçal, N. The Effect of Platelet-Rich Plasma on the Zone of Stasis and Apoptosis in an Experimental Burn Model. Plast. Surg. 2018, 27, 173–181. [Google Scholar] [CrossRef]
- Zhang, J.; Hu, D.-H.; Chen, G.; Bai, X.-Z.; Tang, C.-W. Protective effect of melatonin on oxidative stress inducing hair follicle injury in scald rat. Chin. J. Burn. 2009, 25, 129–132. [Google Scholar]
- Ipaktchi, K.; Mattar, A.; Niederbichler, A.D.; Hoesel, L.M.; Hemmila, M.R.; Su, G.; Remick, D.; Wang, S.C.; Arbabi, S. Topical p38MAPK inhibition reduces dermal inflammation and epithelial apoptosis in burn wounds. Shock 2006, 26, 201–209. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Intervention | Effect | Burn Model | Timing | Reference |
|---|---|---|---|---|
| Human amniotic MSC | Decreased TUNEL staining | mouse | postburn | Li et al. [69] |
| Inhibitor of c-jun | Decreased cell death | mouse | potburn | Giles et al. [70] |
| Rapamycin | Decreased TUNEL staining | rat | postburn | Xiao et al. [71] |
| Methylene Blue | Reduced necrosis | rat | postburn | Rosique et al. [67] |
| Granzyme B inhibitor | Improved healing | mouse | postburn | Shen et al. [64] |
| Hsp90α fragment F-5 | Reduced caspase 3 staining | pig | postburn | Bhatia et al. [72] |
| Platelet Rich Plasma | Reduced TUNEL | rabbits | postburn | Uraloğlu et al. [73] |
| Astaxanthin | Reduced TUNEL staining, reduced cytochrome C and caspase 9 | postburn | Fang et al. [68] | |
| Melatonin | Reduced caspase 3 staining | rat | postburn | Zhang et al. [74] |
| p38MAPK inhibition | Reduced TUNEL | rat | postburn | Ipaktchi et al. [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rennekampff, H.-O.; Alharbi, Z. Burn Injury: Mechanisms of Keratinocyte Cell Death. Med. Sci. 2021, 9, 51. https://doi.org/10.3390/medsci9030051
Rennekampff H-O, Alharbi Z. Burn Injury: Mechanisms of Keratinocyte Cell Death. Medical Sciences. 2021; 9(3):51. https://doi.org/10.3390/medsci9030051
Chicago/Turabian StyleRennekampff, Hans-Oliver, and Ziyad Alharbi. 2021. "Burn Injury: Mechanisms of Keratinocyte Cell Death" Medical Sciences 9, no. 3: 51. https://doi.org/10.3390/medsci9030051
APA StyleRennekampff, H.-O., & Alharbi, Z. (2021). Burn Injury: Mechanisms of Keratinocyte Cell Death. Medical Sciences, 9(3), 51. https://doi.org/10.3390/medsci9030051