Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis

Abstract
1. Diagnosis of Idiopathic Pulmonary Fibrosis
1.1. Clinician Assessment
1.2. Radiological Diagnosis of Idiopathic Pulmonary Fibrosis
1.2.1. High-Resolution Computed Tomography Patterns
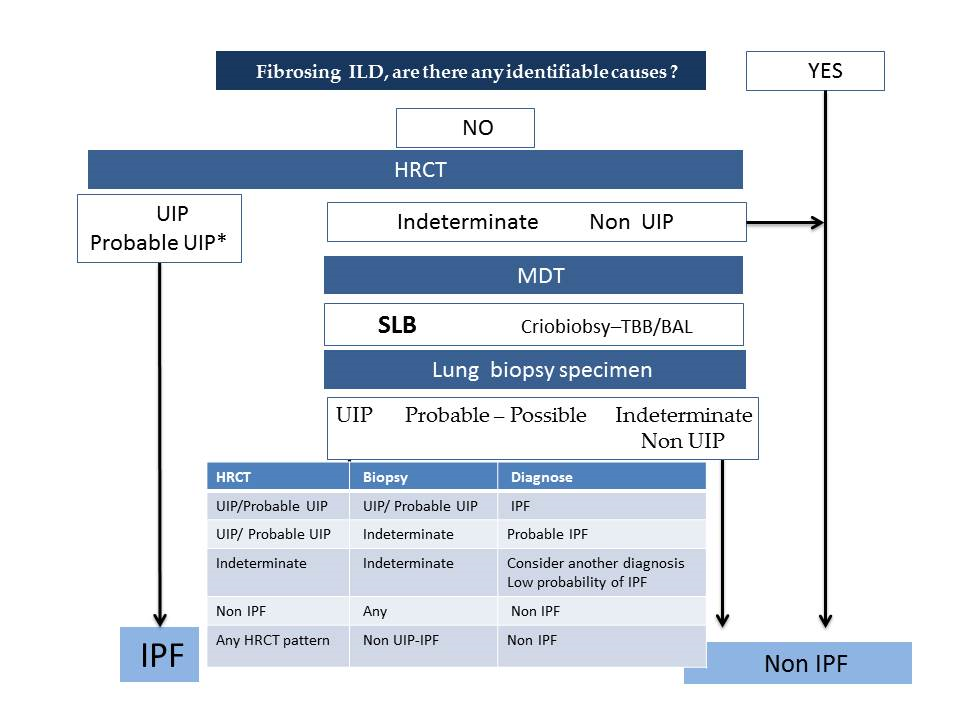
- According to the latest official evidence-based guideline for the diagnosis of IPF [4], this disease is associated with a histopathological and/or radiological pattern of UIP. The radiological UIP pattern was defined by the presence of a reticular pattern and honeycombing, with or without visible traction bronchiectasis, and with a peripheral subpleural and predominantly basal distribution, in the absence of findings inconsistent with UIP (extensive ground glass opacities, micronodules, air trapping, non-honeycomb cysts, consolidation, or a non-basal or non-peripheral predominant distribution). Consensus has established three radiological patterns that IPF can present in HRCT: UIP, possible UIP (UIP-like pattern, but without honeycombing), and inconsistent with UIP (with non-peripheral or non-basal subpleural distribution, or with any finding inconsistent with UIP). Furthermore, it has been established that, in a patient with the appropriate clinical context, the presence of a UIP pattern in HRCT in the absence of a known cause for lung fibrosis (e.g., occupational and environmental exposures, systemic autoimmune disease, or drug toxicity) leads to an IPF diagnosis without the need to perform a lung biopsy, as the probability for such a pattern corresponding to a histopathological UIP pattern is 90–100% [16] (positive predictive value 97.3%, 95% confidence interval 92.3–99.4, in a subsequent study by Raghu et al. 2014 [17]). One of the key findings of this pattern is the honeycombing, which is an indicator of end-stage pulmonary fibrosis. The radiological UIP CT pattern allows, therefore, an IPF diagnosis in the advanced stages of the disease and is observed in less than 50% of patients. When the two other patterns appear in HRCT (possible UIP and inconsistent with UIP), according to the guidelines, it is necessary to do a pulmonary biopsy, and the final diagnosis will depend upon the combination of the different radio-pathological findings.
- Recently, in 2018, the Fleischner Society published the latest IPF diagnostic criteria based on bibliographic review and experts’ opinions [5]. Four types of patterns can be observed as follows:
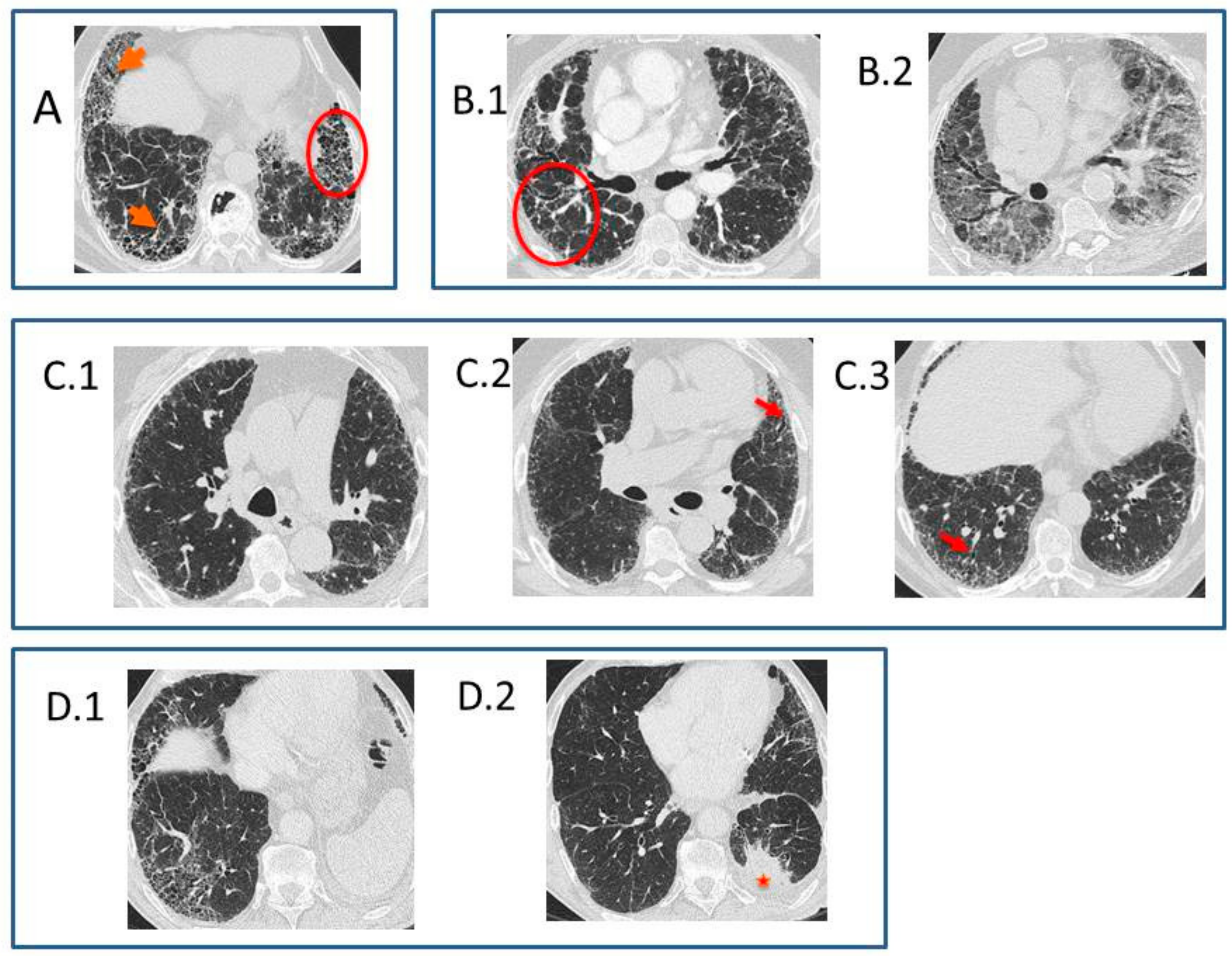
- Typical UIP CT pattern: Reticular opacities and honeycombing, with peripheral traction bronchiectasis and subpleural and basal-predominant distribution. Furthermore, there must be no other findings suggesting an alternative diagnosis (see below, Figure 2A). The fibrosis distribution can be asymmetric. This corresponds to the 2011 UIP pattern guidelines. To make an IPF diagnosis, a lung biopsy is not required in the correct clinical context and in the absence of a known cause of pulmonary fibrosis.
- Probable UIP CT pattern: Replaces the possible UIP pattern in the 2011 guidelines. It includes the same findings as the UIP pattern, although without honeycombing. A lung biopsy is not required in the correct clinical context and in the absence of a known cause of pulmonary fibrosis.
- CT pattern indeterminate for UIP: Appears when the fibrosis presents a variable or diffuse distribution or when there are inconspicuous findings suggesting a non-UIP pattern (Figure 2B). A lung biopsy is required to diagnose IPF in these patients.
- CT features most consistent with a non-IPF diagnosis: Appears when the pulmonary fibrosis is predominant in the upper or middle areas, it is peribronchovascular, it respects the subpleural area, or in any of the following features: predominant consolidation, extensive ground glass opacity without acute exacerbation, extensive mosaic pattern with air trapping on expiration, and nodules or cysts other than in a honeycomb formation. For example, fibrosis with peribronchovascular distribution predominantly in the upper areas, ground glass opacity, and air trapping in non-fibrotic areas, all suggest fibrotic hypersensitivity pneumonitis.
1.2.2. Difficulties Interpreting the High-Resolution Computed Tomography
1.2.3. From Possible Usual Interstitial Pneumonia Pattern (ATS/ERS/JRS/ALAT 2011) to Probable Usual Interstitial Pneumonia Pattern (Fleischner 2018) in High-Resolution Computed Tomography
1.2.4. Identification of Comorbidities and Complications of Idiopathic Pulmonary Fibrosis in High-Resolution Computed Tomography
1.3. Histhological Diagnosis of Idiopathic Pulmonary Fibrosis
2. Differential Diagnosis
2.1. Differential Diagnosis with Other Idiopathic Interstitial Pneumonias
2.1.1. Idiopathic Nonspecific Interstitial Pneumonia
2.1.2. Smoking-Related Idiopathic Interstitial Pneumonia’s
2.1.3. Respiratory Bronchiolitis–Interstitial Lung Disease
2.1.4. Desquamative Interstitial Pneumonia
2.1.5. Acute or Subacute Idiopathic Interstitial Pneumonia’s
Cryptogenic Organizing Pneumonia
Acute Interstitial Pneumonia
2.1.6. Rare Idiopathic Interstitial Pneumonia’s
Idiopathic Lymphoid Interstitial Pneumonia
Idiopathic Pleuroparenchymal Fibroelastosis
2.2. Differential Diagnosis with Known Causes of Usual Interstitial Pneumonia Pattern
2.2.1. Chronic Hypersensitivity Pneumonitis
2.2.2. Connective Tissue Disease in Interstitial Lung Diseases
- Epidemiology: CTDs have are more common in non-smokers and young women with a NSIP pattern [42]. RA is an exception, affecting mostly elderly, smokers, and men. A NIU pattern in a young (<50 year-old) woman should prompt the suspicion of an occult CTD.
- Radiology: NSIP is the most frequent pattern [56]. However, a UIP pattern can be found, especially in an established CTD, such as RA and SSc. Rheumatoid arthritis, remarkably, lacks honeycombing [57]. The HRCT RA-UIP pattern has a worse survival than RA-NSIP. The HRCT RA-UIP pattern has a worse survival than RA-NSIP.
- Histology: RA-UIP has less fibroblast foci, smaller honeycombing cysts, and more pronounced lymphoid hyperplasia than IPF-UIP [57]. Lymphoid aggregates with germinal centers have statistical significance. SSc-related fibrosis has temporal and spatial homogeneity, possibly being pleural fibrosis as well. Lymphocytic infiltrates without fibrosis appear in less than 10% of SSc patients. Lymphocytic interstitial infiltrate is more prominent in Sjögren syndrome.
2.2.3. Exposure-Related Lung Disease: Asbestosis
2.2.4. Drug-Related and Radiation-Related Lung Disease
2.2.5. Microaspiration-Related Lung Disease: Gastroesophageal Reflux
2.2.6. Hermansky–Pudlak Syndrome
3. Conclusions
Funding
Conflicts of Interest
References
- Strongman, H.; Kausar, I.; Maher, T.M. Incidence, prevalence, and survival of patients with idiopathic pulmonary fibrosis in the UK. Adv. Ther. 2018, 35, 724–736. [Google Scholar] [CrossRef] [PubMed]
- Lamas, D.J.; Kawut, S.M.; Bagiella, E.; Philip, N.; Arcasoy, S.M.; Lederer, D.J. Delayed access and survival in idiopathic pulmonary fibrosis: A cohort study. Am. J. Respir. Crit. Care Med. 2011, 184, 842–847. [Google Scholar] [CrossRef] [PubMed]
- National Clinical Guideline Centre. Diagnosis and Management of Suspected Idiopathic Pulmonary Fibrosis: Idiopathic Pulmonary Fibrosis; Royal College of Physicians: London, UK, 2013. [Google Scholar]
- Raghu, G.; Collard, H.R.; Egan, J.J.; Martinez, F.J.; Behr, J.; Brown, K.K.; Colby, T.V.; Cordier, J.F.; Flaherty, K.R.; Lasky, J.A.; et al. An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011, 183, 788–824. [Google Scholar] [PubMed]
- Lynch, D.A.; Sverzellati, N.; Travis, W.D.; Brown, K.K.; Colby, T.V.; Galvin, J.R.; Goldin, J.G.; Hansell, D.M.; Inoue, Y.; Johkoh, T.; et al. Diagnostic criteria for idiopathic pulmonary fibrosis: A Fleischner Society White Paper. Lancet Respir. Med. 2018, 6, 138–153. [Google Scholar] [CrossRef]
- Raghu, G.; Mageto, Y.N.; Lockhart, D.; Schmidt, R.A.; Wood, D.E.; Godwin, J.D. The accuracy of the clinical diagnosis of new-onset idiopathic pulmonary fibrosis and other interstitial lung disease: A prospective study. Chest 1999, 116, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- De Sadeleer, L.J.; Meert, C.; Yserbyt, J.; Slabbynck, H.; Verschakelen, J.A.; Verbeken, E.K.; Weynand, B.; De Langhe, E.; Lenaerts, J.L.; Nemery, B.; et al. Diagnostic ability of a dynamic multidisciplinary discussion in interstitial lung diseases: A retrospective observational study of 938 cases. Chest 2018, 6, 1416–1423. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.F.; Wells, A.U.; Desai, S.R.; Poletti, V.; Piciucchi, S.; Dubini, A.; Nunes, H.; Valeyre, D.; Brillet, P.Y.; Kambouchner, M.; et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: A case-cohort study. Lancet Respir. Med. 2016, 4, 557–565. [Google Scholar] [CrossRef]
- Walsh, S.L.F.; Maher, T.M.; Kolb, M.; Poletti, V.; Nusser, R.; Richeldi, L.; Vancheri, C.; Wilsher, M.L.; Antoniou, K.M.; Behr, J.; et al. IPF Project Consortium. Diagnostic accuracy of a clinical diagnosis of idiopathic pulmonary fibrosis: An international case-cohort study. Eur. Respir. J. 2017, 50, 1700936. [Google Scholar] [PubMed]
- Salisbury, M.L.; Myers, J.L.; Belloli, E.A.; Kazerooni, E.A.; Martinez, F.J.; Flaherty, K.R. Diagnosis and treatment of fibrotic hypersensitivity pneumonia where we stand and where we need to go. Am. J. Respir. Crit. Care Med. 2017, 196, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Vasakova, M.; Morell, F.; Walsh, S.; Leslie, K.; Raghu, G. Hypersensitivity pneumonitis Perspectives in diagnosis and management. Am. J. Respir. Crit. Care Med. 2017, 196, 690–699. [Google Scholar]
- Fernández Pérez, E.R.; Swigris, J.J.; Forsseén, A.V.; Tourin, O.; Solomon, J.J.; Huie, T.J.; Olson, A.L.; Brown, K.K. Identifying an inciting antigen is associated with improved survival in patients with chronic hypersensitivity pneumonitis. Chest 2013, 144, 1644–1651. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Antoniou, K.M.; Brown, K.K.; Cadranel, J.; Corte, T.J.; du Bois, R.M.; Lee, J.S.; Leslie, K.O.; Lynch, D.A.; Matteson, E.L.; et al. An official European Respiratory Society/American Thoracic Society research statement: Interstitial pneumonia with autoimmune features. Eur. Respir. J. 2015, 46, 976–987. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V.; Crestani, B.; Valeyre, D.; Wallaert, B.; Cadranel, J.; Dalphin, J.C.; Delaval, P.; Israel-Biet, D.; Kessler, R.; Reynaud-Gaubert, M. Diagnosis and management of idiopathic pulmonary fibrosis: French practical guidelines. Eur. Respir. Rev. 2014, 23, 193–214. [Google Scholar] [CrossRef] [PubMed]
- Beigelman-Aubry, C.; Hill, C.; Guibal, A.; Savatovsky, J.; Grenier, P.A. Multi-detector row CT and postprocessing techniques in the assessment of diffuse lung disease. Radiographics 2005, 25, 1639–1652. [Google Scholar] [CrossRef] [PubMed]
- Hunninghake, G.W.; Zimmerman, M.B.; Schwartz, D.A.; King, T.E., Jr.; Lynch, J.P., III; Hegele, R.; Waldron, J.; Colby, T.; Muller, N.; Lynch, D.; et al. Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2001, 164, 193–196. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Lynch, D.; Godwin, J.D.; Webb, R.; Colby, T.V.; Leslie, K.O.; Behr, J.; Brown, K.K.; Egan, J.J.; Flaherty, K.R.; et al. Diagnosis of idiopathic pulmonary fibrosis with high-resolution CT in patients with little or no radiological evidence of honeycombing: Secondary analysis of a randomised, controlled trial. Lancet Respir. Med. 2014, 2, 277–284. [Google Scholar] [CrossRef]
- Hansell, D.M.; Bankier, A.A.; MacMahon, H.; McLoud, T.C.; Müller, N.L.; Remy, J. Fleischner Society: Glossary of terms for thoracic imaging. Radiology 2008, 246, 697–722. [Google Scholar] [CrossRef] [PubMed]
- Johkoh, T.; Sakai, F.; Noma, S.; Akira, M.; Fijimoto, K.; Watadani, T.; Sugiyama, Y. Honeycombing on CT: Its definition, pathologic correlation and future direction of its diagnosis. Eur. J. Radiol. 2014, 83, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, H.; Honma, K. Honeycomb lung: History and current concepts. Am. J. Roentgenol. 2011, 196, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Walsh, S.L.; Calandriello, L.; Sverzellati, N.; Wells, A.U.; Hansell, D.M. UIP Observer Consort. Interobserver agreement for the ATS/ERS/JRS/ALAT criteria for a UIP pattern on CT. Thorax 2016, 71, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Watadani, T.; Sakai, F.; Johkoh, T.; Noma, S.; Akira, M.; Fujimoto, K.; Bankier, A.A.; Lee, K.S.; Müller, N.L.; Song, J.-W.; et al. Interobserver variability in the CT assessment of honeycombing in the lungs. Radiology 2013, 266, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Staats, P.; Kligerman, S.; Todd, N.; Tavora, F.; Xu, L.; Allen Burke, A. A comparative study of honeycombing on high resolution computed tomography with histologic lung remodeling in explants with usual interstitial pneumonia. Pathol. Res. Pract. 2015, 211, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Brownell, R.; Moua, T.; Henry, T.S.; Elicker, B.M.; White, D.; Vittinghoff, E.; Jones, K.D.; Urisman, A.; Aravena, C.; Johannson, K.A.; et al. The use of pretest probability increases the value of high-resolution CT in diagnosing usual interstitial pneumonia. Thorax 2017, 72, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Richeldi, L.; du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Wells, A.U.; Nicholson, A.G.; Richeldi, L.; Flaherty, K.R.; Le Maulf, F.; Stowasser, S.; Schlenker-Herceg, R.; Hansell, D.M. Effect of nintedanib in subgroups of idiopathic pulmonary fibrosis by diagnostic criteria. Am. J. Respir. Crit. Care Med. 2017, 195, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Gruden, J.F.; Panse, P.M.; Leslie, K.O.; Tazelaar, H.D.; Colby, T.V. UIP diagnosed at surgical lung biopsy, 2000–2009: HRCT patterns and proposed classification system. Am. J. Roentgenol. 2013, 200, 458–467. [Google Scholar] [CrossRef] [PubMed]
- Gruden, J.F.; Panse, P.M.; Gotway, M.B.; Jensen, E.A.; Wellnitz, C.V.; Wesselius, L. Diagnosis of usual interstitial pneumonitis in the absence of honeycombing: Evaluation of specific CT criteria with clinical follow-up in 38 patients. Am. J. Roentgenol. 2016, 206, 472–480. [Google Scholar] [CrossRef] [PubMed]
- Johkoh, T.; Müller, N.L.; Ichicado, K.; Yoshida, S.; Honda, O.; Mihara, N.; Higashi, M.; Tomiyama, N.; Nakamura, H.; Nagareda, T. Respiratory change in size of honeycombing: Inspiratory and expiratory spiral volumetric CT analysis of 97 cases. J. Comput. Assist. Tomogr. 1999, 23, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Mai, C.; Verleden, S.E.; McDonough, J.E.; Willems, S.; De Weber, W.; Coolen, J.; Dubbeldam, A.; Van Raemdonck, D.E.; Verbeken, E.K.; Verleden, G.M.; et al. Thin-section CT features of idiopathic pulmonary fibrosis correlated with micro-CT and histologic analysis. Radiology 2017, 283, 252–263. [Google Scholar] [CrossRef] [PubMed]
- Cottin, V. Combined pulmonary fibrosis and emphysema: Bad and ugly all the same? Eur. Respir. J. 2017, 50, 1700846. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.D.; Chung, J.H.; Kanne, J.P. Idiopathic pulmonary fibrosis. J. Thorac. Imaging 2016, 31, 127–139. [Google Scholar] [CrossRef] [PubMed]
- Collard, H.R.; Ryerson, C.J.; Corte, T.J.; Jenkins, G.; Kondoh, Y.; Lederer, D.J.; Lee, J.S.; Maher, T.M.; Wells, A.U.; Antoniou, K.M.; et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am. J. Respir. Crit. Care Med. 2016, 194, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Ichikado, K. High-resolution computed tomography findings of acute respiratory distress syndrome, acute interstitial pneumonia, and acute exacerbation of idiopathic pulmonary fibrosis. Semin. Ultrasound CT MRI 2014, 35, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Rush, B.; Wiskar, K.; Berger, L.; Griesdale, D. The use of mechanical ventilation in patients with idiopathic pulmonary fibrosis in the United States: A nationwide retrospective cohort analysis. Respir. Med. 2016, 111, 72–76. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, J.P.; McKeever, T.M.; Fogarty, A.W.; Navaratnam, V.; Hubbard, R.B. Surgical lung biopsy for the diagnosis of interstitial lung disease in England: 1997–2008. Eur. Resp. J. 2016, 48, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Pajares, V.; Puzo, C.; Castillo, D.; Lerma, E.; Montero, M.A.; Ramos-Barbón, D.; Amor-Carro, O.; Gil de Bernabé, A.; Franquet, T.; Plaza, V.; et al. Diagnostic yield of transbronchial cryobiopsy in interstitial lung disease: A randomized trial. Respirology 2014, 19, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Tomassetti, S.; Wells, A.U.; Costabel, U.; Cavazza, A.; Colby, T.V.; Rossi, G.; Sverzellati, N.; Carloni, A.; Carretta, E.; Buccioli, M.; et al. Bronchoscopic lung cryobiopsy increases diagnostic confidence in the multidisciplinary diagnosis of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2016, 193, 745–752. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.L.; Myers, J.L. Idiopathic pulmonary fibrosis: Clinical relevance of pathologic classification. Am. J. Respir. Crit. Care Med. 1998, 157, 1301–1315. [Google Scholar] [CrossRef] [PubMed]
- Flaherty, K.R.; Travis, W.D.; Colby, T.V.; Toews, G.B.; Kazerooni, E.A.; Gross, B.H.; Jain, A.; Strawderman, R.L.; Flint, A.; Lynch, J.P.; et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2001, 164, 1722–1727. [Google Scholar] [CrossRef] [PubMed]
- Katzenstein, A.L.; Zisman, D.A.; Litzky, L.A.; Nguyen, B.T.; Kotloff, R.M. Usual interstitial pneumonia: Histologic study of biopsy and explant specimens. Am. J. Surg. Pathol. 2002, 26, 1567–1577. [Google Scholar] [CrossRef] [PubMed]
- Song, J.W.; Do, K.H.; Kim, M.Y. ; Jang. S.J.; Colby, T.V.; Kim, D.S. Pathologic and radiologic differences between idiopathic and collagen vascular disease-related usual interstitial pneumonia. Chest 2009, 136, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Roglli, L.V.; Gibbs, R.A.; Attanoos, R.; Churg, A.; Popper, H.; Cagle, P.; Corrin, B.; Franks, T.J.; Galateau-Salle, F.; Galvin, J.; et al. Pathology of asbestosis. An update of the diagnostic criteria. Arch. Pathol. Lab. Med. 2010, 134, 462–480. [Google Scholar]
- Travis, W.D.; Costabel, U.; Hansell, D.M.; King, T.E., Jr.; Lynch, D.A.; Nicholson, A.G.; Ryerson, C.J.; Ryu, J.H.; Selman, M.; Wells, A.U.; et al. ATS/ERS Committee on Idiopathic Interstitial Pneumonias. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am. J. Respir. Crit. Care Med. 2013, 188, 733–748. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, S.L.; Rubens, M.B.; Hansell, D.M.; Copley, S.J.; Desai, S.R.; du Bois, R.M.; Nicholson, A.G.; Colby, T.V.; Wells, A.U. Nonspecific interstitial pneumonia and usual interstitial pneumonia: Comparative appearances at and diagnostic accuracy of thin-section CT. Radiology 2001, 221, 600–605. [Google Scholar] [CrossRef] [PubMed]
- Akira, M.; Inoue, Y.; Arai, T.; Okuma, T.; Kawata, Y. Long-term follow-up high-resolution CT findings in non-specific interstitial pneumonia. Thorax 2011, 66, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Travis, W.D.; Matsui, K.; Moss, J.; Ferrans, V.J. Idiopathic nonspecific interstitial pneumonia: Prognostic significance of cellular and fibrosing patterns: Survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am. J. Surg. Pathol. 2000, 24, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Park, I.N.; Jegal, Y.; Kim, D.S.; Do, K.H.; Yoo, B.; Shim, T.S.; Lim, S.D.; Lee, Y.; Koh, W.S.; Kim, W.D.; et al. Clinical course and lung function change of idiopathic nonspecific interstitial pneumonia. Eur. Respir. J. 2009, 33, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Fraig, M.; Shreesha, U.; Savici, D.; Katzenstein, A.L. Respiratory bronchiolitis: A clinicopathologic study in current smokers, ex-smokers, and never-smokers. Am. J. Surg. Pathol. 2002, 26, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, A.; Franquet, T.; Gimenez, A.; Bordes, R.; Pineda, R.; Madrid, M. Smoking-related interstitial lung diseases: Radiologic-pathologic correlation. Eur. Radiol. 2006, 16, 2463–2470. [Google Scholar] [CrossRef] [PubMed]
- Portnoy, J.; Veraldi, K.L.; Schwarz, M.I.; Cool, C.D.; Curran-Everett, D.; Cherniack, R.M.; et al. Respiratory bronchiolitis-interstitial lung disease: Long-term outcome. Chest 2007, 131, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Kligerman, S.; Franks, T.J.; Galvin, J.R. Clinical-radiologic-pathologic correlation of smoking-related diffuse parenchymal lung disease. Radiol. Clin. North. Am. 2016, 54, 1047–1063. [Google Scholar] [CrossRef] [PubMed]
- Johkoh, T.; Muller, N.L.; Taniguchi, H.; Kondoh, Y.; Akira, M.; Ichikado, K.; Ando, M.; Nakamura, H. Acute interstitial pneumonia: Thin-section CT findings in 36 patients. Radiology 1999, 211, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Cha, S.I.; Fessler, M.B.; Cool, C.D.; Schwarz, M.I.; Brown, K.K. Lymphoid interstitial pneumonia: Clinical features, associations and prognosis. Eur. Respir. J. 2006, 28, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Takemura, T.; Akashi, T.; Kamiya, H.; Ikushima, S.; Ando, T.; Oritsu, M.; Sawahata, M.; Ogura, T. Pathological differentiation of chronic hypersensitivity pneumonitis from idiopathic pulmonary fibrosis/usual interstitial pneumonia. Histopathology 2012, 61, 1026–1035. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; du Bois, R. Interstitial lung disease in connective tissue disorders. Lancet 2012, 380, 689–698. [Google Scholar] [CrossRef]
- Wuyts, W.A.; Cavazza, A.; Rossi, G.; Bonella, F.; Sverzellati, N.; Spagnolo, P. Differencial diagnosis of usual interstitial pneumonia: When is it truly idiopathic? Eur. Respir. Rev. 2014, 23, 308–319. [Google Scholar] [CrossRef] [PubMed]
- Alhamad, E.H.; Cal, J.G.; AlBoukai, A.A.; Shaik, S.A.; Omair, M.A. Autoimmune symptoms in idiopathic pulmonary fibrosis: Clinical significance. Clin. Respir. J. 2016, 10, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Akira, M.; Morinaga, K. The comparison of hight-resolution computed tomography findings in asbestosis and idiopathic pulmonary fibrosis. Am. J. Ind. Med. 2016, 59, 301–306. [Google Scholar] [CrossRef] [PubMed]
- Attanoos, R.L.; Alchamis, F.S.; Pooley, F.D.; Gibbs, A.R. Usual interstitial pneumonia is asbestos-exposed cohorts-concurrent idiopathic pulmonary fibrosis or atypical asbestosis? Histopathology 2016, 69, 492–498. [Google Scholar] [CrossRef] [PubMed]
- Bonniad, P.; Georges, M.; Favrolt, N.; Camus, P. Drug-induced interstitial lung diseases. Rev. Prat. 2014, 64, 951–956. [Google Scholar]
- Giridhar, P.; Mallick, S.; Rath, G.K.; Julka, P.K. Radiation induced lung injury: Prediction, assessment and management. Asian Pac. J. Cancer Prev. 2015, 16, 2613–2617. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Lee, J.S.; Pianosi, P.T.; Ryu, J.H. Aspiration-related pulmonary syndromes. Chest 2014, 147, 926–934. [Google Scholar] [CrossRef] [PubMed]
- Vicary, G.W.; Vergne, Y.; Cornier, A.S.; Young, L.R.; Roman, J. Pulmonary fibrosis in Hermansky-Pudlak syndrome. Ann. Am. Thorac. Soc. 2016, 13, 1839–1846. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| ATS-ERS-JRS-ALAT Guideline 2011 | |||||
|---|---|---|---|---|---|
| UIP Pattern (All Four Criteria) | Probable UIP | Possible UIP (All Three Criteria) | Not UIP Pattern (Any of Them) | ||
| - Evidence of marked fibrosis/architectural distortion, honeycombing in a predominantly subpleural/paraseptal distribution - Presence of patchy involvement of lung parenchyma by fibrosis - Presence of FF - Absence of features against a diagnosis of UIP suggesting an alternate diagnosis. | - Evidence of marked fibrosis /architectural distortion, honeycombing - Absence of either patchy involvement or FF, but not both - Absence of features against a diagnosis of UIP suggesting an alternate diagnosis or Honeycomb changes only. | - Patchy or diffuse involvement of lung parenchyma by fibrosis, with or without interstitial inflammation - Absence of other criteria for UIP - Absence of features against a diagnosis of UIP suggesting an alternate diagnosis | - Hyaline membranes - Organizing pneumonia - Granulomas - Marked interstitial inflammatory cell infiltrate away from honeycombing - Predominant airway centered changes - Other features suggestive of an alternate diagnosis | ||
| Fleishner Society White Paper | |||||
| UIP-IPF Pattern (All Four Criteria) | Probable UIP-IPF (Not All Four Criteria) | Indeterminate for UIP-IPF | Features Most Consistent with an Alternative Diagnosis | ||
| Patients show features with all four criteria, and do not show features that might suggest an alternative diagnosis (e.g., non-UIP) | Honeycomb fibrosis only or; dense fibrosis causing architecture remodeling with frequent honeycombing; patchy lung involvement by fibrosis; FF at the edge of dense scars may or may not be present | Possible-UIP disappears in Fleischner White paper | Less compelling histological changes than those classified by the final column (e.g., occasional foci of centrilobular injury or scarring, rare granulomas or giant cells, only a minor degree of lymphoid hyperplasia or diffuse inflammation, or diffuse homogenous fibrosis favouring fNSIP); | Non-UIP pattern: features of other fibrotic disorders—e.g., fHP, fNSIP, fOP, PPFE, pulmonary Langerhans cell histiocytosis, or smoking-related interstitial fibrosis; UIP pattern with ancillary features strongly suggesting an alternative diagnosis: e.g., prominent DAD or OP (consider acute exacerbation of UIP), granulomas, (consider HP, sarcoid, infection), marked interstitial inflammatory cell infiltrate away from areas of UIP (consider HP). | |
| Unclassifiable Idiopathic Interstitial Pneumonias |
|---|
| Major Idiopathic Interstitial Pneumonias |
| Idiopathic pulmonary fibrosis (IPF) |
| Idiopathic nonspecific interstitial pneumonia (NSIP) |
| Respiratory bronchiolitis–interstitial lung disease (RBILD) |
| Desquamative interstitial pneumonia (DIP) |
| Acute interstitial pneumonia (AIP) |
| Cryptogenic organizing pneumonia (COP) |
| Rare idiopathic interstitial pneumonias |
| Idiopathic lymphoid interstitial pneumonia (ILIP) |
| Idiopathic pleuroparenchymal fibroelastosis (IPPFE) |
| Chronic hypersensitivity pneumonitis (CHP) |
| Connective tissue disease (CTD)-related ILD |
| Exposure-related lung disease: Asbestosis |
| Drug-related and radiation-related lung disease |
| Microaspiration-related lung disease: gastroesophageal reflux (GER) |
| Hermansky–Pudlak syndrome |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aburto, M.; Herráez, I.; Iturbe, D.; Jiménez-Romero, A. Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis. Med. Sci. 2018, 6, 73. https://doi.org/10.3390/medsci6030073
Aburto M, Herráez I, Iturbe D, Jiménez-Romero A. Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis. Medical Sciences. 2018; 6(3):73. https://doi.org/10.3390/medsci6030073
Chicago/Turabian StyleAburto, Myriam, Inmaculada Herráez, David Iturbe, and Ana Jiménez-Romero. 2018. "Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis" Medical Sciences 6, no. 3: 73. https://doi.org/10.3390/medsci6030073
APA StyleAburto, M., Herráez, I., Iturbe, D., & Jiménez-Romero, A. (2018). Diagnosis of Idiopathic Pulmonary Fibrosis: Differential Diagnosis. Medical Sciences, 6(3), 73. https://doi.org/10.3390/medsci6030073