The Gut Microbiome in Multiple Sclerosis: A Potential Therapeutic Avenue


Abstract
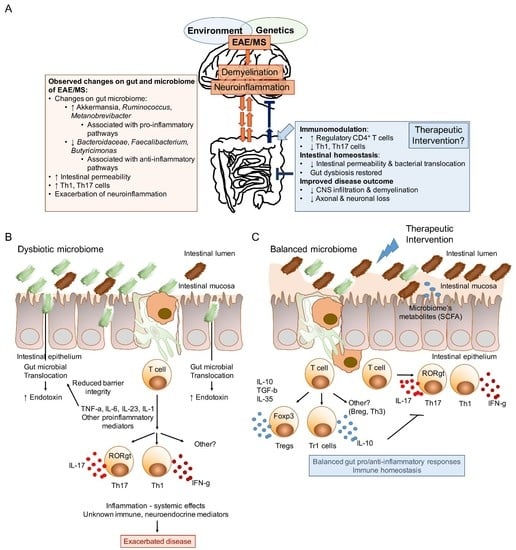
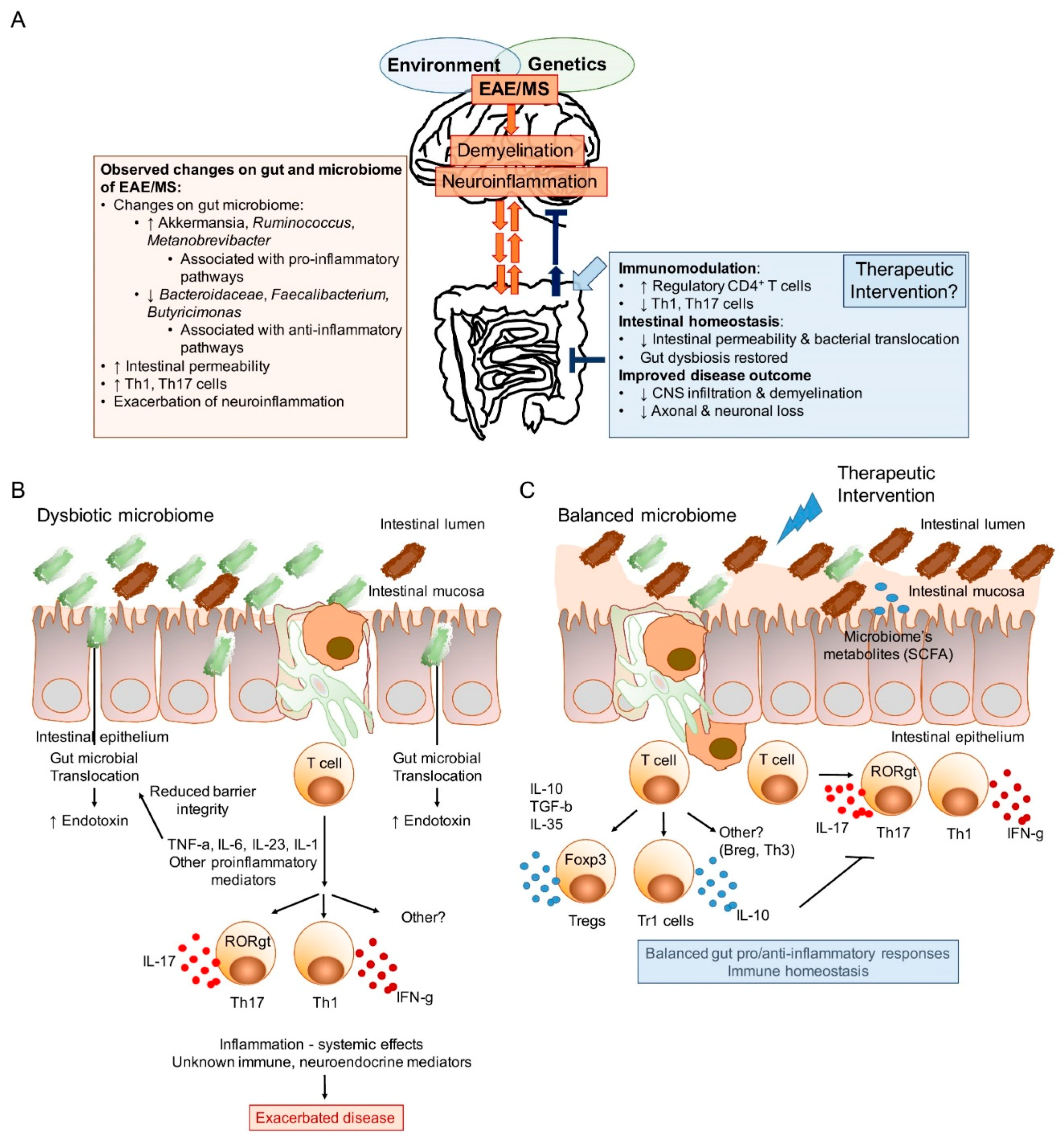
1. Introduction
2. The Gut Microbiome
2.1. The Anatomy of the Gut Epithelium
2.2. The Gut/Immune System Nexus
3. Multiple Sclerosis and Autoimmunity
3.1. The Gut Microbiome and Multiple Sclerosis
3.2. Targeting the Gut Microbiome with Therapeutic Options
3.2.1. Antibiotic Therapy
3.2.2. Phage Therapy
3.2.3. Fecal Microbiota Transplantation
3.2.4. Dietary Supplementation
3.2.5. Probiotics and Immunomodulatory Factors Derived from Gut Microbes
4. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- Fallingborg, J. Intraluminal pH of the human gastrointestinal tract. Dan. Med. Bull. 1999, 46, 183–196. [Google Scholar] [PubMed]
- Hansson, G.C. Role of mucus layers in gut infection and inflammation. Curr. Opin. Microbiol. 2012, 15, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, G.P.; Lee, S.M.; Mazmanian, S.K. Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 2016, 14, 20–32. [Google Scholar] [CrossRef] [PubMed]
- Koropatkin, N.M.; Cameron, E.A.; Martens, E.C. How glycan metabolism shapes the human gut microbiota. Nat. Rev. Microbiol. 2012, 12, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Bevins, C.L.; Salzman, N.H. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011, 9, 356–368. [Google Scholar] [CrossRef] [PubMed]
- Needham, B.D.; Trent, M.S. Fortifying the barrier: The impact of lipid A remodeling on bacterial pathogenesis. Nat. Rev. Microbiol. 2013, 11, 467–481. [Google Scholar] [CrossRef] [PubMed]
- Peterson, D.A.; McNulty, N.P.; Guruge, J.L.; Gordon, J.I. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe 2007, 2, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Palm, N.W.; de Zoete, M.R.; Cullen, T.W.; Barry, N.A.; Stefanowski, J.; Hao, L.; Degnan, P.H.; Hu, J.; Peter, I.; Zhang, W.; et al. Immunoglobulin A coating identifies colitogenic bacteria inflammatory bowel disease. Cell 2014, 158, 1000–1010. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA. 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Joscelyn, J.; Kasper, L.H. Digesting the emerging role for the gut microbiome in central nervous system demyelination. Mult. Scler. 2014, 20, 1553–1559. [Google Scholar] [CrossRef] [PubMed]
- Montague, L.; Piel, C.; Lallès, J.P. Effect of diet on mucin kinetics and composition: Nutrition and health implications. Nutr. Rev. 2004, 62, 105–114. [Google Scholar] [CrossRef]
- Wells, J.M.; Brummer, R.J.; Derrien, M.; MacDonald, T.T.; Troost, F.; Cani, P.D.; Theodorou, V.; Dekker, J.; Méheust, A.; de Vos, W.M.; et al. Homeostasis of the gut barrier and potential biomarkers. Am. J. Physiol. Gastrointest Liver Physiol. 2017, 312, G171–G193. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; van Es, J.H.; Snippert, H.J.; Strange, D.E.; Vries, R.G.; van den Born, M.; Barker, N.; Shroyer, N.F.; van de Wetering, M.; Clevers, H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature 2011, 469, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability—A new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [PubMed]
- Alhasson, F.; Das, S.; Seth, R.; Dattaroy, D.; Chandrashekaran, V.; Ryan, C.N.; Chan, L.S.; Testerman, T.; Burch, J.; Hofseth, L.J.; et al. Altered gut microbiome in a mouse model of Gulf War Illness causes neuroinflammation and intestinal injury via leaky gut and TLR4 activation. PLoS ONE 2017, 12, e0172914. [Google Scholar] [CrossRef] [PubMed]
- Pham, O.H.; O’Donnell, H.; Al-Shamkhani, A.; Kerrinnes, T.; Tsolis, R.M.; McSorley, S.J. T cell expression of IL-18R and DR3 is essential for non-cognate stimulation of Th1 cells and optimal clearance of intracellular bacteria. PLoS Pathog. 2017, 13, e1006566. [Google Scholar] [CrossRef] [PubMed]
- Domingues-Villar, M.; Gautron, A.S.; de Marcken, M.; Keller, M.J.; Hafler, D.A. TLR7 induces anergy in human CD4+ T cells. Nat. Immunol. 2014, 16, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Van den Hoogen, W.J.; Laman, J.D.; ‘t Hart, B.A. Modulation of Multiple Sclerosis and Its Animal Model Experimental Autoimmune Encephalomyelitis by Food and Gut Microbiota. Front. Immunol. 2017, 8, 1081. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U.; Joshua, V.; Artacho, A.; Abdolaahi-Roodsaz, S.; Öckinger, J.; Kullberg, S.; Sköld, M.; Eklund, A.; Grunewald, A.; Clemente, J.C.; et al. The lung microbiota in early rheumatoid arthritis and autoimmunity. Microbiome 2016, 4, 60. [Google Scholar] [CrossRef] [PubMed]
- Petersen, C.; Round, J.L. Defining Dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Espinoza, J.L.; Minami, M. Sensing Bacterial-Induced DNA Damaging Effects via Natural Killer Group 2 Member D Immune Receptor: From Dysbiosis to Autoimmunity and Carcinogenesis. Front. Immunol. 2018, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Platone, D.; De Angelis, F.; Doshi, A.; Chataway, J. Secondary Progressive Multiple Sclerosis: Definition and Measurement. CNS Drugs 2016, 30, 517–526. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, E.; Patti, F.; Zanghì, A.; Zappia, M. A Personalized Approach in Progressive Multiple Sclerosis: The Current Status of Disease Modifying Therapies (DMTs) and Future Perspectives. Int. J. Mol. Sci. 2016, 17, 1725. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of Immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat. Genet. 2013, 45, 1353–1360. [Google Scholar] [CrossRef] [PubMed]
- Frischer, J.M.; Bramow, S.; Dal-Bianco, A.; Lucchinetti, C.F.; Rauschka, H.; Schmidbauer, M.; Laursen, H.; Sorensen, P.S.; Lassmann, H. The regulation between inflammation and neurodegeneration in multiple sclerosis brains. Brain 2009, 132, 1175–1189. [Google Scholar] [CrossRef] [PubMed]
- Palanichamy, A.; Apeltsin, L.; Kuo, T.C.; Sirota, M.; Wang, S.; Pitts, S.J.; Sundar, P.D.; Telman, D.; Zhao, L.Z.; Derstine, M.; et al. Immunoglobulin class-switched B cells form an active immune axis between CNS and periphery in multiple sclerosis. Sci. Transl. Med. 2014, 6, 248ra106. [Google Scholar] [CrossRef] [PubMed]
- Friese, M.A.; Schatting, B.; Fugger, L. Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat. Rev. Neurol. 2014, 10, 225–238. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.; Hellings, N.; Broekmans, T.; Hensen, K.; Rummens, J.L.; Stinissen, P. Natural naïve CD4+CD25+CD127low regulatory T cell (Treg) development and function are disturbed in multiple sclerosis patients: Recovery of memory Treg homeostasis during disease progression. J. Immunol. 2008, 180, 6411–6420. [Google Scholar] [CrossRef] [PubMed]
- Venken, K.; Hellings, N.; Thewissen, M.; Somers, V.; Hensen, K.; Rummens, J.; Medaer, R.; Hupperts, R.; Stinissen, P. Compromised CD4+CD25high regulatory T-cell function in patients with relapsing-remitting multiple sclerosis is correlated with reduced frequency of FOXP3 positive cells and reduced FOXP3 expression at the single-cell level. Immunology 2008, 123, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflamm. 2017, 14, 117. [Google Scholar] [CrossRef] [PubMed]
- Bates, J.M.; Akerlund, J.; Mittge, E.; Guillmin, K. Intestinal alkaline phosphatase detoxifies lipopolysaccharide and prevents inflammation in zebrafish in response to the gut microbiota. Cell Host Microbe 2007, 2, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Mirza, A.; Mao-Draayer, Y. The gut microbiome and microbial translocation in multiple sclerosis. Clin. Immunol. 2017, 183, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Shamriz, O.; Mizrahi, H.; Werbner, M.; Shoenfeld, Y.; Avni, O.; Koren, O. Microbiota at the crossroads of autoimmunity. Autoimmun. Rev. 2016, 15, 859–869. [Google Scholar] [CrossRef] [PubMed]
- Gomes-Neto, J.C.; Kittana, H.; Mantz, S.; Segura Munoz, R.R.; Schmaltz, R.J.; Bindels, L.B.; Clarke, J.; Hostetter, J.M.; Benson, A.K.; Walter, J.; et al. A gut pathobioant synergizes with the microbiota to instigate inflammatory bowel disease marked by immunoreactivity against other symbioants but not itself. Sci. Rep. 2017, 7, 17707. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Ditrio, L.E.; Burroughs, A.R.; Foureau, D.M.; Haque-Begum, S.; Kasper, L.H. Role of Gut Commensal Microflora in the Development of Experimental Autoimmune Encephalomyelitis. J. Immunol. 2009, 183, 6041–6050. [Google Scholar] [CrossRef] [PubMed]
- Yokote, H.; Miyake, S.; Croxford, J.L.; Oki, S.; Mizusawa, H.; Yamamura, T. NKT cell-dependent amelioration of a mouse model of multiple sclerosis by altering gut flora. Am. J. Pathol. 2008, 173, 1714–1723. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Jin, P.; Zhang, X.; Wong, F.S.; Wen, L. Different immunological responses to early-life antibiotic exposure affecting autoimmune diabetes development in NOD mice. J. Autoimmun. 2016, 72, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.S.; Bloom, S.M.; Norian, L.A.; Geske, M.J.; Flavell, R.A.; Stappenbecl, T.S.; Allen, P.M. An Antibiotic-Responsove Mouse Model of Fulminant Ulcerative Colitis. PLoS Med. 2008, 5, e41. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.K.; Metea, C.; Karstens, L.; Asquith, M.; Gruner, H.; Moscibrocki, C.; Lee, I.; Brislaw, C.J.; Jansson, J.K.; Rosenbaum, J.T.; et al. Gut Microbial Alterations Associated with Protection from Autoimmune Uveitis. Investig. Ophthalmol. Vis. Sci. 2016, 57, 3747–3758. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, S.L.; Kasper, E.J.; Keever, A.; Liljenberg, C.; Kirby, T.; Magori, K.; Kasper, L.H.; Ochoa-Reparaz, J. A Bidirectional Association Between the Gut Microbiota and CNS Disease in a Biphasic Murine Model of Multiple Sclerosis. Gut Microbes 2017, 8, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. The Gut Microbiota Shapes Intestinal Immune Responses During Health and Disease. Nat. Rev. Immunol. 2009, 9, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms Underlying the Resistance to Diet-Induced Obesity in Germ-Free Mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Sellon, R.K.; Tonkonogy, S.; Schultz, M.; Dieleman, L.A.; Grenther, W.; Balish, E.; Rennick, D.M.; Sartor, R.B. Resident Enteric Bacteria Are Necessary for Development of Spontaneous Colitis and Immune System Activation in Interleukin-10-Deficient Mice. Infect. Immun. 1998, 66, 5224–5231. [Google Scholar] [PubMed]
- Wu, H.-J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-Residing Segmented Filamentous Bacteria Drive Autoimmune Arthritis via T Helper 17 Cells. Immunity 2016, 32, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Mues, M.; Koutrolos, M.; Rasbi, Z.A.; Boziki, M.; Johner, C.; Wekerle, H.; Krishnamoorthy, G. Commensal Microbiota and Myelin Autoantigen Cooperate to Trigger Autoimmune Demyelination. Nature 2011, 479, 538–541. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-Cell Responses to Gut Microbiota Promote Experimental Autoimmune Encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108 Suppl. 1, 4615–4622. [Google Scholar] [CrossRef]
- Miyake, S.; Kim, S.; Suda, W.; Oshima, K.; Nakamura, M.; Matsuoka, T.; Chihara, N.; Tomita, A.; Sato, W.; Kim, S.-W.; et al. Dysbiosis in the Gut Microbiota of Patients with Multiple Sclerosis, with a Striking Depletion of Species Belonging to Clostridia XIVa and IV Clusters. PLoS ONE 2015, 10, e0137429. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Chia, N.; Kalari, K.R.; Yao, J.Z.; Novotna, M.; Paz Soldan, M.M.; Luckey, D.H.; Marietta, E.V.; Jeraldo, P.R.; Chen, X.; et al. Multiple sclerosis patients have a distinct gut microbiota compared to healthy controls. Sci. Rep. 2016, 6, 28484. [Google Scholar] [CrossRef] [PubMed]
- Jangi, S.; Gandhi, R.; Cox, L.M.; Li, N.; von Glehn, F.; Yan, R.; Patel, B.; Mazzola, M.A.; Liu, S.; Glanz, B.L.; et al. Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 2016, 7, 12015. [Google Scholar] [CrossRef] [PubMed]
- Tremlett, H.; Bauer, K.C.; Appel-Cresswell, S.; Finlay, B.B.; Waubant, E. The gut microbiome in human neurological disease: A review. Ann. Neurol. 2017, 81, 369–382. [Google Scholar] [CrossRef] [PubMed]
- Cree, B.A.; Spencer, C.M.; Varrin-Doyer, M.; Baranzini, S.E.; Zamvil, S.S. Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann. Neurol. 2016, 80, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Scheperjans, F.; Aho, V.; Pereira, P.A.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic bacterial composition in Parkinson’s disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, S.; Mohajeri, M.H. Changes of Colonic Bacterial Composition in Parkinson’s Disease and Other Neurodegenerative Diseases. Nutrients 2018, 10, 708. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota Modulate Behavioral and Physiological Abnormalities Associated with Neurodevelopmental Disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.R.; Frost, E.L.; Bolte, A.C.; Paysour, M.J.; Shaw, M.E.; Bellinger, C.E.; Weigel, T.K.; Zunder, E.R.; Lukens, J.R. Cutting Edge: Critical Roles for Microbiota-Mediated Regulation of the Immune System in a Prenatal Immune Activation Model of Autism. J. Immunol. 2018, 201, Ji1701755. [Google Scholar] [CrossRef] [PubMed]
- Tabouy, L.; Getselter, D.; Ziv, O.; Karpuj, M.; Tabouy, T.; Lukic, I.; Maayouf, R.; Werbner, N.; Ben-Amram, H.; Nuriel-Ohayon, M.; et al. Dysbiosis of microbiome and probiotic treatment in a genetic model of autism spectrum disorders. Brain Behav. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bercik, P.; Denou, E.; Collins, J.; Jackson, W.; Lu, J.; Jury, J.; Deng, Y.; Blennerhassett, P.; Macri, J.; McCoy, K.D.; et al. The Intestinal Microbiota Affect Central Levels of Brain-Derive Neurotropic Factor and Behavior in Mice. Gastroenterology 2011, 141, 599–609. [Google Scholar] [CrossRef] [PubMed]
- Sylvia, K.E.; Jewell, C.P.; Rendon, N.M.; St John, E.A.; Demas, G.E. Sex-specific modulation of the gut microbiome and behavior in Siberian hamsters. Brain Behav. Immun. 2017, 60, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Sylvia, K.E.; Deyoe, J.E.; Demas, G.E. Early-life sickness may predispose Siberian hamsters to behavioral changes following alterations of the gut microbiome in adulthood. Brain Behav. Immun. 2018. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Fan, W.; Zhou, X.; Liu, Y.; Ma, P.; Qi, S.; Gu, B. Probiotics decrease depressive behaviors induced by constipation via activating the AKT signaling pathway. Metab. Brain Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Magori, K.; Kasper, L.H. The chicken or the egg dilemma: Intestinal dysbiosis in multiple sclerosis. Ann. Transl. Med. 2017, 5, 145. [Google Scholar] [CrossRef] [PubMed]
- Berer, K.; Gerdes, L.A.; Cekanaviciute, E.; Jia, X.; Xiao, L.; Xia, Z.; Liu, C.; Klotz, L.; Stauffer, U.; Baranzini, S.E.; et al. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc. Natl. Acad. Sci. USA 2017, 114, 10719–10724. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; KAnner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbates symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Lebouvier, T.; Lardeux, B.; Biraud, M.; Rouaud, T.; Pouclet, H.; Coron, E.; Bruley des Varannes, S.; Naveilhan, P.; Nguyen, J.M.; et al. Colonic inflammation in Parkinson’s disease. Neurobiol. Dis. 2013, 50, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rub, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Verbaan, D.; Marinus, J.; Visser, M.; van Rooden, S.M.; Stiggelbout, A.M.; van Hilten, J.J. Patient-reported autonomic symptoms in Parkinsons’s disease. Neurology 2007, 69, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Grandinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Knoop, F.C.; Owens, M.; Crocker, I.C. Clostridium difficile: Clinical disease and diagnosis. Clin. Microbiol. Rev. 1993, 6, 251–265. [Google Scholar] [CrossRef]
- Ross, C.L.; Spinler, J.K.; Savidge, T.C. Structural and functional changes within the gut microbiota and susceptibility to Clostridium difficile infection. Anaerobe 2016, 41, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Yassour, M.; Vatanen, T.; Siljander, H.; Hämäläinen, A.; Härkönen, T.; Ryhänen, S.J.; Franzosa, E.A.; Vlamakis, H.; Huttenhower, C.; Gevers, D.; et al. Natural history of the infant gut microbiome and impact of antibiotic treatments on strain-level diversity and stability. Sci. Transl. Med. 2017, 8, 343ra81. [Google Scholar] [CrossRef] [PubMed]
- Bruttin, A.; Brüssow, H. Human volunteers receiving Escherichia coli phage T4 orally: A safety test of phage therapy. Antimicrob. Agents Chemother. 2005, 49, 2874–2878. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Pirnay, J.P.; Verbeken, G.; Chanishvili, N.; Tediashvili, M.; Lashkhi, N.; Glonti, T.; Krylov, V.; Mast, J.; Van Parys, L.; et al. Quality-controlled small-scale production of a well-defined bacteriophage cocktail for use in human clinical trials. PLoS ONE 2009, 4, e4944. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.W.; Roach, J.; Azcarate-Peril, M.A. Emerging Technologies for Gut Microbiome Research. Trends Microbiol. 2016, 24, 887–901. [Google Scholar] [CrossRef] [PubMed]
- Breitbart, M.; Hewson, I.; Felts, B.; Mahaffy, J.M.; Nulton, J.; Salamon, P.; Rohwer, F. Metagenomic analyses of an uncultured viral community from human feces. J. Bacteriol. 2003, 85, 6220–6223. [Google Scholar] [CrossRef]
- Mills, S.; Shanahan, F.; Stanton, C.; Hill, C.; Coffey, A.; Ross, R.P. Movers and shakers: Influence of bacteriophage in shaping the mammalian gut microbiota. Gut Microbes 2013, 4, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Modi, S.R.; Lee, H.H.; Spina, C.S.; Collins, J.J. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature 2013, 499, 219–222. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.M.; Koskella, B.; Lin, H.C. Phage therapy: An alternative to antibiotics in the age of multi-drug resistance. World J. Gastrointest. Pharmacol. Ther. 2017, 8, 162–173. [Google Scholar] [CrossRef] [PubMed]
- Regeimbal, J.M.; Jacobs, A.C.; Corey, B.W.; Henry, M.S.; Thompson, M.G.; Pavlicek, R.L.; Quinones, J.; Hannah, R.M.; Ghebremedhin, M.; Crane, N.J.; et al. Personalized Therapeutic Cocktail of Wild Environmental Phages Rescues Mice from Acinetobacter baumannii Wound Infections. Antimicrob. Agents Chemother. 2016, 60, 5806–5816. [Google Scholar] [CrossRef] [PubMed]
- Bamba, S.; Nishida, A.; Imaeda, H.; Inatomi, O.; Sasaki, M.; Sugimoyo, M.; Andoh, A. Successful treatment by fecal microbiota transplantation for Japanese patients with refractory Clostridium difficile infection: A prospective case series. J. Microbiol. Immunol Infect. 2017. [Google Scholar] [CrossRef] [PubMed]
- Friedman-Korn, T.; Livovsky, D.M.; Maharshak, N.; Aviv Cohen, N.; Paz, K.; Bar-Gil Shitri, A.; Goldin, E.; Koslowsky, B. Fecal Transplantation for Treatment of Clostridium Difficile Infection in Elderly and Debilitated Patients. Dig. Dis. Sci. 2018, 63, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Konturek, P.C.; Haziri, D.; Brzozowski, T.; Hess, T.; Heyman, S.; Kwiecien, S.; Konturek, S.J.; Koziel, J. Emerging role of fecal microbiota therapy in the treatment of gastrointestinal and extra-gastrointestinal diseases. J. Physiol. Pharmacol. 2015, 66, 483–491. [Google Scholar] [PubMed]
- Matsuoka, K.; Kanai, T. The gut microbiota and inflammatory bowel disease. Semin. Immunopathol. 2015, 37, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Colman, R.J.; Rubin, D.T. Fecal microbiota transplantation as therapy for inflammatory bowel disease: A systematic review and meta-analysis. J. Crohns Colitis 2014, 8, 1569–1581. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; van der Spek, M.J.; Tijssen, J.G.; Hartman, J.H.; Duflou, A.; Löwenberg, M.; van der Brink, G.R.; Mathus-Vilegen, E.M.; de Vos, W.M.; et al. Findings from a Randomized Controlled Trial of Fecal Transplantation for Patients with Ulcerative Colitis. Gastroenetrology 2015, 149, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Borody, T.J.; Leis, S.M.; Campbell, J.; Torres, M.; Nowak, A. Fecal microbiota transplantation (FMT) in multiple sclerosis (MS) [abstract]. Am. J. Gastroenterol. 2011, 106, S352. [Google Scholar]
- Makkawi, S.; Camara-Lemarroy, C.; Metz, L. Fecal microbiota transplantation associated with 10 years of stability in a patient with SPMS. Neurol. Neuroimmunol. Neuroinflamm. 2018, 5, e459. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Xu, M.; Wang, W.; Cao, X.; Piao, M.; Khan, S.; Yan, F.; Cao, H.; Wang, B. Systematic Review: Adverse Events of Fecal Microbiota Transplantation. PLoS ONE 2016, 11, e0161174. [Google Scholar] [CrossRef] [PubMed]
- Totsch, S.K.; Quinn, T.L.; Strath, L.J.; McMeekin, L.J.; Cowell, R.M.; Gower, B.A.; Sorge, R.E. The impact of the Standard American Diet in rats: Effects on behavior, physiology, and recovery from inflammatory injury. Scand. J. Pain 2017, 17, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Thorburn, A.N.; Macia, L.; Mackay, C.R. Diet, metabolites, and “western-lifestyle” inflammatory diseases. Immunity 2014, 40, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Braniste, V.; Al-Asmakh, M.; Kowal, C.; Anuar, F.; Abbaspour, A.; Tóth, M.; Korecka, A.; Bakocevic, N.; Ng, L.G.; Kundu, P.; et al. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263ra158. [Google Scholar] [CrossRef] [PubMed]
- Riccio, P.; Rossano, R. Diet, Gut Microbiota, and Vitamins D + A in Multiple Sclerosis. Neurotherapeutics 2018, 15, 75–91. [Google Scholar] [CrossRef] [PubMed]
- Shoemaker, T.J.; Mowry, E.M. A review of vitamin D supplementation as disease-modifying therapy. Mult. Scler. 2018, 24, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Zhang, H.; Wu, H.; Li, H.; Liu, L.; Guo, J.; Li, C.; Shih, D.Q.; Zhang, X. Protective role of 1,25(OH)2 vitamin D3 in the mucosal injury and epithelial barrier disruption in DSS-induced acute colitis in mice. BMC Gastroenterol. 2012, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Sun, J. VDR/vitamin D receptor regulates autophagic activity through ATG16L1. Autophagy 2016, 12, 1057–1058. [Google Scholar] [CrossRef] [PubMed]
- Rotstein, D.L.; Healy, B.C.; Malik, M.T.; Carruthers, R.L.; Musallam, A.J.; Kivisakk, P.; Weiner, H.L.; Glanz, B.; Chitnis, T. Effect of vitamin D on MS activity by disease-modifying therapy class. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e167. [Google Scholar] [CrossRef] [PubMed]
- Tankou, S.K.; Regev, K.; Healy, B.C.; Cox, L.M.; Tjon, E.; Kivisakk, P.; Vanande, I.P.; Cook, S.; Gandhi, R.; Glanz, B.; et al. Investigation of probiotics in multiple sclerosis. Mult. Scler. 2018, 24, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Shapira, L.; Ayalon, S.; & Brenner, T. Effects of Porphyromonas gingivalis on the central nervous system: Activation of glial cells and exacerbation of experimental autoimmune encephalomyelitis. J. Periodontology 2002, 73, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Nichols, F.C.; Housley, W.J.; O’Conor, C.A.; Manning, T.; Wu, S.; & Clark, R.B. Unique lipids from a common human bacterium represent a new class of Toll-like receptor 2 ligands capable of enhancing autoimmunity. Am. J. Pathol. 2009, 175, 2430–2438. [Google Scholar] [CrossRef] [PubMed]
- Ezendam, J.; de Klerk, A.; Gremmer, E.R.; van Loveren, H. Effects of Bifidobacterium Animalis Administered During Lactation on Allergic and Autoimmune Responses in Rodents. Clin. Exp. Immunol. 2008, 154, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Lavasani, S.; Dzhambazov, B.; Nouri, M.; Fåk, F.; Buske, S.; Molin, G.; Thorlacius, H.; Alenfall, J.; Jeppsson, B.; Weström, B. A Novel Probiotic Mixture Exerts a Therapeutic Effect on Experimental Autoimmune Encephalomyelitis Mediated by IL-10 Producing Regulatory T Cells. PLoS ONE 2010, 5, e9009–e9011. [Google Scholar] [CrossRef] [PubMed]
- Rezende, R.M.; Oliveira, R.P.; Medeiros, S.R.; Gomes-Santos, A.C.; Alves, A.C.; Loli, F.G.; Guimarães, M.A.; Amaral, S.S.; da Cunha, A.P.; Weiner, H.L.; et al. Hsp65-producing Lactococcus lactis prevents experimental autoimmune encephalomyelitis in mice by inducing CD4+LAP+ regulatory T cells. J. Autoimmun 2013, 40, 45–57. [Google Scholar] [CrossRef] [PubMed]
- Takata, K.; Kinoshita, M.; Okuno, T.; Moriya, M.; Kohda, T.; Honorat, J.A.; Sugimoto, T.; Kumanogoh, A.; Kayama, H.; Takeda, K.; et al. The lactic acid bacterium Pediococcus acidilactici suppresses autoimmune encephalomyelitis by inducing IL-10-producing regulatory T cells. PLoS ONE 2011, 6, e27644. [Google Scholar] [CrossRef] [PubMed]
- Mangalam, A.; Shahi, S.K.; Luckey, D.; Karau, M.; Marietta, E.; Luo, N.; Choung, R.S.; Ju, J.; Sompallae, R.; Gibson-Corley, K.; et al. Human Gut-Derived Commensal Bacteria Suppress CNS Inflammatory and Demyelinating Disease. Cell Rep. 2017, 20, 1269–1277. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Wang, Y.; Begum-Haque, S.; Dasgupta, S.; Kasper, D.L.; Kasper, L.H. A Polysaccharide from the Human Commensal Bacteroides Fragilis Protects Against CNS Demyelinating Disease. Mucosal Immunol. 2010, 3, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Ochoa-Repáraz, J.; Mielcarz, D.W.; Ditrio, L.E.; Burroughs, A.R.; Begum-Haque, S.; Dasgupta, S.; Kasper, D.L.; Kasper, L.H. Central nervous system demyelinating disease protection by the human commensal Bacteroides fragilis depends on polysaccharide A expression. J. Immunol. 2010, 185, 4101–4108. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Telesford, K.M.; Ochoa-Reparaz, J.; Haque-Begum, S.; Christy, M.; Kasper, E.J.; Wang, L.; Wu, Y.; Robson, S.C.; Kasper, D.L.; et al. An Intestinal Commensal Symbiosis Factor Controls Neuroinflammation via TLR2-Mediated CD39 Signalling. Nat. Commun. 2014, 5, 4432. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Begum-Haque, S.; Telesford, K.M.; Ochoa-Reparaz, J.; Christy, M.; Kasper, E.J.; Kasper, D.L.; Robson, S.C.; Kasper, L.H. A Commensal Bacterial Product Elicits and Modulates Migratory Capacity of CD39+ CD4 T Regulatory Subsets in the Suppression of Neuroinflammation. Gut Microbes 2014, 5, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Liu, C.H.; Tzianabos, A.O.; Kasper, D.L. An Immunomodulatory Molecule of Symbiotic Bacteria Directs Maturation of the Host Immune System. Cell 2005, 122, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A Microbial Symbiosis Factor Prevents Intestinal Inflammatory Disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.L.; Jones, M.B.; Cobb, B.A. Bacterial Capsular Polysaccharide Prevents the Onset of Asthma Through T-Cell Activation. Glycobiology 2015, 25, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, J.M.; Lonergan, R.; Costelloe, L.; Kinsella, K.; Moran, B.; O’Farrelly, C.; Tubridy, N.; Mills, K.H.G. CD39+Foxp3+ Regulatory T Cells Suppress Pathogenic Th17 Cells and Are Impaired in Multiple Sclerosis. J. Immunol. 2009, 183, 7602–7610. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Erturk-Hasdemir, D.; Ochoa-Reparaz, J.; Reinecker, H.C.; Kasper, D.L. Plasmacytoid dendritic cells mediate anti-inflammatory responses to a gut commensal molecule via both innate and adaptive mechanisms. Cell Host Microbe 2014, 15, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Telesford, K.M.; Yan, W.; Ochoa-Reparaz, J.; Pant, A.; Kircher, C.; Christy, M.A.; Begum-Haque, S.; Kasper, D.L.; Kasper, L.H. A Commensal Symbiotic Factor Derived from Bacteroides Fragilis Promotes Human CD39+Foxp3+ T Cells and Treg Function. Gut Microbes 2015, 6, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Burgess, J.N.; Pant, A.B.; Kasper, L.H.; Colpitts Brass, S. CD4+ T cells from multiple sclerosis patients respond to a commensal-derived antigen. Ann. Clin. Transl. Neurol. 2017, 4, 825–829. [Google Scholar] [CrossRef] [PubMed]
- Torkildsen, Ø.; Myhr, K.-M.; Bø, L. Disease-modifying treatments for multiple sclerosis—A review of approved medications. Eur. J. Neurol. 2016, 23, 18–27. [Google Scholar] [CrossRef] [PubMed]


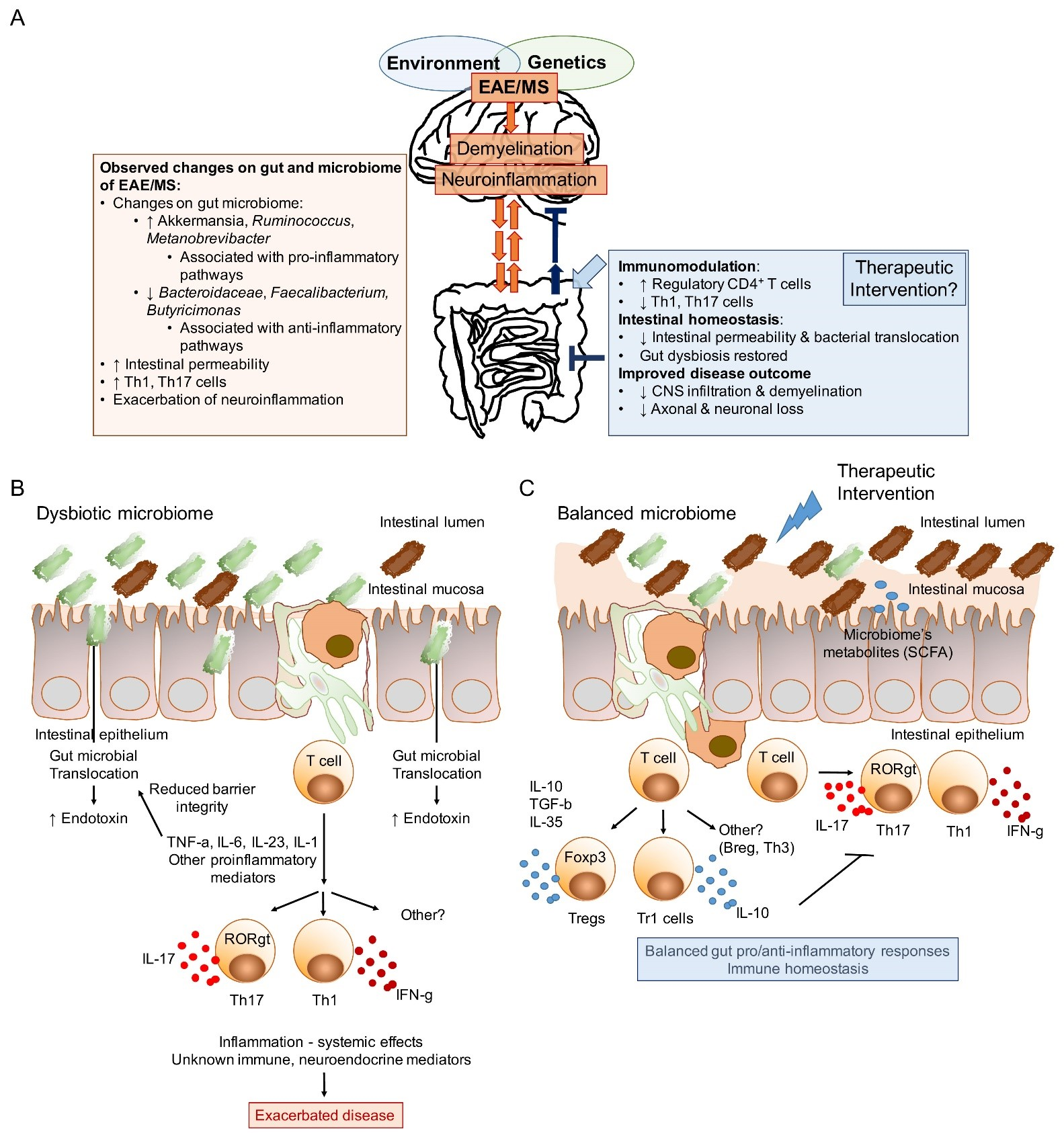{kind=link}
{kind=link}
| Microbial Intervention | Model | Experimental Outcome | Proposed Mechanism of Action | Refs |
|---|---|---|---|---|
| Colonization studies in germ-free (GF) animals or mice treated with antibiotics | ||||
| Monocolonization of GF with segmented filamentous bacteria (SFB) | Mouse, EAE | Disease susceptibility restored | GF mice: Reduced peripheral Th17 cell and increased Treg frequencies and anti-inflammatory cytokines. Reconstitution with SFB-induced Th17 cells. | [51] |
| Monocolonization of GF with specific pathogen-free microbiota | Mouse (MOG-specific TCR Tg), EAE | Disease susceptibility restored | GF mice: Deficit in Th17 cell in lamina propria and Peyer’s patches. Lack of autoimmune T cells and B cell recruitment and autoantibody production reduced. SPF microbiota restores susceptibility | [50] |
| Antibiotics + PSA-producing Bacteroides fragilis | Mouse, EAE | Disease severity restored (PSA-production dependent) | Antibiotics treated: disease reduction [38,39]. PSA-deficient, but not PSA-producing B. fragilis restores EAE susceptibility by promoting IL-17-producing and interferon-gamma proinflammatory cells | [113] |
| Colonization of GF mice with gut microbiota of MS patients | Mouse, EAE | Restores disease susceptibility | MS gut microbiota reduces proportions and function of IL-10+ Tregs | [70] |
| Colonization of GF mice with gut microbiota of MS patients | Mouse (MOG-specific TCR Tg), EAE | Restores disease susceptibility | MS gut microbiota promotes Treg dysfunction and reduces immunoregulation by IL-10 | [69] |
| Colonization studies in conventionally housed animals | ||||
| Colonization with Lactococcus spp. | Mouse, EAE | Reduced severity | Induction of IL-10-producing Tregs | [108] |
| Colonization with Bifidobacterium animalis | Lewis rats, EAE | Reduces EAE severity | Proposed changes in Th1/Th2 balances | [107] |
| Colonization with Pediococcus acidilactici R037 | Mouse, EAE | Reduces EAE severity | Induction of IL-10-producing Tr1 cells | [110] |
| Colonization with Lactococcus lactis Hsp65 | Mouse, EAE | Reduces the severity of EAE | Induction of Tregs cells and LAP+ CD4+ Tregs | [109] |
| Colonization with Prevotella histicola | HLA class II Tg mouse, EAE | Reduces the severity of EAE | Induction of Tregs, reduction of Th1 and Th17 cells function | [111] |
| Treatment with purified symbiont factor isolated from gut microbiota | ||||
| Oral treatment with purified PSA produced by B. fragilis | Mouse, EAE | Reduces the severity of EAE | Prophylactic and therapeutic protection by IL-10-producing CD39+ and Tregs | [113,114,115] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirby, T.O.; Ochoa-Repáraz, J. The Gut Microbiome in Multiple Sclerosis: A Potential Therapeutic Avenue. Med. Sci. 2018, 6, 69. https://doi.org/10.3390/medsci6030069
Kirby TO, Ochoa-Repáraz J. The Gut Microbiome in Multiple Sclerosis: A Potential Therapeutic Avenue. Medical Sciences. 2018; 6(3):69. https://doi.org/10.3390/medsci6030069
Chicago/Turabian StyleKirby, Trevor O., and Javier Ochoa-Repáraz. 2018. "The Gut Microbiome in Multiple Sclerosis: A Potential Therapeutic Avenue" Medical Sciences 6, no. 3: 69. https://doi.org/10.3390/medsci6030069
APA StyleKirby, T. O., & Ochoa-Repáraz, J. (2018). The Gut Microbiome in Multiple Sclerosis: A Potential Therapeutic Avenue. Medical Sciences, 6(3), 69. https://doi.org/10.3390/medsci6030069