The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Obesity and Type 2 Diabetes Mellitus
- Abdominal fat distribution, determined by an abdominal circumference of over 102 cm in men or over 88 cm in women (Caucasian).
- Serum triglycerides greater than 150 mg/dL (>1.7 mmol/L), or therapy already initiated to reduce triglycerides.
- High density lipoprotein (HDL) cholesterol ≤ 40 mg/dL (<1.05 mmol/L) in men or <50 mg/dL (1.25 mmol/L) in women.
- Blood pressure of 130/85 mmHg or more, or already initiated therapy to reduce hypertension.
- Fasting blood sugar ≥ 110 mg/dL (5.6 mmol/L), or type 2 diabetes.
2.1. The Role and Composition of Gut Microbiota in Developing Elements of the Metabolic Syndrome with Special Regard to Obesity
2.2. The Role and Composition of Gut Microbiota in Developing Elements of the Metabolic Syndrome with Special Regard to Diabetes Mellitus Type 2
2.3. Possible Pathophysiologic Action of Gut Microbiota in the Development of Obesity and Diabetes Mellitus Type 2
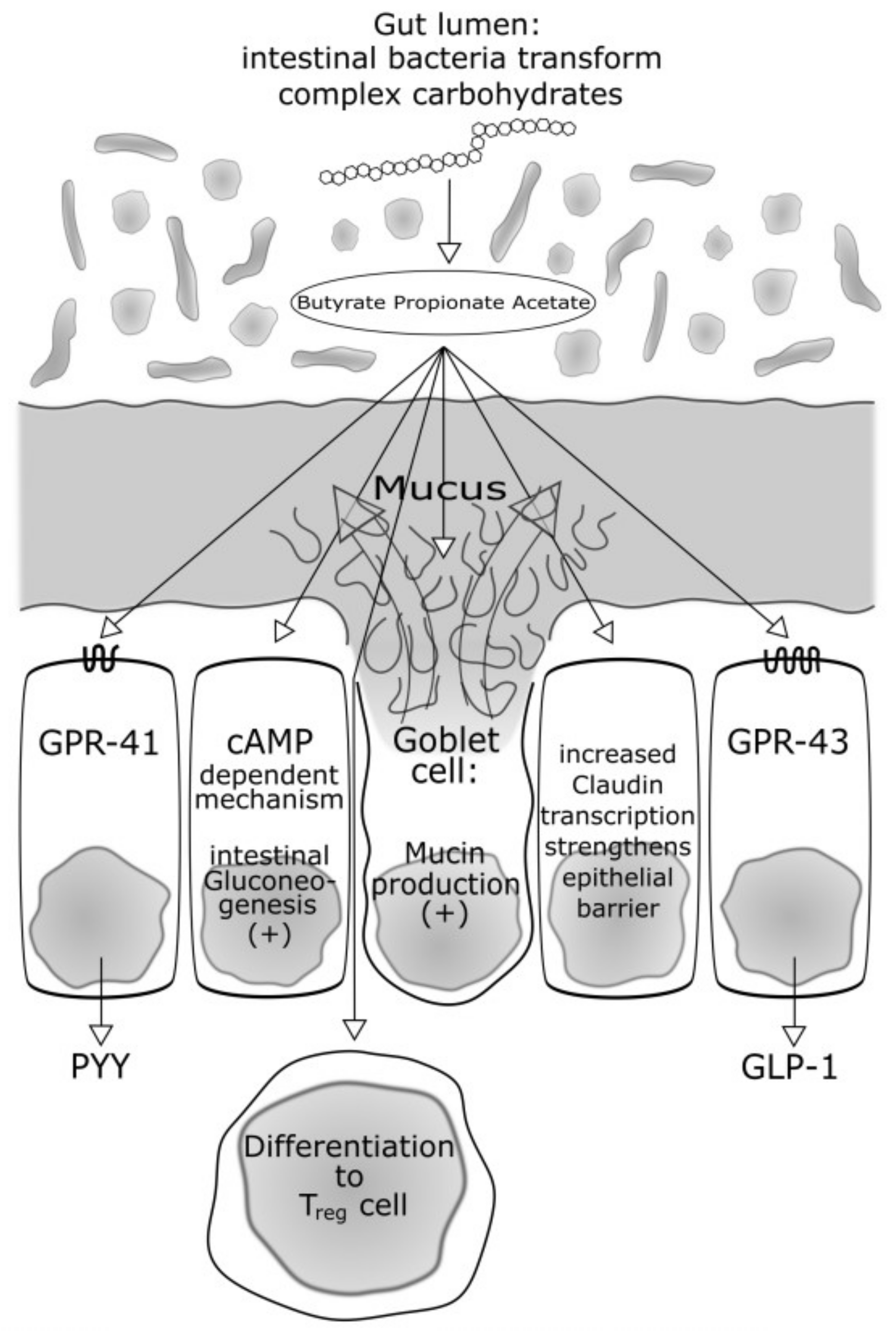
2.3.1. Bacteria Producing Butyrate and Other Short-Chain Fatty Acids: Role in Energy Intake, Food Regulation and Insulin Sensitivity
2.3.2. Microbiome with an Increased Capacity for Energy Harvest in Obesity and in Diabetes
2.3.3. Gut Microbiota and Bile Acids
2.3.4. Influence on the Immune System—Low-Grade Metabolic Inflammation (I): Short-Chain Fatty Acids and the Inflammasome
2.3.5. Influence on the Immune System—Low-Grade Metabolic Inflammation (II): Endotoxinemia and the Lipopolysaccharides
2.3.6. Influence on the Immune System—Low-Grade Metabolic Inflammation (III): Metabolic Infection
3. Type 1 Diabetes Mellitus
3.1. The Role and Composition of Gut Microbiota with Special Regard of Diabetes Mellitus Type 1
3.2. Possible Action and Pathophysiology of Gut Microbiota in the Development of Diabetes Mellitus Type 1
3.2.1. Impact of Gut Microbiota on Toll-Like Receptors
3.2.2. Impact of Sex Hormones on the Development of Gut Microbiota Promoting or Preventing Autoimmunity
3.2.3. Endotoxinemia and the Lipopolysaccharides—Role in Autoimmunity
3.2.4. Butyrate—Also a Protective Role in Autoimmunity?
4. Therapy
4.1. Metformin
4.2. Metabolic Surgery
4.3. Probiotics
4.4. Faecal Microbiota Transplantation
5. Summary and Perspectives
Acknowledgments
Conflicts of Interest
References
- Lederberg, J.; McCray, A. Ome sweet omics—A genealogical treasury of words. Scientist 2001, 15, 8. [Google Scholar]
- Ursell, L.K.; Metcalf, J.L.; Parfrey, L.W.; Knight, R. Defining the human microbiome. Nutr. Rev. 2012, 70 (Suppl. 1), 38–44. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S. Shotgun DNA sequencing using cloned DNase I-generated fragments. Nucleic Acids Res. 1981, 9, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. The Human Microbiome Project Consortium: Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health National Heart, Lung, and Blood Institute. National Cholesterol Education Program (NCEP): Third Report of the Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (ATP III Final Report); NIH Publication No. 02-5215; NIH: Bethesda, MD, USA, 2002. [Google Scholar]
- Vazquez, G.; Duval, S.; Jacobs, D.R., Jr.; Silventoinen, K. Comparison of body mass index, waist circumference, and waist/hip ratio in predicting incident diabetes: A meta-analysis. Epidemiol. Rev. 2007, 115, 2007–2029. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1031. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Cheng, J.; Duncan, A.E.; Kau, A.L.; Griffin, N.W.; Lombard, V.; Henrissat, B.; Bain, J.R.; et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 2013, 341, 1241214. [Google Scholar] [CrossRef] [PubMed]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef] [PubMed]
- Swidsinski, A.; Weber, J.; Loening-Baucke, V.; Hale, L.P.; Lochs, H. Spatial organization and composition of the mucosal flora in patients with inflammatory bowel disease. J. Clin. Microbiol. 2005, 43, 3380–3389. [Google Scholar] [CrossRef] [PubMed]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Microbial ecology: Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef] [PubMed]
- Furet, J.P.; Kong, L.C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Mariat, D.; Corthier, G.; Doré, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weigh tloss: Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Cotillard, A.; Kennedy, S.P.; Kong, L.C.; Prifti, E.; Pons, N.; Le Chatelier, E.; Almeida, M.; Quinquis, B.; Levenez, F.; Galleron, N.; et al. Dietary intervention: Impact on gut microbial gene richness. Nature 2013, 500, 585–588. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.; Li, Y.; Cai, Z.; Li, S.; Zhu, J.; Zhang, F.; Liang, S.; Zhang, W.; Guan, Y.; Shen, D.; et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012, 490, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Biagi, E.; Nylund, L.; Candela, M.; Ostan, R.; Bucci, L.; Pini, E.; Nikkïla, J.; Monti, D.; Satokari, R.; Franceschi, C.; et al. Through ageing and beyond: Gut microbiota and inflammatory status in seniors and centenarians. PLoS ONE 2010, 5, e10667. [Google Scholar] [CrossRef]
- Hur, K.Y.; Lee, M.-S. Gut microbiota and metabolic disorders. Diabetes Metab. J. 2015, 39, 198–203. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Shi, J.; Mao, X. Safety and efficacy of acarbose in the treatment of diabetes in Chinese patients. Ther. Clin. Risk Manag. 2014, 10, 505–511. [Google Scholar] [PubMed]
- Zhang, X.; Fang, Z.; Zhang, C.; Xia, H.; Jie, Z.; Han, X.; Chen, Y.; Ji, L. Effects of Acarbose on the gut microbiota of prediabetic patients: A randomized, double-blind, controlled crossover trial. Diabetes Ther. 2017, 8, 293–307. [Google Scholar] [CrossRef] [PubMed]
- Shin, N.R.; Lee, J.C.; Lee, H.Y.; Kim, M.S.; Whon, T.W.; Lee, M.S.; Bae, J.W. An increase in the Akkermansia spp. population induced by metformin treatment improves glucose homeostasis in diet-induced obese mice. Gut 2014, 63, 727–735. [Google Scholar] [CrossRef] [PubMed]
- De la Cuesta-Zuluaga, J.; Mueller, N.T.; Corrales-Agudelo, V.; Velásquez-Mejía, E.P.; Carmona, J.A.; Abad, J.M.; Escobar, J.S. Metformin Is associated with higher relative abundance of mucin-degrading Akkermansia muciniphila and several short-chain fatty acid-producing microbiota in the gut. Diabetes Care 2017, 40, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A.R. Microbiota and diabetes: An evolving relationship. Gut 2014, 63, 1513–1521. [Google Scholar] [CrossRef] [PubMed]
- Kimura, I.; Ozawa, K.; Inoue, D.; Imamura, T.; Kimura, K.; Maeda, T.; Terasawa, K.; Kashihara, D.; Hirano, K.; Tani, T.; et al. The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat. Commun. 2013, 4, 1829. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Tazoe, H.; Hayashi, H.; Kashiwabara, H.; Tooyama, K.; Suzuki, Y.; Kuwahara, A. Expression of the short-chain fatty acid receptor, GPR43, in the human colon. J. Mol. Histol. 2008, 39, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Tazoe, H.; Otomo, Y.; Kaji, I.; Tanaka, R.; Karaki, S.I.; Kuwahara, A. Roles of short-chain fatty acids receptors, GPR41 and GPR43 on colonic functions. J. Physiol. Pharmacol. 2008, 59 (Suppl. 2), 251–262. [Google Scholar] [PubMed]
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 2012, 6, 364–371. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Goncalves, D.; Vinera, J.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-generated metabolites promote metabolic benefits via gut-brain neural circuits. Cell 2014, 156, 84–96. [Google Scholar] [CrossRef] [PubMed]
- De Vadder, F.; Kovatcheva-Datchary, P.; Zitoun, C.; Duchampt, A.; Bäckhed, F.; Mithieux, G. Microbiota-produced succinate improves glucose homeostasis via intestinal gluconeogenesis. Cell Metab. 2016, 24, 151–157. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Sina, C.; Gavrilova, O.; Förster, M.; Till, A.; Derer, S.; Hildebrand, F.; Raabe, B.; Chalaris, A.; Scheller, J.; Rehmann, A.; et al. G protein-coupled receptor 43 is essential for neutrophil recruitment during intestinal inflammation. J. Immunol. 2009, 183, 7514–7522. [Google Scholar] [CrossRef] [PubMed]
- Vinolo, M.A.; Ferguson, G.J.; Kulkarni, S.; Damoulakis, G.; Anderson, K.; Bohlooly, Y.M.; Stephens, L.; Hawkins, P.T.; Curi, R. SCFAs induce mouse neutrophil chemotaxis through the GPR43 receptor. PLoS ONE 2011, 6, e21205. [Google Scholar] [CrossRef] [PubMed]
- Neel, J.V. Diabetes Mellitus: A “thrifty” genotype rendered detrimental by “progress”? Am. J. Hum. Genet. 1962, 14, 353–362. [Google Scholar] [PubMed]
- Swann, J.R.; Want, E.J.; Geier, F.M.; Spagou, K.; Wilson, I.D.; Sidaway, J.E.; Nicholson, J.K.; Holmes, E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4523–4530. [Google Scholar] [CrossRef] [PubMed]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Houten, S.M.; Mataki, C.; Christoffolete, M.A.; Kim, B.W.; Sato, H.; Messaddeq, N.; Harney, J.W.; Ezaki, O.; Kodama, T.; et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature 2006, 439, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Hirokane, H.; Nakahara, M.; Tachibana, S.; Shimizu, M.; Sato, R. Bile acid reduces the secretion of very low density lipoprotein by repressing microsomal triglyceride transfer protein gene expression mediated by hepatocyte nuclear factor-4. J. Biol. Chem. 2004, 279, 45685–45692. [Google Scholar] [CrossRef] [PubMed]
- Kast, H.R.; Nguyen, C.M.; Sinal, C.J.; Jones, S.A.; Laffitte, B.A.; Reue, K.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: A molecular mechanism linking plasma triglyceride levels to bile acids. Mol. Endocrinol. 2001, 15, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Klüppelholz, B.; Thorand, B.; Koenig, W.; de Las Heras Gala, T.; Meisinge, C.; Huth, C.; Giani, G.; Franks, P.W.; Roden, M.; Rathmann, W.; et al. Association of subclinical inflammation with deterioration of glycaemia before the diagnosis of type 2 diabetes: The KORA S4/F4 study. Diabetologia 2015, 58, 2269–2277. [Google Scholar] [CrossRef] [PubMed]
- Trompette, A.; Gollwitzer, E.S.; Yadava, K.; Sichelstiel, A.K.; Sprenger, N.; Ngom-Bru, C.; Blanchard, C.; Junt, T.; Nicod, L.P.; Harris, N.L.; et al. Gut microbiota metabolism of dietary fibre influences allergic airway disease and haematopoiesis. Nat. Med. 2014, 20, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Elinav, E.; Strowig, T.; Kau, A.L.; Henao-Mejia, J.; Thaiss, C.A.; Booth, C.J.; Peaper, D.R.; Bertin, J.; Eisenbarth, S.C.; Gordon, J.I.; et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 2011, 145, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kallio, K.A.E.; Buhlin, K.; Jauhiainen, M.; Keva, R.; Tuomainen, A.M.; Klinge, B.; Gustafsson, A.; Pussinen, P.J. Lipopolysaccharide associates with pro-atherogenic lipoproteins in periodontitis patients. Innate Immun. 2008, 14, 247–253. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Amar, J.; Iglesias, M.A.; Poggi, M.; Knauf, C.; Bastelica, D.; Neyrinck, A.M.; Fava, F.M.; Tuohy, K.M.; Chabo, C.; et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007, 56, 1761–1772. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, C.; Gallagher, E.; Horton, F.; Ellis, R.J.; Ijaz, U.Z.; Wu, H.; Jaiyeola, E.; Diribe, O.; Duparc, T.; Cani, P.D.; et al. Host-microbiome interactions in human type 2 diabetes following prebiotic fibre (galacto-oligosaccharide) intake. Br. J. Nutr. 2016, 116, 1869–1877. [Google Scholar] [CrossRef] [PubMed]
- Lassenius, M.I.; Pietiläinen, K.H.; Kaartinen, K.; Pussinen, P.J.; Syrjänen, J.; Forsblom, C.; Pörsti, I.; Rissanen, A.; Kaprio, J.; Mustonen, J.; et al. Bacterial endotoxin activity in human serum is associated with dyslipidaemia, insulin resistance, obesity, and chronic inflammation. Diabetes Care 2011, 34, 1809–1815. [Google Scholar] [CrossRef] [PubMed]
- Pussinen, P.J.; Havulinna, A.S.; Lehto, M.; Sundvall, J.; Salomaa, V. Endotoxemia is associated with an increased risk of incident diabetes. Diabetes Care 2011, 34, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Munford, R.S. Endotoxemia-menace, marker, or mistake? J. Leukoc. Biol. 2016, 100, 687–698. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Serino, M.; Lange, C.; Chabo, C.; Iacovoni, J.; Mondot, S.; Lepage, P.; Klopp, C.; Mariette, J.; Bouchez, O.; et al. Involvement of tissue bacteria in the onset of diabetes in humans: Evidence for a concept. Diabetologia 2011, 54, 3055–3061. [Google Scholar] [CrossRef] [PubMed]
- Burcelin, R.; Serino, M.; Chabo, C.; Garidou, L.; Pomié, C.; Courtney, M.; Amar, J.; Bouloumié, A. Metagenome and metabolism: The tissue microbiota hypothesis. Diabetes Obes. Metab. 2013, 15 (Suppl. 3), 61–70. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Chabo, C.; Waget, A.; Klopp, P.; Vachoux, C.; Bermúdez-Humarán, L.G.; Smirnova, N.; Bergé, M.; Sulpice, T.; Lahtinen, S.; et al. Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: Molecular mechanisms and probiotic treatment. EMBO Mol. Med. 2011, 3, 559–572. [Google Scholar] [CrossRef] [PubMed]
- Amar, J.; Lange, C.; Payros, G.; Garret, C.; Chabo, C.; Lantieri, O.; Courtney, M.; Marre, M.; Charles, M.A.; Balkau, B.; et al. Blood microbiota dysbiosis is associated with the onset of cardiovascular events in a large general population: The DESIR study. PLoS ONE 2013, 8, e54461. [Google Scholar] [CrossRef] [PubMed]
- American Diabetes Association. Classification and diagnosis of diabetes. Sec. 2. In Standards of Medical Care in Diabetes—2017. Diabetes Care 2017, 40 (Suppl. 1), S11–S24. [Google Scholar]
- Chillarón, J.J.; Benaiges, D.; Mañé, L.; Pedro-Botet, J.; Flores Le-Roux, J.A. Obesity and type 1 diabetes mellitus management. Minerva Endocrinol. 2015, 40, 53–60. [Google Scholar] [PubMed]
- Paun, A.; Yau, C.; Danska, J.S. The Influence of the Microbiome on Type 1 Diabetes. J. Immunol. 2017, 590–595. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Calcinaro, F.; Dionisi, S.; Marinaro, M.; Candeloro, P.; Bonato, V.; Marzotti, S.; Corneli, R.B.; Ferretti, E.; Gulino, A.; Grasso, F.; et al. Oral probiotic administration induces interleukin-10 production and prevents spontaneous autoimmune diabetes in the non-obese diabetic mouse. Diabetologia 2005, 48, 1565–1575. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Nagata, Y.; Kado, S.; Uchida, K.; Kato, I.; Hashimoto, S.; Yokokura, T. Prevention of onset in an insulin-dependent diabetes mellitus model, NOD mice, by oral feeding of Lactobacillus casei. APMIS 1997, 105, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Brugmann, S.; Klatter, F.A.; Visser, J.T.J.; Wildeboer-Veloo, A.C.M.; Harmsen, H.J.M.; Rozinmg, J. Antibiotic treatment partially protects against type 1 diabetes in the bio-breeding diabetes-prone rat. Is the gut flora involved in the development of type 1 diabetes? Diabetologia 2006, 49, 2105–2108. [Google Scholar] [CrossRef] [PubMed]
- Wicker, L.S.; Miller, B.J.; Coker, L.Z.; Mcnally, S.E.; Scott, S.; Mullen, Y. Genetic control of diabetes and insulinitis in the nonobese diabetic mouse. J. Exp. Med. 1987, 165, 1639–1654. [Google Scholar] [CrossRef] [PubMed]
- Roesch, L.F.; Lorca, G.L.; Casella, G.; Giongo, A.; Naranjo, A.; Pionzio, A.M.; Li, N.; Mai, V.; Wasserfall, C.H.; Schatz, D.; et al. Culture-independent identification of gut bacteria correlated with the onset of diabetes in a rat model. ISME J. 2009, 3, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Novelo, L.L.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; Hyöty, H.; et al. Toward defining the autoimmune microbiome for type 1 diabetes. ISME J. 2011, 5, 82–91. [Google Scholar] [CrossRef] [PubMed]
- De Goffau, M.C.; Luopajärvi, K.; Knip, M.; Ilonen, J.; Ruohtula, T.; Härkönen, T.; Orivuori, L.; Hakala, S.; Welling, G.W.; Harmsen, H.J.; et al. Faecal microbiota composition differs between children with β-cell autoimmunity and those without. Diabetes 2013, 62, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, D.; zu Castell, W.; Ardissone, A.; Davis-Richardson, A.G.; Achenbach, P.; Hagen, M.; Pflueger, M.; Gano, K.A.; Fagen, J.R.; Drew, J.C.; et al. Compromised gut microbiota networks in children with anti-islet cell autoimmunity. Diabetes 2014, 63, 2006–2014. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.D.; Gevers, D.; Siljander, H.; Vatanen, T.; Hyötyläinen, T.; Hämäläinen, A.M.; Peet, A.; Tillmann, V.; Pöhö, P.; Mattila, I.; et al. The dynamics of the human infant gut microbiome in development and in progression toward type 1 diabetes. Cell Host Microbe 2015, 17, 260–273. [Google Scholar] [CrossRef] [PubMed]
- De Groot, P.F.; Belzer, C.; Aydin, Ö.; Levin, E.; Levels, J.H.; Aalvink, S.; Boot, F.; Holleman, F.; van Raalte, D.H.; Scheithauer, T.P.; et al. Distinct faecal and oral microbiota composition in human type 1 diabetes, an observational study. PLoS ONE 2017, 12, e0188475. [Google Scholar] [CrossRef] [PubMed]
- Qi, C.J.; Zhang, Q.; Yu, M.; Xu, J.P.; Zheng, J.; Wang, T.; Xiao, X.H. Imbalance of Faecal Microbiota at Newly Diagnosed Type 1 Diabetes in Chinese Children. Chin. Med. J. (Engl.) 2016, 129, 1298–1304. [Google Scholar] [PubMed]
- Pellegrini, S.; Sordi, V.; Bolla, A.M.; Saita, D.; Ferrarese, R.; Canducci, F.; Clementi, M.; Invernizzi, F.; Mariani, A.; Bonfanti, R.; et al. Duodenal Mucosa of Patients With Type 1 Diabetes Shows Distinctive Inflammatory Profile and Microbiota. J. Clin. Endocrinol. Metab. 2017, 102, 1468–1477. [Google Scholar] [CrossRef] [PubMed]
- Beutler, B.A. TLRs and innate immunity. Blood 2009, 113, 1399–1407. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Ley, R.E.; Volchkov, P.Y.; Stranges, P.B.; Avanesyan, L.; Stonebraker, A.C.; Hu, C.; Wong, F.S.; Szot, G.L.; Bluestone, J.A.; et al. Innate immunity and intestinal microbiota in the development of Type 1 diabetes. Nature 2008, 455, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Peng, J.; Narasimhan, S.; Marchesi, J.R.; Benson, A.; Wong, F.S.; Wen, L. Long term effect of gut microbiota transfer on diabetes development. J. Autoimmun. 2014, 53, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Burrows, M.P.; Volchkov, P.; Kobayashi, K.S.; Chervonsky, A.V. Microbiota regulates type 1 diabetes through Toll-like receptors. Proc. Natl. Acad. Sci. USA 2015, 112, 9973–9977. [Google Scholar] [CrossRef] [PubMed]
- Pozzilli, P.; Signore, A.; Williams, A.J.; Beales, P.E. NOD mouse colonies around the world–recent facts and figures. Immunol. Today 1993, 14, 193–196. [Google Scholar] [CrossRef]
- Markle, J.G.; Fish, E.N. SeXX matters in immunity. Trends Immunol. 2014, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Mortin-Toth, S.; Robertson, C.E.; Feazel, L.M.; Rolle-Kampczyk, U.; von Bergen, M.; McCoy, K.D.; Macpherson, A.J.; Danska, J.S. Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 2013, 339, 1084–1088. [Google Scholar] [CrossRef] [PubMed]
- Markle, J.G.; Frank, D.N.; Adeli, K.; von Bergen, M.; Danska, J.S. Microbiome manipulation modifies sex-specific risk for autoimmunity. Gut Microbes 2014, 5, 485–493. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yurkovetskiy, L.; Burrows, M.; Khan, A.A.; Graham, L.; Volchkov, P.; Becker, L.; Antonopoulos, D.; Umesaki, Y.; Chervonsky, A.V. Gender bias in autoimmunity is influenced by microbiota. Immunity 2013, 39, 400–412. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, A.; Reunanen, A.; Romanov, A.; Karvonen, A.; Viskari, H.; Vesikari, T.; Ilonen, J.; Knip, M.; Hyöty, H. A six-fold gradient in the incidence of type 1 diabetes at the eastern border of Finland. Ann. Med. 2005, 37, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Mustonen, N.; Siljander, H.; Peet, A.; Tillmann, V.; Härkönen, T.; Ilonen, J.; Hyöty, H.; Knip, M. DIABIMMUNE Study Group. Early childhood infections precede development of beta-cell autoimmunity and type 1 diabetes in children with HLA-conferred disease risk. Pediatr. Diabetes 2017. [Google Scholar] [CrossRef]
- Vatanen, T.; Kostic, A.D.; d’Hennezel, E.; Siljander, H.; Franzosa, E.A.; Yassour, M.; Kolde, R.; Vlamakis, H.; Arthur, T.D.; Hämäläinen, A.M.; et al. Variation in Microbiome LPS Immunogenicity Contributes to Autoimmunity in Humans. Cell 2016, 165, 842–853. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Park, B.S.; Kim, J.I.; Kim, S.E.; Lee, J.; Oh, S.C.; Enkhbayar, P.; Matsushima, N.; Lee, H.; Yoo, O.J.; et al. Crystal structure of the TLR4-MD-2 complex with bound endotoxin antagonist Eritoran. Cell 2007, 130, 906–917. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.T.; Davis-Richardson, A.G.; Giongo, A.; Gano, K.A.; Crabb, D.B.; Mukherjee, N.; Casella, G.; Drew, J.C.; Ilonen, J.; Knip, M.; et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011, 6, e25792. [Google Scholar] [CrossRef] [PubMed]
- Endesfelder, D.; Engel, M.; Davis-Richardson, A.G.; Ardissone, A.N.; Achenbach, P.; Hummel, S.; Winkler, C.; Atkinson, M.; Schatz, D.; Triplett, E.; et al. Towards a functional hypothesis relating anti-islet cell autoimmunity to the dietary impact on microbial communities and butyrate production. Microbiome 2016, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Hummel, S.; Pflüger, M.; Hummel, M.; Bonifacio, E.; Ziegler, A.G. Primary dietary intervention study to reduce the risk of islet autoimmunity in children at increased risk for type 1 diabetes: The BABYDIET study. Diabetes Care 2011, 34, 1301–1305. [Google Scholar] [CrossRef] [PubMed]
- Bosi, E.; Molteni, L.; Radaelli, M.G.; Folini, L.; Fermo, I.; Bazzigaluppi, E.; Piemonti, L.; Pastore, M.R.; Paroni, R. Increased intestinal permeability precedes clinical onset of type 1 diabetes. Diabetologia 2006, 49, 2824–2827. [Google Scholar] [CrossRef] [PubMed]
- Burger-van Paassen, N.; Vincent, A.; Puiman, P.J.; van der Sluis, M.; Bouma, J.; Boehm, G.; van Goudoever, J.B.; van Seuningen, I.; Renes, I.B. The regulation of intestinal mucin MUC2 expression by short-chain fatty acids: Implications for epithelial protection. Biochem. J. 2009, 420, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef] [PubMed]
- Scheppach, W.; Weiler, F. The butyrate story: Old wine in new bottles? Curr. Opin. Clin. Nutr. Metab. Care 2004, 7, 563–567. [Google Scholar] [CrossRef] [PubMed]
- Menzel, T.; Lührs, H.; Zirlik, S.; Schauber, J.; Kudlich, T.; Gerke, T.; Gostner, A.; Neumann, M.; Melcher, R.; Scheppach, W. Butyrate inhibits leukocyte adhesion to endothelial cells via modulation of VCAM-1. Inflamm. Bowel Dis. 2004, 10, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Siljander, H. The role of the intestinal microbiota in type 1 diabetes mellitus. Nat. Rev. Endocrinol. 2016, 12, 154–167. [Google Scholar] [CrossRef] [PubMed]
- Holman, R. Metformin as first choice in oral diabetes treatment: The UKPDS experience. Journ. Annu. Diabetol. Hotel Dieu 2007, 13–20. [Google Scholar] [PubMed]
- Liou, A.P.; Paziuk, M.; Luevano, J.M., Jr.; Machineni, S.; Turnbaugh, P.J.; Kaplan, L.M. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci. Transl. Med. 2013, 5, 178ra41. [Google Scholar] [CrossRef] [PubMed]
- López-Cepero, A.A.; Palacios, C. Association of the Intestinal Microbiota and Obesity. P R Health Sci. J. 2015, 34, 60–64. [Google Scholar] [PubMed]
- Zhang, H.; DiBaise, J.K.; Zuccolo, A.; Kudrna, D.; Braidotti, M.; Yu, Y.; Parameswaran, P.; Crowell, M.D.; Wing, R.; Rittmann, B.E.; et al. Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. USA 2009, 106, 2365–2370. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.; Guarner, F.; Reid, G.; Gibson, G.R.; Merenstein, D.J.; Pot, B.; Morelli, L.; Canani, R.B.; Flint, H.J.; Salminen, S.; et al. Expert consensus document. The International Scientific Association for Probiotics and Prebiotics consensus statement on the scope and appropriate use of the term probiotic. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Roberfroid, M.B. Prebiotics and synbiotics: Concepts and nutritional properties. Br. J. Nutr. 1998, 80, 197–202. [Google Scholar]
- Patterson, E.; Ryan, P.M.; Cryan, J.F.; Dinan, T.G.; Ross, R.P.; Fitzgerald, G.F.; Stanton, C. Gut microbiota, obesity and diabetes. Postgrad. Med. J. 2016, 92, 286–300. [Google Scholar] [CrossRef] [PubMed]
- Ejtahed, H.S.; Mohtadi-Nia, J.; Homayouni-Rad, A.; Niafar, M.; Asghari-Jafarabadi, M.; Mofid, V. Probiotic yogurt improves antioxidant status in type 2 diabetic patients. Nutrition 2012, 28, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Akbari, V.; Hendijani, F. Effects of probiotic supplementation in patients with type 2 diabetes: Systematic review and meta-analysis. Nutr. Rev. 2016, 74, 774–784. [Google Scholar] [CrossRef] [PubMed]
- Samah, S.; Ramasamy, K.; Lim, S.M.; Neoh, C.F. Probiotics for the management of type 2 diabetes mellitus: A systematic review and meta-analysis. Diabetes Res. Clin. Pract. 2016, 118, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Hampe, C.S.; Roth, C.L. Probiotic strains and mechanistic insights for the treatment of type 2 diabetes. Endocrine 2017, 58, 207–227. [Google Scholar] [CrossRef] [PubMed]
- Knip, M.; Honkanen, J. Modulation of Type 1 Diabetes Risk by the Intestinal Microbiome. Curr. Diabetes Rep. 2017, 17, 105. [Google Scholar] [CrossRef] [PubMed]
- Valladares, R.; Sankar, D.; Li, N.; Williams, E.; Lai, K.K.; Abdelgeliel, A.S.; Gonzalez, C.F.; Wasserfall, C.H.; Larkin, J.; Schatz, D.; et al. Lactobacillus johnsonii N6.2 mitigates the development of type 1 diabetes in BB-DP rats. PLoS ONE 2010, 5, e10507. [Google Scholar] [CrossRef] [PubMed]
- Lau, K.; Benitez, P.; Ardissone, A.; Wilson, T.D.; Collins, E.L.; Lorca, G.; Li, N.; Sankar, D.; Wasserfall, C.; Neu, J.; et al. Inhibition of type 1 diabetes correlated to a Lactobacillus johnsonii N6.2-mediated Th17 bias. J. Immunol. 2011, 186, 3538–3546. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Scheen, A.J. Pharmacological management of type 2 diabetes: What’s new in 2017? Expert Rev. Clin. Pharmacol. 2017, 10, 1383–1394. [Google Scholar] [CrossRef] [PubMed]
- Van Raalte, D.H.; Verchere, C.B. Glucagon-Like Peptide-1 Receptor Agonists: Beta-Cell Protection or Exhaustion? Trends Endocrinol. Metab. 2016, 27, 442–445. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.; Curtis, K.L.; March, J.C. Secretion of insulinotropic proteins by commensal bacteria: Rewiring the gut to treat diabetes. Appl. Environ. Microbiol. 2008, 74, 7437–7438. [Google Scholar] [CrossRef] [PubMed]
- Duan, F.F.; Liu, J.H.; March, J.C. Engineered commensal bacteria reprogram intestinal cells into glucose-responsive insulin-secreting cells for the treatment of diabetes. Diabetes 2015, 64, 1794–1803. [Google Scholar] [CrossRef] [PubMed]
- Ryan, P.M.; Patterson, E.; Kent, R.M.; Stack, H.; O’Connor, P.M.; Murphy, K.; Peterson, V.L.; Mandal, R.; Wishart, D.S.; Dinan, T.G.; et al. Recombinant Incretin-Secreting Microbe Improves Metabolic Dysfunction in High-Fat Diet Fed Rodents. Sci. Rep. 2017, 7, 13523. [Google Scholar] [CrossRef] [PubMed]
- Udayappan, S.D.; Hartstra, A.V.; Dallinga-Thie, G.M.; Nieuwdorp, M. Intestinal microbiota and faecal transplantation as treatment modality for insulin resistance and type 2 diabetes mellitus. Clin. Exp. Immunol. 2014, 177, 24–29. [Google Scholar] [CrossRef] [PubMed]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012, 143, 913–916. [Google Scholar] [CrossRef] [PubMed]
- Richey, J.M.; Woolcott, O. Re-visiting the Endocannabinoid System and Its Therapeutic Potential in Obesity and Associated Diseases. Curr. Diabetes Rep. 2017, 17, 99. [Google Scholar] [CrossRef] [PubMed]
- Muccioli, G.G.; Naslain, D.; Bäckhed, F.; Reigstad, C.S.; Lambert, D.M.; Delzenne, N.M.; Cani, P.D. The endocannabinoid system links gut microbiota to adipogenesis. Mol. Syst. Biol. 2010, 6, 92. [Google Scholar] [CrossRef] [PubMed]
- Soltani, N.; Qiu, H.; Aleksic, M.; Glinka, Y.; Zhao, F.; Liu, R.; Li, Y.; Zhang, N.; Chakrabarti, R.; Ng, T.; et al. GABA exerts protective and regenerative effects on islet beta cells and reverses diabetes. Proc. Natl. Acad. Sci. USA 2011, 108, 11692–11697. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Dang, H.; Chen, Z.; Guan, A.; Jin, Y.; Atkinson, M.A.; Kaufman, D.L. γ-Aminobutyric acid regulates both the survival and replication of human β-cells. Diabetes 2013, 62, 3760–3765. [Google Scholar] [CrossRef] [PubMed]
- Kootte, R.S.; Levin, E.; Salojärvi, J.; Smits, L.P.; Hartstra, A.V.; Udayappan, S.D.; Hermes, G.; Bouter, K.E.; Koopen, A.M.; Holst, J.J.; et al. Improvement of Insulin Sensitivity after Lean Donor Faeces in Metabolic Syndrome Is Driven by Baseline Intestinal Microbiota Composition. Cell Metab. 2017, 611–619. [Google Scholar] [CrossRef] [PubMed]
- Atıcı, S.; Soysal, A.; Karadeniz Cerit, K.; Yılmaz, Ş.; Aksu, B.; Kıyan, G.; Bakır, M. Catheter-related Saccharomyces cerevisiae Fungemia Following Saccharomyces boulardii Probiotic Treatment: In a child in intensive care unit and review of the literature. Med. Mycol. Case Rep. 2017, 15, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Lammi, N.; Karvonen, M.; Tuomilehto, J. Do microbes have a causal role in type 1 diabetes? Med. Sci. Monit. 2005, 11, 63–69. [Google Scholar]
- De Groot, P.F.; Frissen, M.N.; de Clercq, N.C.; Nieuwdorp, M. Faecal microbiota transplantation in metabolic syndrome: History, present and future. Gut Microbes 2017, 8, 253–267. [Google Scholar] [CrossRef] [PubMed]
- Alang, N.; Kelly, C.R. Weight gain after faecal microbiota transplantation. Open Forum Infect. Dis. 2015, 2, ofv004. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Harsch, I.A.; Konturek, P.C. The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Med. Sci. 2018, 6, 32. https://doi.org/10.3390/medsci6020032
Harsch IA, Konturek PC. The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Medical Sciences. 2018; 6(2):32. https://doi.org/10.3390/medsci6020032
Chicago/Turabian StyleHarsch, Igor Alexander, and Peter Christopher Konturek. 2018. "The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases" Medical Sciences 6, no. 2: 32. https://doi.org/10.3390/medsci6020032
APA StyleHarsch, I. A., & Konturek, P. C. (2018). The Role of Gut Microbiota in Obesity and Type 2 and Type 1 Diabetes Mellitus: New Insights into “Old” Diseases. Medical Sciences, 6(2), 32. https://doi.org/10.3390/medsci6020032