
Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Anti-Tumor Responses



{kind=link}
{kind=link}
Abstract
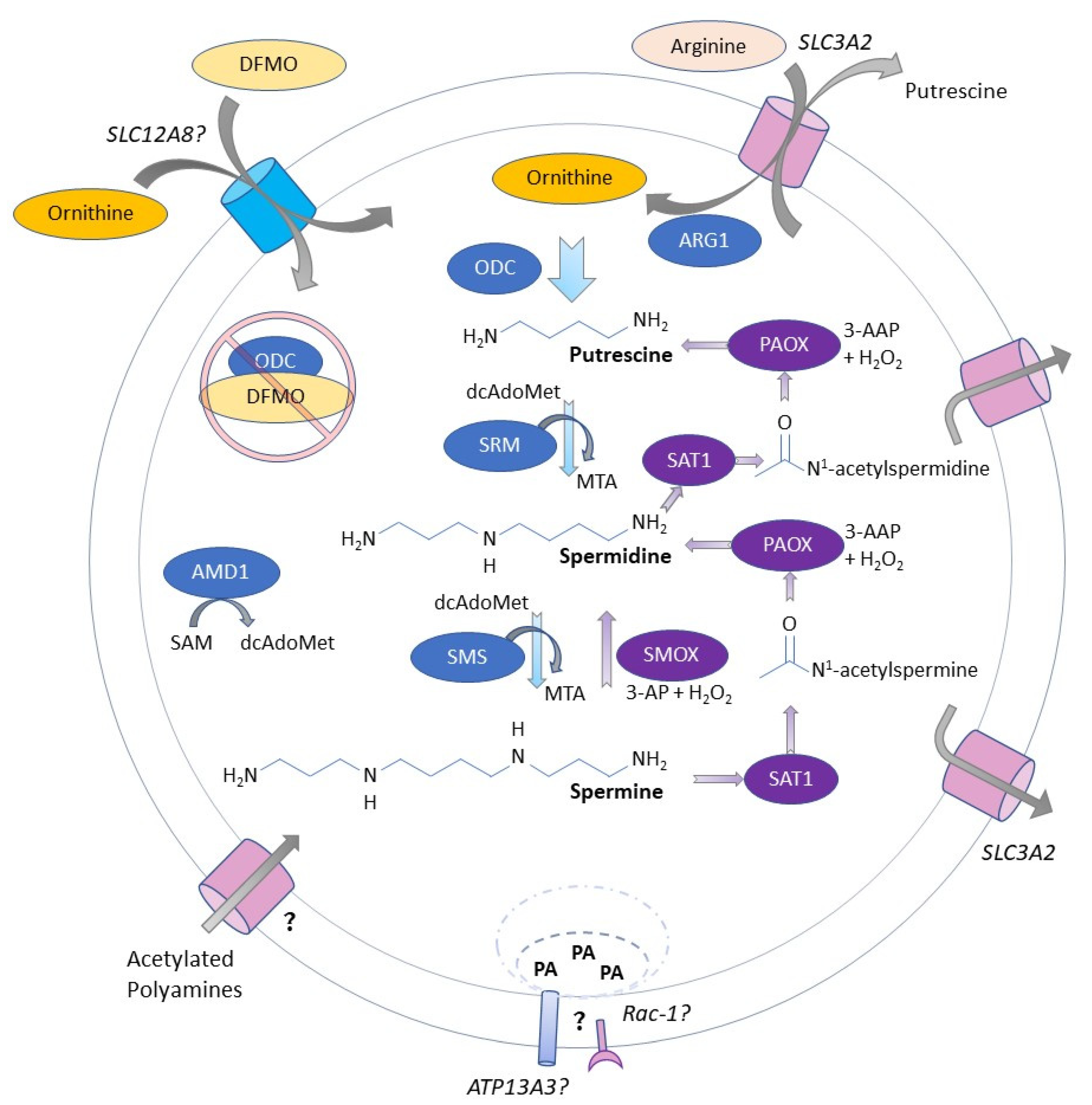
:1. Introduction
2. New Evidence Indicates That Polyamines Are Critical for Immune Cell Specification and Function
2.1. Deficiency in Polyamine Biosynthesis in CD4+ T Cells Confers Aberrant Function and Delayed Proliferation but Enhances IFNγ Expression
2.2. Spermidine Potentiates Foxp3-Expressing Tregs
2.3. Polyamine Transport Can Compensate for Deficiency in Biosynthesis in CD4+ T Cells
2.4. Odc Deletion in Myeloid Cells Enhances Pro-Inflammatory Response to Bacterial Infection
2.5. Microbiota-Derived Polyamines Are Important for Modulating Macrophage Function
2.6. The Role of Hypusine Biosynthesis in Specifying Macrophage Polarization Is Unclear
2.7. Immune Cell-Intrinsic Deficiencies in Polyamine Biosynthesis Induced by Odc Deletion or Pharmacological Inhibition via DFMO Can Restrict Proliferation and Alter Function
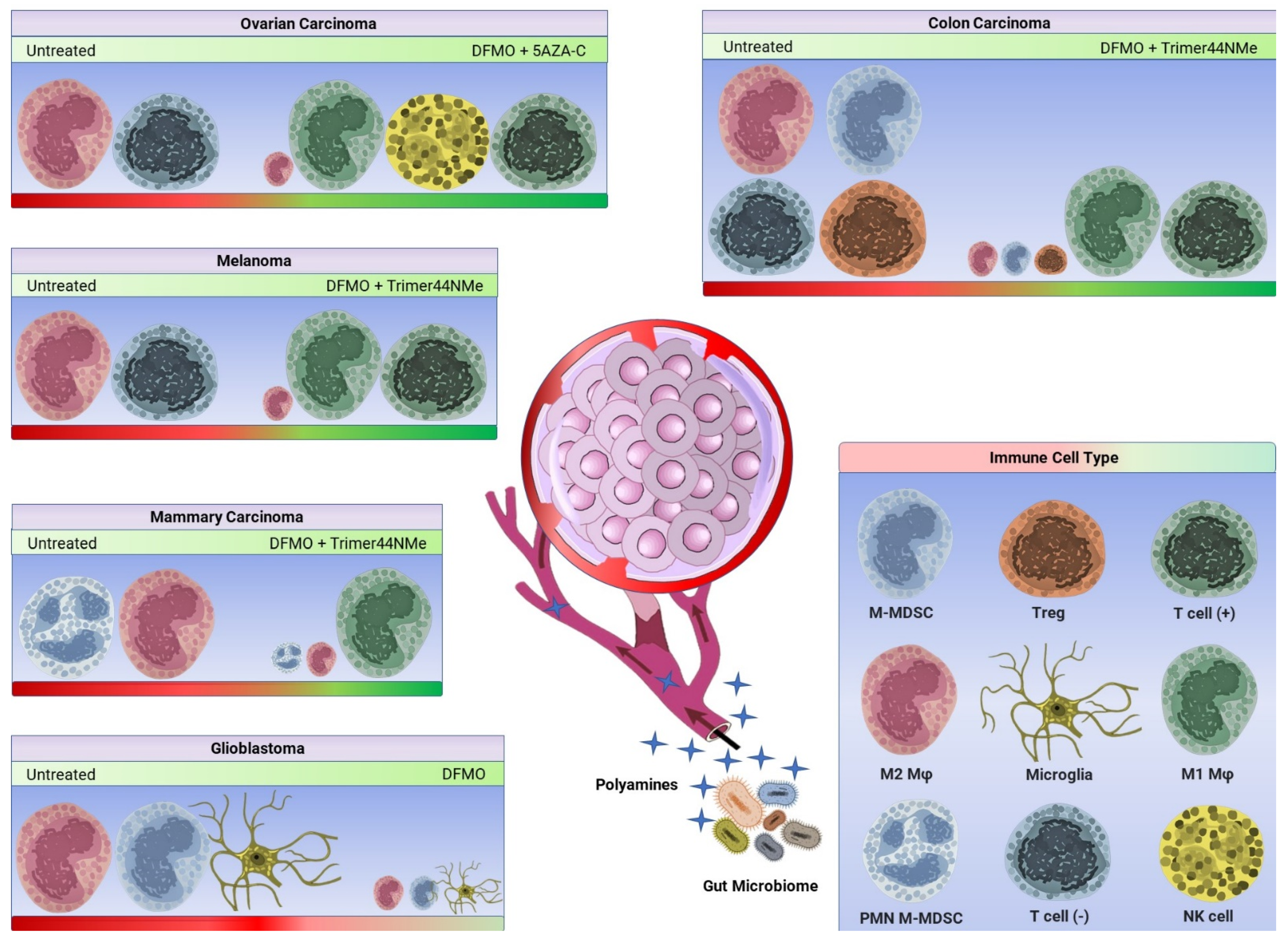
3. Immunomodulatory Effect of DFMO in the Context of Polyamine Blocking Therapy
3.1. DFMO and AMXT 1501 Reduce Tumor Growth in Immunocompetent Mice
3.2. DFMO Alone Can Reduce Immunosuppression in the Tumor Microenvironment
3.3. DFMO and Trimer44NMe Reduce Immunosuppression and Sensitize Tumors to α-PD-1
3.4. DFMO and 5-Azacytidine Reverse Immunosuppression and Enhance Pro-Inflammatory Myeloid-Derived Cells
4. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Casero, R.A., Jr.; Marton, L.J. Targeting polyamine metabolism and function in cancer and other hyperproliferative diseases. Nat. Rev. Drug Discov. 2007, 6, 373–390. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A., Jr.; Murray Stewart, T.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef] [PubMed]
- Murray Stewart, T.; Dunston, T.T.; Woster, P.M.; Casero, R.A., Jr. Polyamine catabolism and oxidative damage. J. Biol. Chem. 2018, 293, 18736–18745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uemura, T.; Yerushalmi, H.F.; Tsaprailis, G.; Stringer, D.E.; Pastorian, K.E.; Hawel, L., 3rd; Byus, C.V.; Gerner, E.W. Identification and characterization of a diamine exporter in colon epithelial cells. J. Biol. Chem. 2008, 283, 26428–26435. [Google Scholar] [CrossRef] [Green Version]
- Hamouda, N.N.; Van den Haute, C.; Vanhoutte, R.; Sannerud, R.; Azfar, M.; Mayer, R.; Cortes Calabuig, A.; Swinnen, J.V.; Agostinis, P.; Baekelandt, V.; et al. ATP13A3 is a major component of the enigmatic mammalian polyamine transport system. J. Biol. Chem. 2021, 296, 100182. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, V.; Andl, T.; Phanstiel, O.t. ATP13A3 facilitates polyamine transport in human pancreatic cancer cells. Sci. Rep. 2022, 12, 4045. [Google Scholar] [CrossRef] [PubMed]
- McCubbrey, A.L.; McManus, S.A.; McClendon, J.D.; Thomas, S.M.; Chatwin, H.B.; Reisz, J.A.; D’Alessandro, A.; Mould, K.J.; Bratton, D.L.; Henson, P.M.; et al. Polyamine import and accumulation causes immunomodulation in macrophages engulfing apoptotic cells. Cell Rep. 2022, 38, 110222. [Google Scholar] [CrossRef] [PubMed]
- Poulin, R.; Casero, R.A.; Soulet, D. Recent advances in the molecular biology of metazoan polyamine transport. Amino Acids 2012, 42, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Novita Sari, I.; Setiawan, T.; Seock Kim, K.; Toni Wijaya, Y.; Won Cho, K.; Young Kwon, H. Metabolism and function of polyamines in cancer progression. Cancer Lett. 2021, 519, 91–104. [Google Scholar] [CrossRef]
- Pendeville, H.; Carpino, N.; Marine, J.C.; Takahashi, Y.; Muller, M.; Martial, J.A.; Cleveland, J.L. The ornithine decarboxylase gene is essential for cell survival during early murine development. Mol. Cell. Biol. 2001, 21, 6549–6558. [Google Scholar] [CrossRef] [Green Version]
- Gabay, M.; Li, Y.; Felsher, D.W. MYC activation is a hallmark of cancer initiation and maintenance. Cold Spring Harb. Perspect. Med. 2014, 4, a014241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.Z.; Zuo, Z.W.; Zhou, Z.R.; Ji, Y. Polyamine homeostasis-based strategies for cancer: The role of combination regimens. Eur. J. Pharmacol. 2021, 910, 174456. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Baixauli, F.; Sanin, D.E.; Edwards-Hicks, J.; Villa, M.; Kabat, A.M.; Kaminski, M.M.; Stanckzak, M.; Weiss, H.J.; Grzes, K.M.; et al. Polyamine metabolism is a central determinant of helper T cell lineage fidelity. Cell 2021, 184, 4186–4202.e20. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Nishimura, K.; Zanelli, C.F.; Valentini, S.R. Functional significance of eIF5A and its hypusine modification in eukaryotes. Amino Acids 2010, 38, 491–500. [Google Scholar] [CrossRef] [Green Version]
- Park, M.H.; Wolff, E.C. Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 2018, 293, 18710–18718. [Google Scholar] [CrossRef] [Green Version]
- Anderson-Baucum, E.; Pineros, A.R.; Kulkarni, A.; Webb-Robertson, B.J.; Maier, B.; Anderson, R.M.; Wu, W.; Tersey, S.A.; Mastracci, T.L.; Casimiro, I.; et al. Deoxyhypusine synthase promotes a pro-inflammatory macrophage phenotype. Cell Metab. 2021, 33, 1883–1893.e7. [Google Scholar] [CrossRef]
- Carriche, G.M.; Almeida, L.; Stuve, P.; Velasquez, L.; Dhillon-LaBrooy, A.; Roy, U.; Lindenberg, M.; Strowig, T.; Plaza-Sirvent, C.; Schmitz, I.; et al. Regulating T-cell differentiation through the polyamine spermidine. J. Allergy Clin. Immunol. 2021, 147, 335–348.e11. [Google Scholar] [CrossRef]
- Wu, R.; Chen, X.; Kang, S.; Wang, T.; Gnanaprakasam, J.R.; Yao, Y.; Liu, L.; Fan, G.; Burns, M.R.; Wang, R. De novo synthesis and salvage pathway coordinately regulate polyamine homeostasis and determine T cell proliferation and function. Sci. Adv. 2020, 6, eabc4275. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Hardbower, D.M.; Asim, M.; Luis, P.B.; Singh, K.; Barry, D.P.; Yang, C.; Steeves, M.A.; Cleveland, J.L.; Schneider, C.; Piazuelo, M.B.; et al. Ornithine decarboxylase regulates M1 macrophage activation and mucosal inflammation via histone modifications. Proc. Natl. Acad. Sci. USA 2017, 114, E751–E760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakamura, A.; Kurihara, S.; Takahashi, D.; Ohashi, W.; Nakamura, Y.; Kimura, S.; Onuki, M.; Kume, A.; Sasazawa, Y.; Furusawa, Y.; et al. Symbiotic polyamine metabolism regulates epithelial proliferation and macrophage differentiation in the colon. Nat. Commun. 2021, 12, 2105. [Google Scholar] [CrossRef] [PubMed]
- Puleston, D.J.; Buck, M.D.; Klein Geltink, R.I.; Kyle, R.L.; Caputa, G.; O’Sullivan, D.; Cameron, A.M.; Castoldi, A.; Musa, Y.; Kabat, A.M.; et al. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 2019, 30, 352–363.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, S.J.; Ruckerl, D.; Thomas, G.D.; Hewitson, J.P.; Duncan, S.; Brombacher, F.; Maizels, R.M.; Hume, D.A.; Allen, J.E. IL-4 directly signals tissue-resident macrophages to proliferate beyond homeostatic levels controlled by CSF-1. J. Exp. Med. 2013, 210, 2477–2491. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Ito, T.; Fields, L.; Shiraishi, M.; Ichihara, Y.; Sato, N.; Podaru, M.; Kainuma, S.; Tanaka, H.; Suzuki, K. IL-4 as a repurposed biological drug for myocardial infarction through augmentation of reparative cardiac macrophages: Proof-of-concept data in mice. Sci. Rep. 2017, 7, 6877. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.L.; Assmann, N.; O’Connor, E.; Keane, C.; Walls, J.; Choi, C.; Oefner, P.J.; Gardiner, C.M.; Dettmer, K.; Finlay, D.K. De novo polyamine synthesis supports metabolic and functional responses in activated murine NK cells. Eur. J. Immunol. 2021, 51, 91–102. [Google Scholar] [CrossRef]
- Hayes, C.S.; Shicora, A.C.; Keough, M.P.; Snook, A.E.; Burns, M.R.; Gilmour, S.K. Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Miska, J.; Rashidi, A.; Lee-Chang, C.; Gao, P.; Lopez-Rosas, A.; Zhang, P.; Burga, R.; Castro, B.; Xiao, T.; Han, Y.; et al. Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma. Sci. Adv. 2021, 7, eabc8929. [Google Scholar] [CrossRef]
- Muth, A.; Madan, M.; Archer, J.J.; Ocampo, N.; Rodriguez, L.; Phanstiel, O.t. Polyamine transport inhibitors: Design, synthesis, and combination therapies with difluoromethylornithine. J. Med. Chem. 2014, 57, 348–363. [Google Scholar] [CrossRef]
- Alexander, E.T.; Minton, A.; Peters, M.C.; Phanstiel, O.t.; Gilmour, S.K. A novel polyamine blockade therapy activates an anti-tumor immune response. Oncotarget 2017, 8, 84140–84152. [Google Scholar] [CrossRef] [Green Version]
- Gitto, S.B.; Pandey, V.; Oyer, J.L.; Copik, A.J.; Hogan, F.C.; Phanstiel, O.T.; Altomare, D.A. Difluoromethylornithine Combined with a polyamine transport inhibitor is effective against gemcitabine resistant pancreatic cancer. Mol. Pharm. 2018, 15, 369–376. [Google Scholar] [CrossRef]
- Nakkina, S.P.; Gitto, S.B.; Pandey, V.; Parikh, J.G.; Geerts, D.; Maurer, H.C.; Olive, K.P.; Phanstiel, O.t.; Altomare, D.A. Differential expression of polyamine pathways in human pancreatic tumor progression and effects of polyamine blockade on tumor microenvironment. Cancers 2021, 13, 6391. [Google Scholar] [CrossRef]
- Alexander, E.T.; Mariner, K.; Donnelly, J.; Phanstiel, O.t.; Gilmour, S.K. Polyamine blocking therapy decreases survival of tumor-infiltrating immunosuppressive myeloid cells and enhances the antitumor efficacy of PD-1 blockade. Mol. Cancer Ther. 2020, 19, 2012–2022. [Google Scholar] [CrossRef] [PubMed]
- Boivin, G.; Faget, J.; Ancey, P.B.; Gkasti, A.; Mussard, J.; Engblom, C.; Pfirschke, C.; Contat, C.; Pascual, J.; Vazquez, J.; et al. Durable and controlled depletion of neutrophils in mice. Nat. Commun. 2020, 11, 2762. [Google Scholar] [CrossRef] [PubMed]
- Stone, M.L.; Chiappinelli, K.B.; Li, H.; Murphy, L.M.; Travers, M.E.; Topper, M.J.; Mathios, D.; Lim, M.; Shih, I.M.; Wang, T.L.; et al. Epigenetic therapy activates type I interferon signaling in murine ovarian cancer to reduce immunosuppression and tumor burden. Proc. Natl. Acad. Sci. USA 2017, 114, E10981–E10990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Travers, M.; Brown, S.M.; Dunworth, M.; Holbert, C.E.; Wiehagen, K.R.; Bachman, K.E.; Foley, J.R.; Stone, M.L.; Baylin, S.B.; Casero, R.A., Jr.; et al. DFMO and 5-azacytidine increase M1 macrophages in the tumor microenvironment of murine ovarian cancer. Cancer Res. 2019, 79, 3445–3454. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V.; Donthireddy, L.; Marvel, D.; Condamine, T.; Wang, F.; Lavilla-Alonso, S.; Hashimoto, A.; Vonteddu, P.; Behera, R.; Goins, M.A.; et al. Cancer-associated fibroblasts neutralize the anti-tumor effect of CSF1 receptor blockade by inducing PMN-MDSC infiltration of tumors. Cancer Cell 2017, 32, 654–668.e5. [Google Scholar] [CrossRef] [Green Version]
- O’Brien, S.A.; Orf, J.; Skrzypczynska, K.M.; Tan, H.; Kim, J.; DeVoss, J.; Belmontes, B.; Egen, J.G. Activity of tumor-associated macrophage depletion by CSF1R blockade is highly dependent on the tumor model and timing of treatment. Cancer Immunol. Immunother. 2021, 70, 2401–2410. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Li, L.; Starr, T.K.; Subramanian, S. Tumor location impacts immune response in mouse models of colon cancer. Oncotarget 2017, 8, 54775–54787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in M1(LPS+) vs. classically and M2(LPS−) vs. alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef] [PubMed]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5, eaay6017. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Kjallquist, U.; Moliner, A.; Zajac, P.; Fan, J.B.; Lonnerberg, P.; Linnarsson, S. Characterization of the single-cell transcriptional landscape by highly multiplex RNA-seq. Genome Res. 2011, 21, 1160–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merritt, C.R.; Ong, G.T.; Church, S.E.; Barker, K.; Danaher, P.; Geiss, G.; Hoang, M.; Jung, J.; Liang, Y.; McKay-Fleisch, J.; et al. Multiplex digital spatial profiling of proteins and RNA in fixed tissue. Nat. Biotechnol. 2020, 38, 586–599. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chin, A.; Bieberich, C.J.; Stewart, T.M.; Casero, R.A., Jr. Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Anti-Tumor Responses. Med. Sci. 2022, 10, 31. https://doi.org/10.3390/medsci10020031
Chin A, Bieberich CJ, Stewart TM, Casero RA Jr. Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Anti-Tumor Responses. Medical Sciences. 2022; 10(2):31. https://doi.org/10.3390/medsci10020031
Chicago/Turabian StyleChin, Alexander, Charles J. Bieberich, Tracy Murray Stewart, and Robert A. Casero, Jr. 2022. "Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Anti-Tumor Responses" Medical Sciences 10, no. 2: 31. https://doi.org/10.3390/medsci10020031
APA StyleChin, A., Bieberich, C. J., Stewart, T. M., & Casero, R. A., Jr. (2022). Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Anti-Tumor Responses. Medical Sciences, 10(2), 31. https://doi.org/10.3390/medsci10020031