Experimental Determination of Impure CO2 Alteration of Calcite Cemented Cap-Rock, and Long Term Predictions of Cap-Rock Reactivity


Abstract
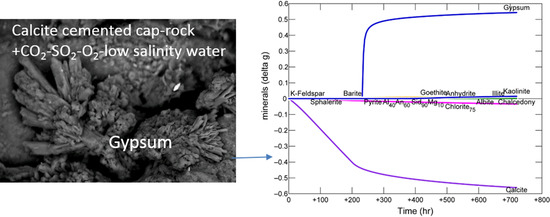
:
1. Introduction
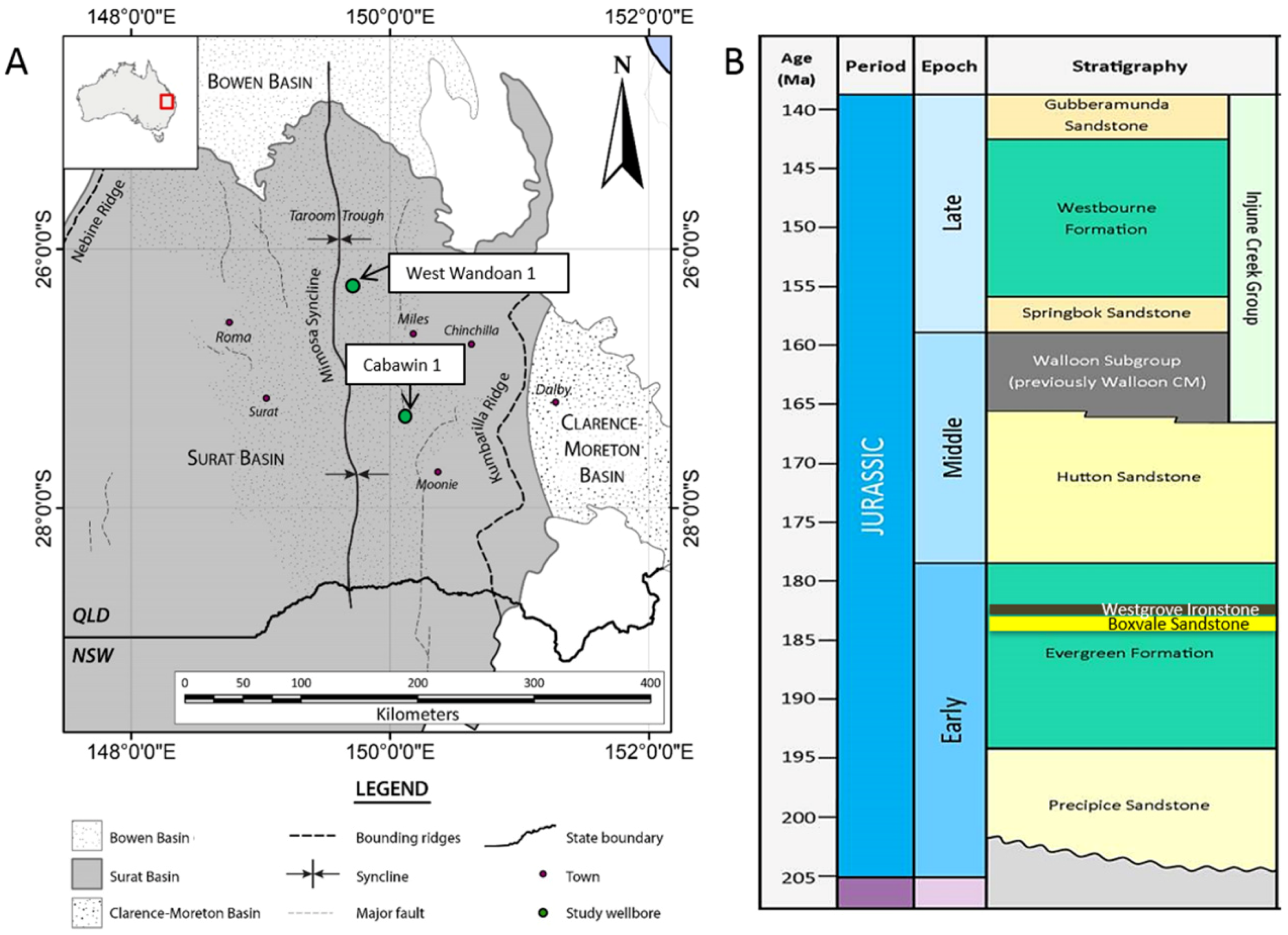
2. Materials and Methods
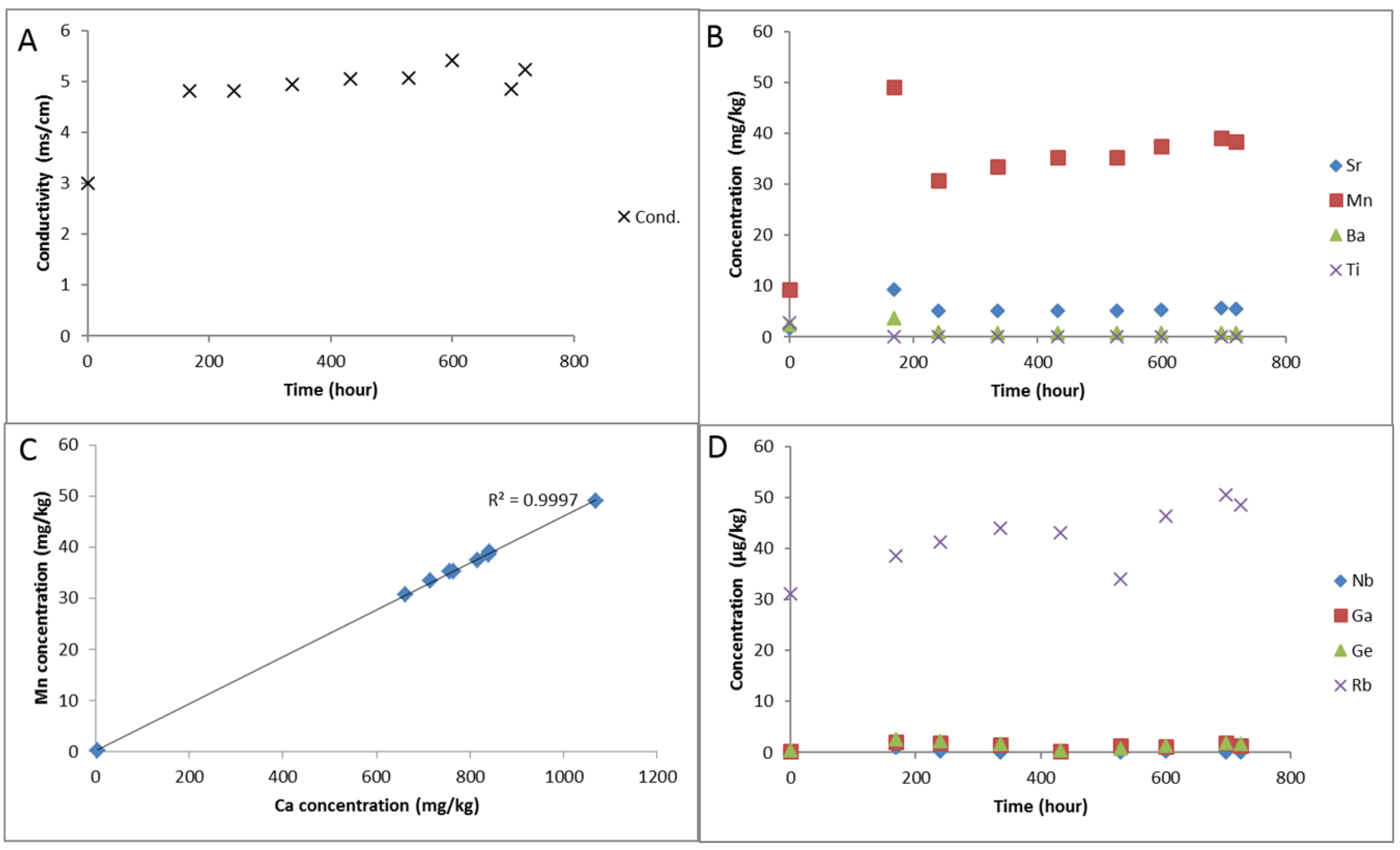
3. Results
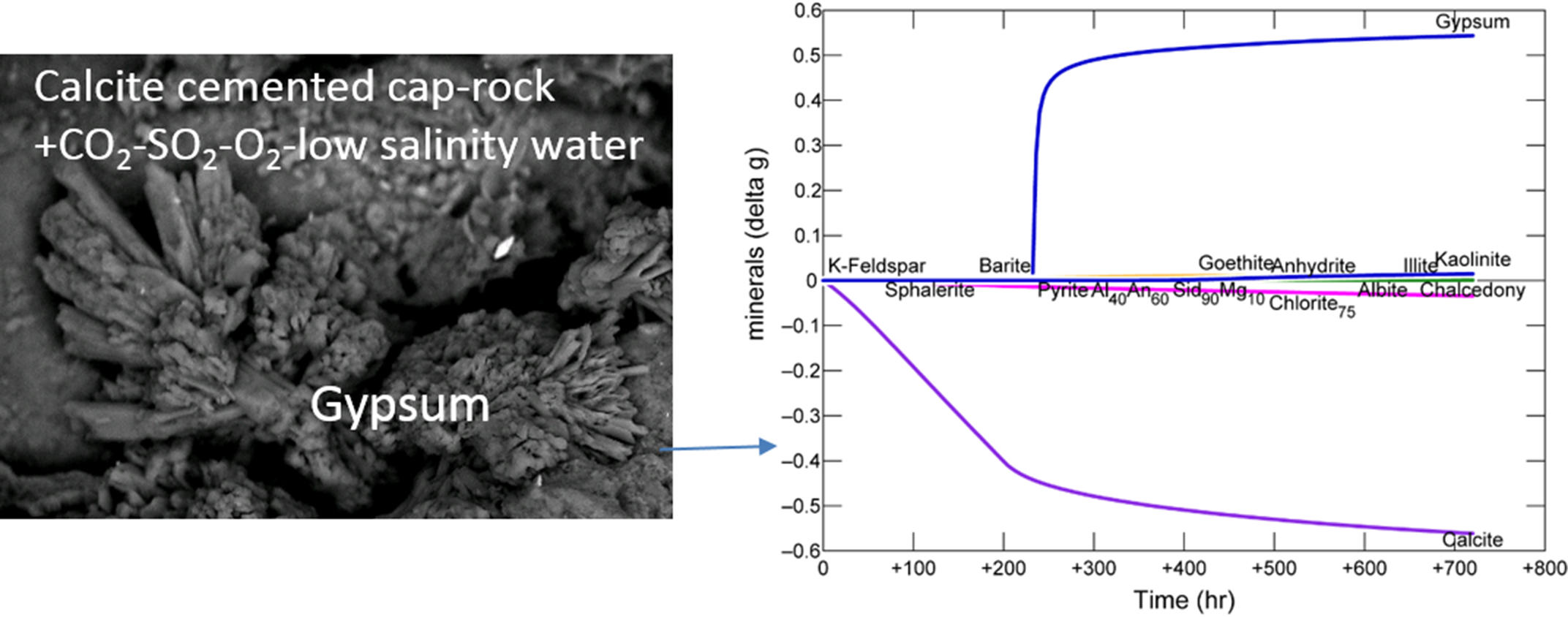
3.1. Calcite Cemented Caprock
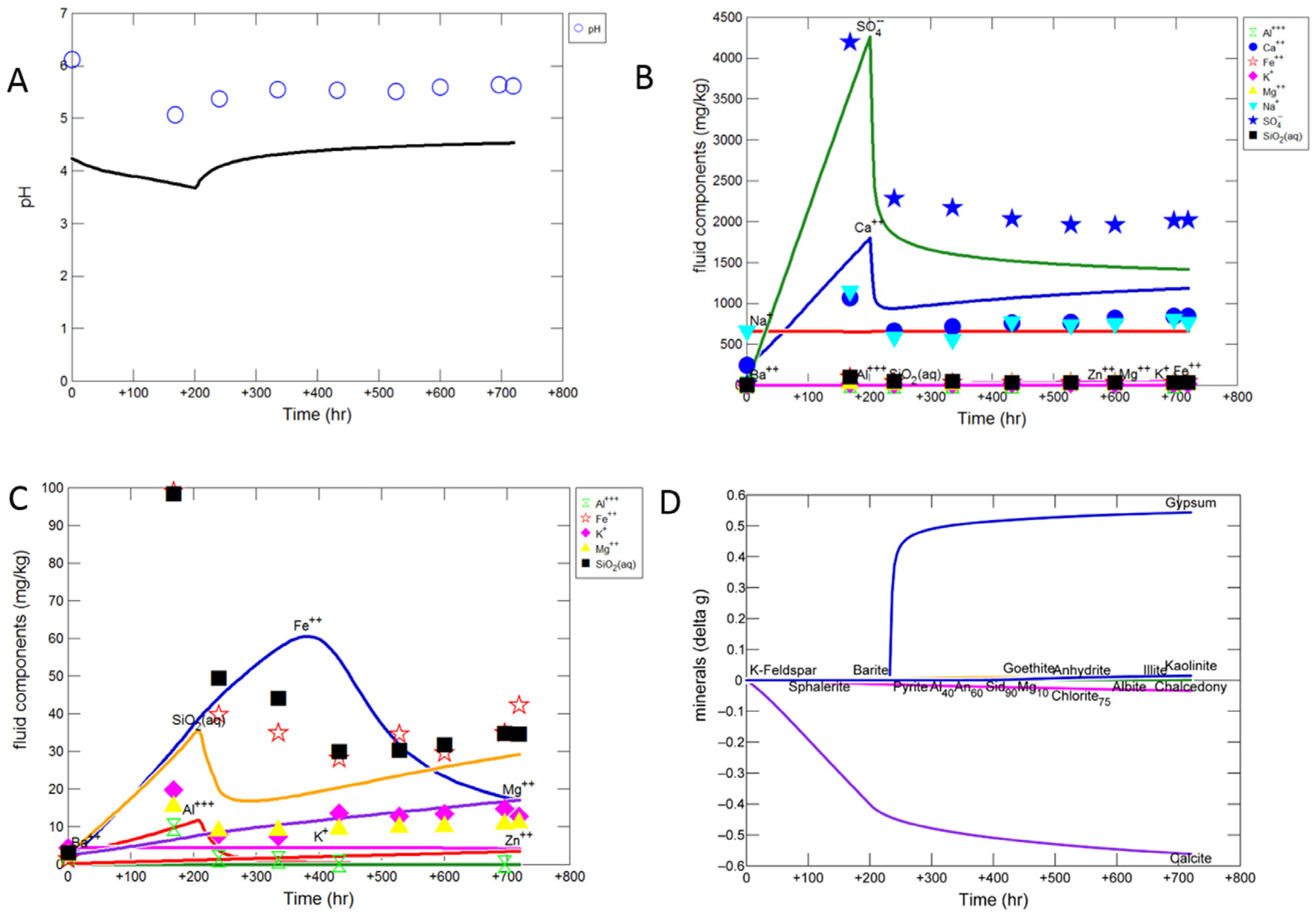
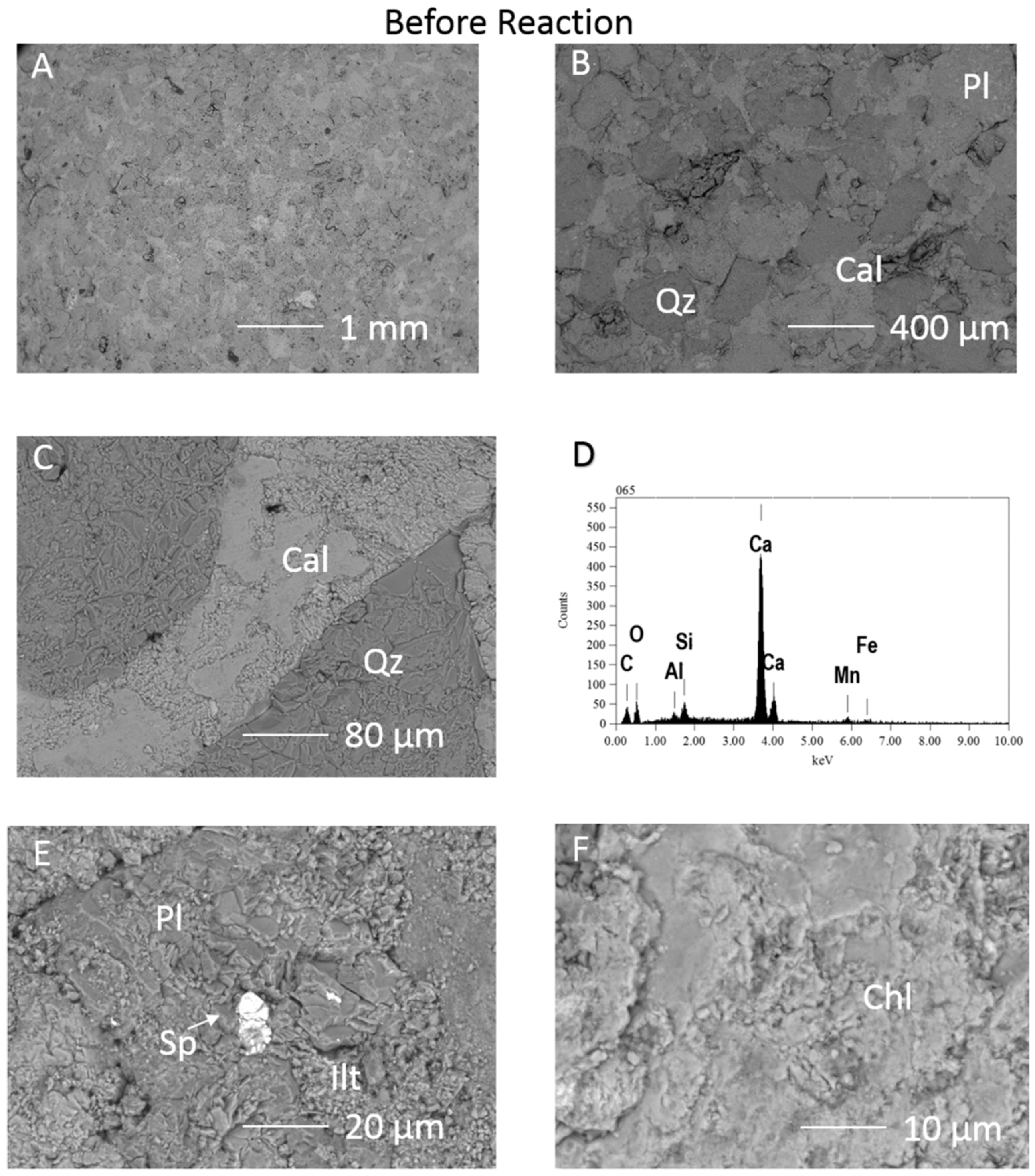
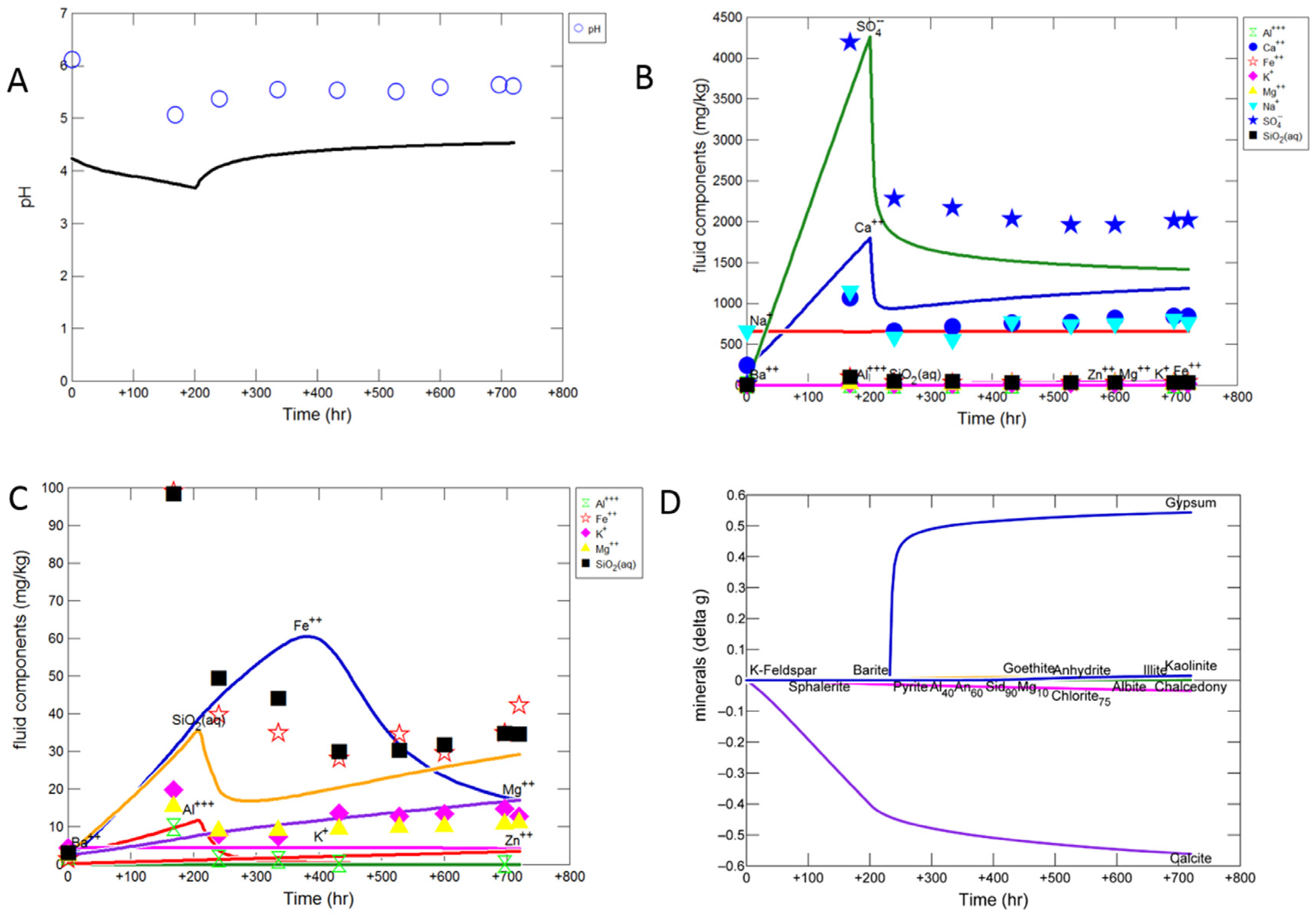
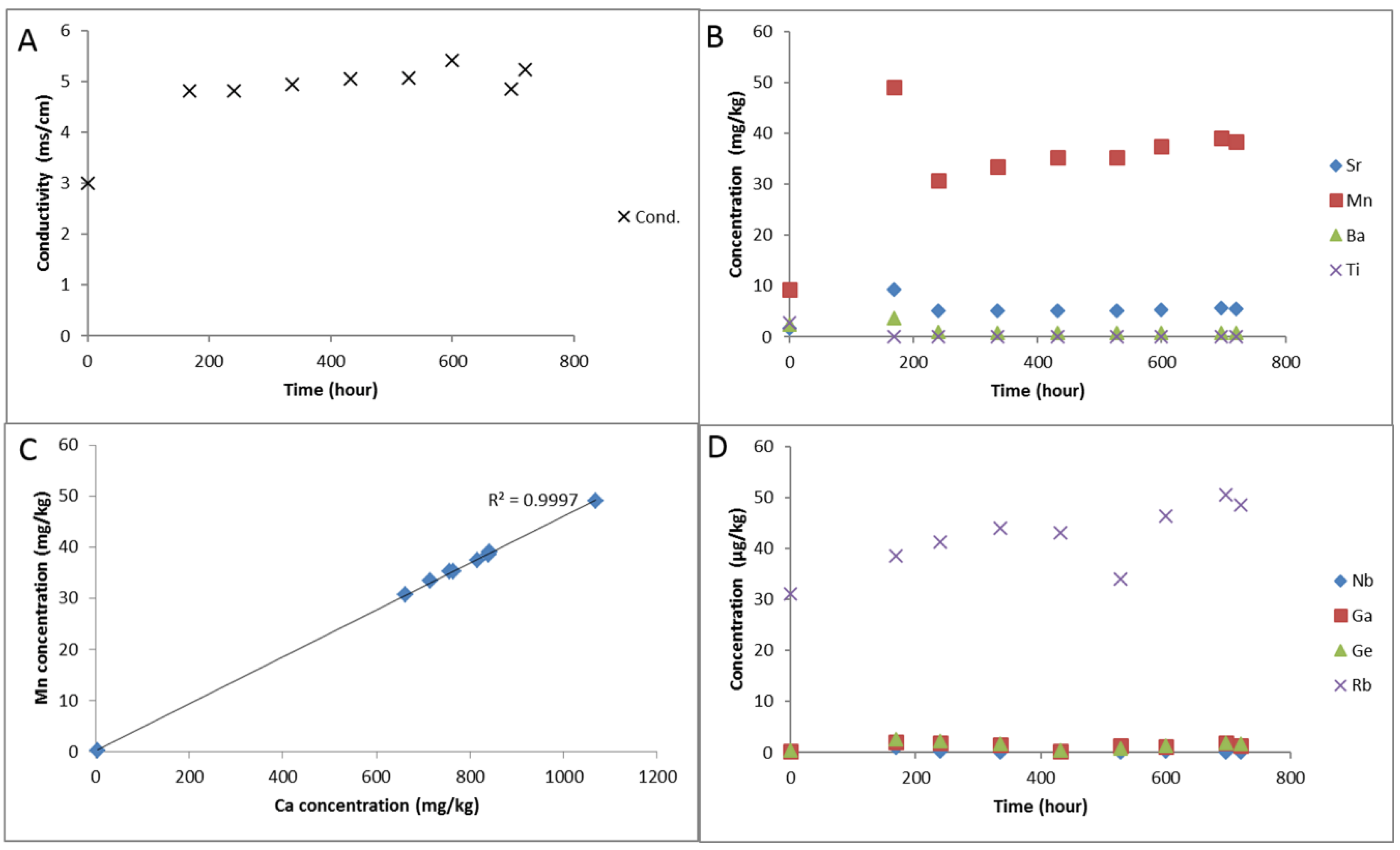
3.1.1. Experimental Results
3.1.2. Modelling the Experiment
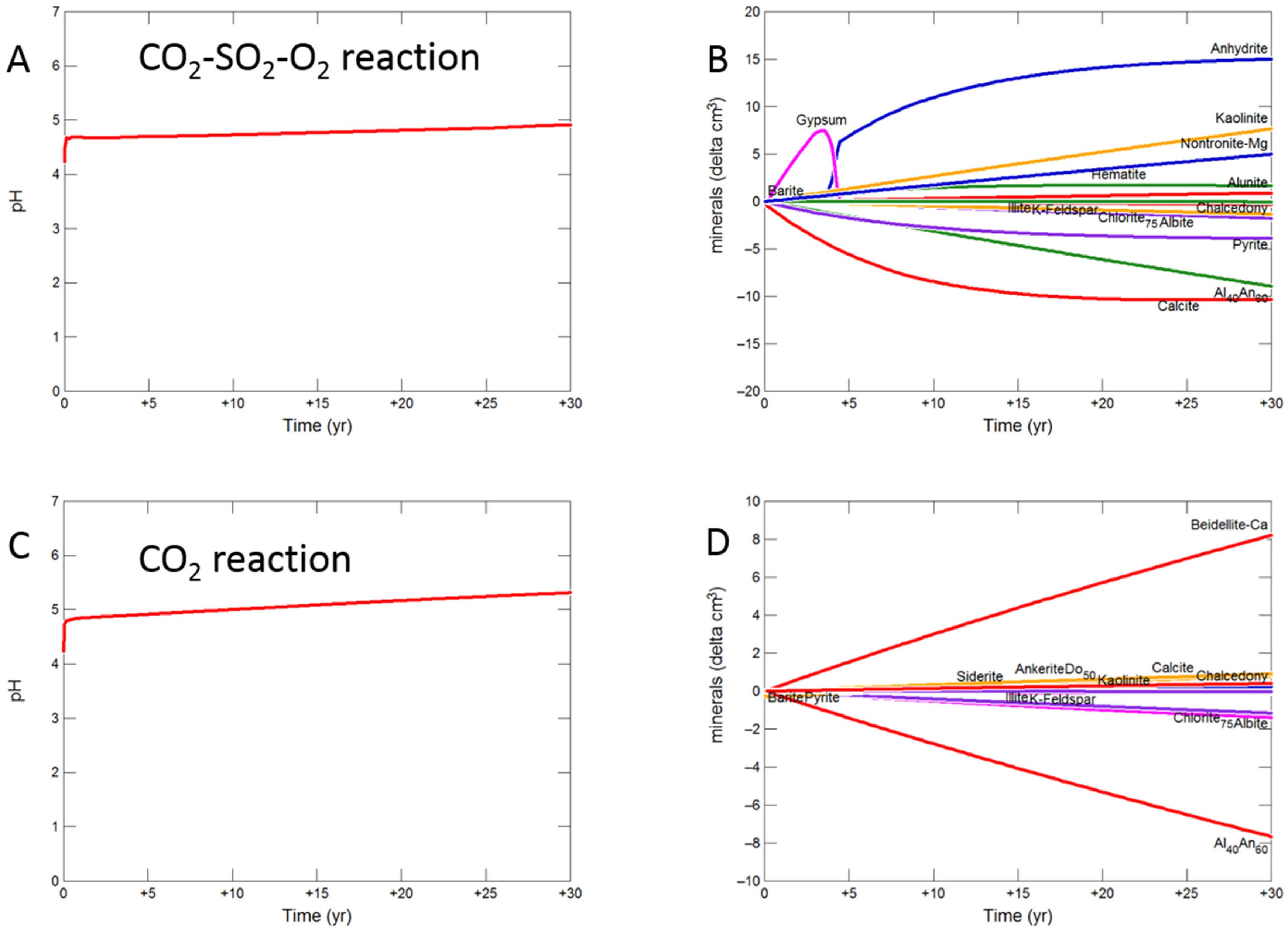
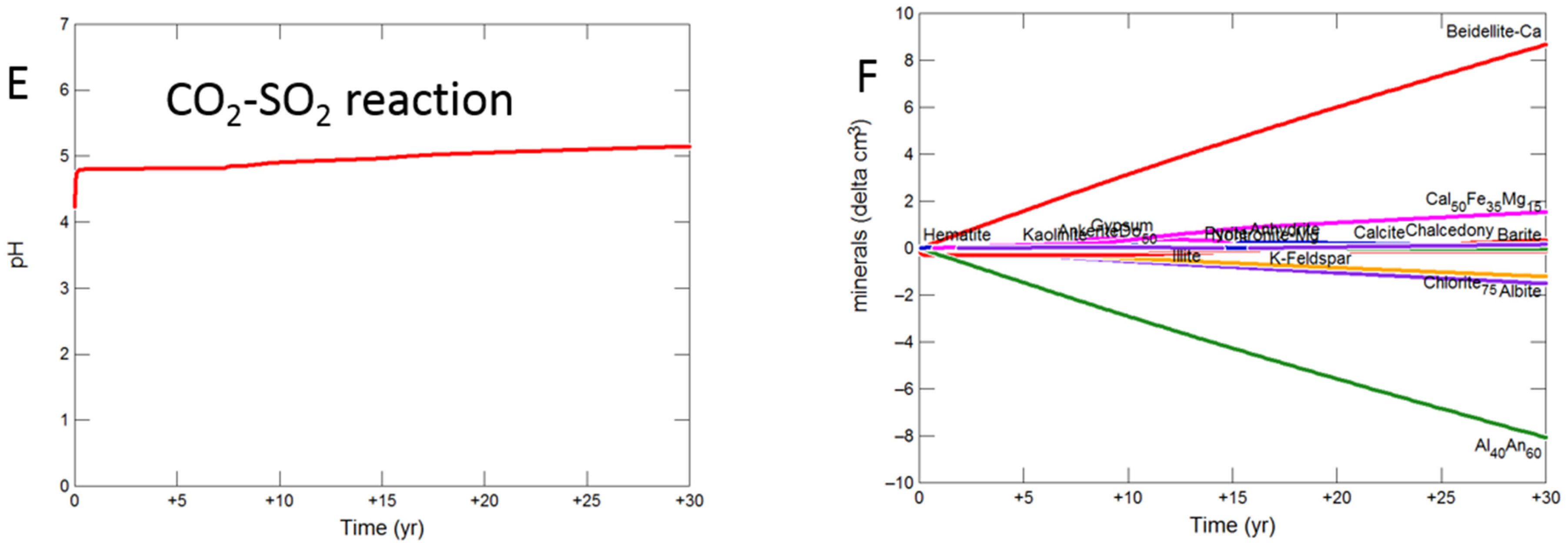
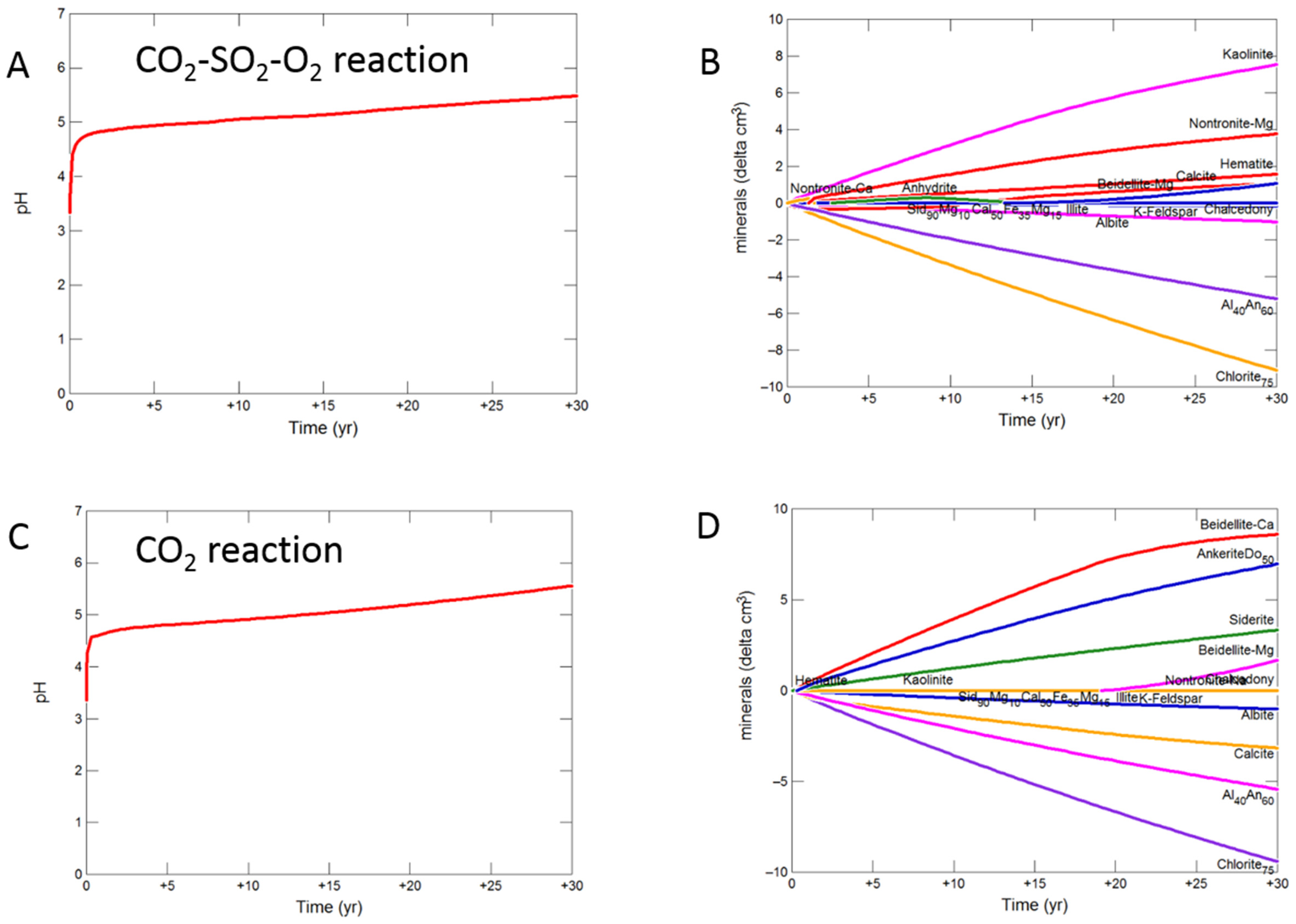
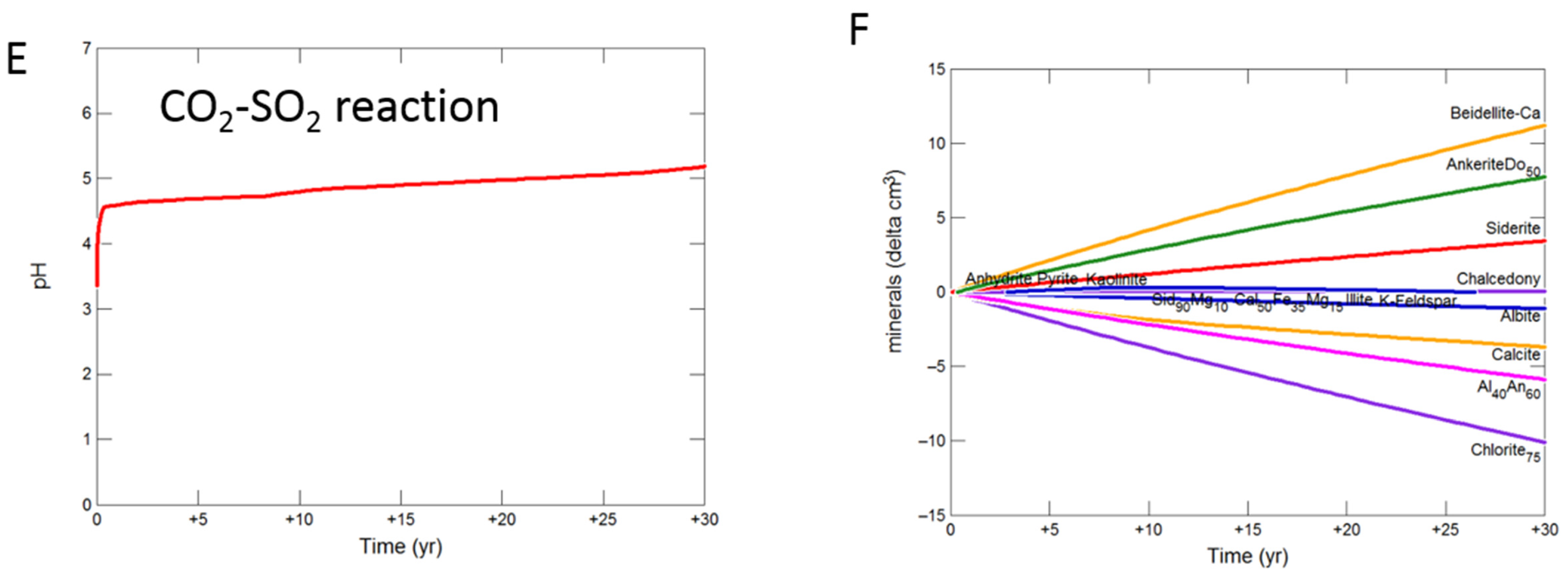
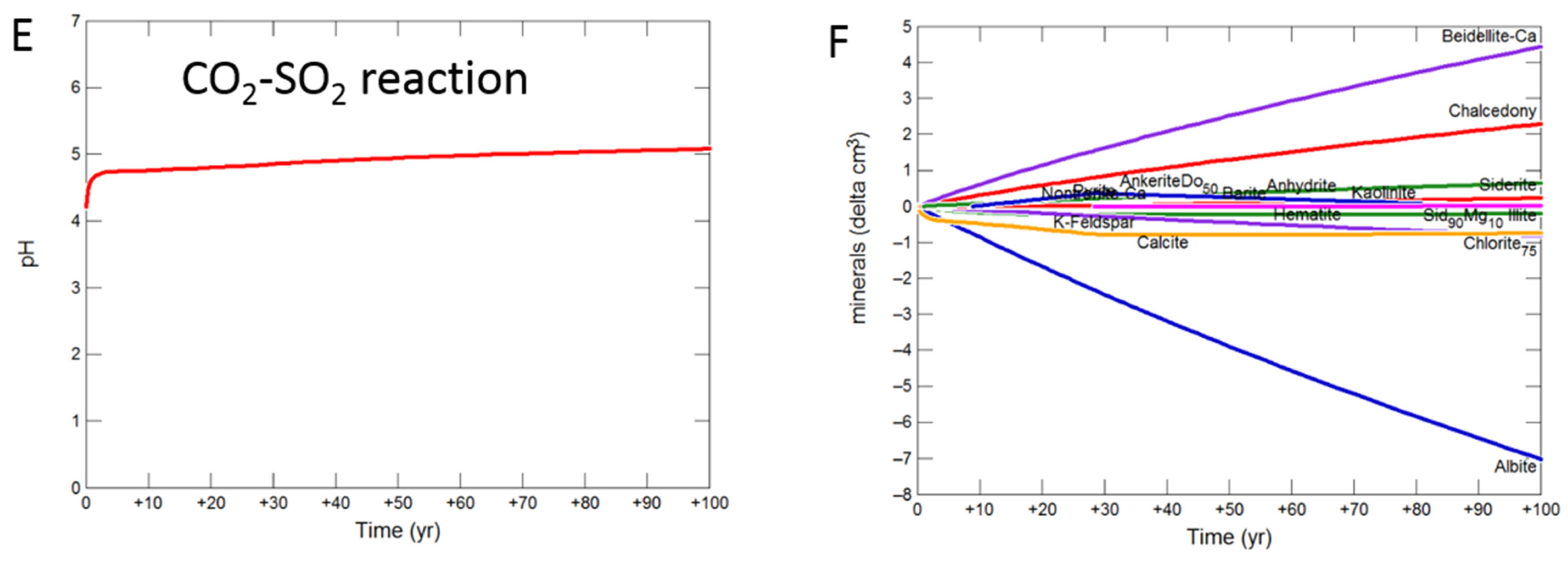
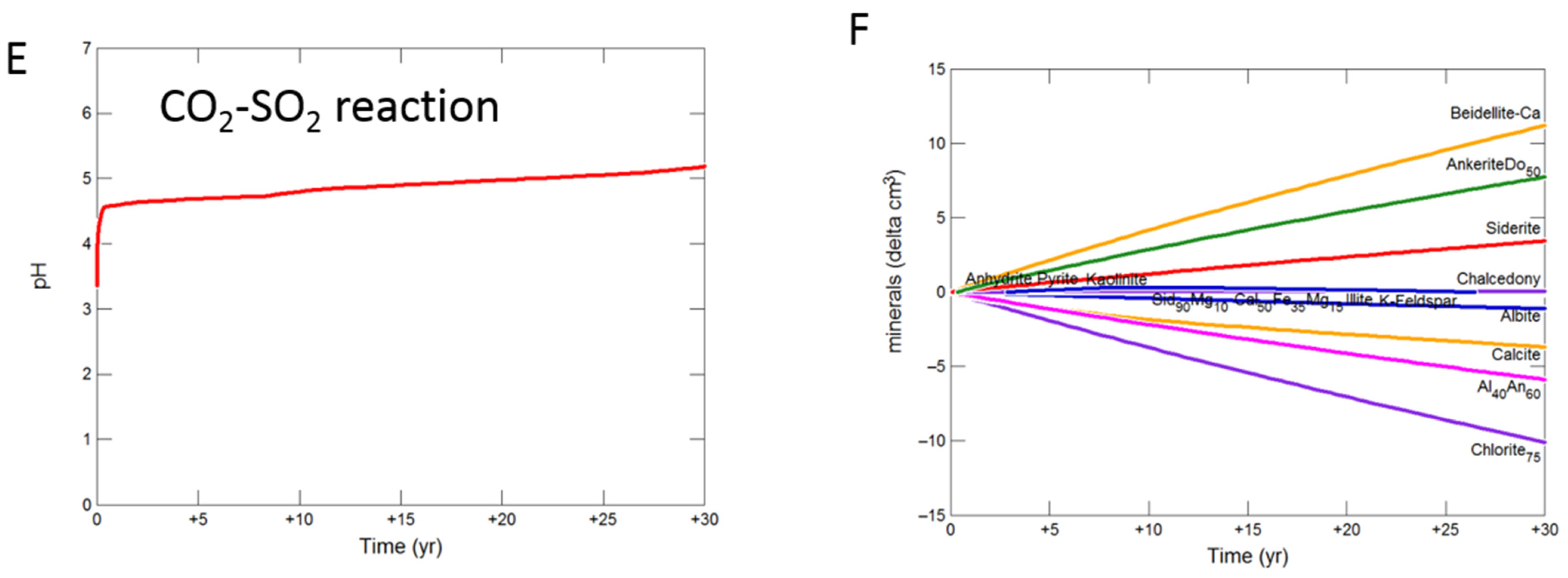
3.2. Geochemical Modelling of Cap-Rock
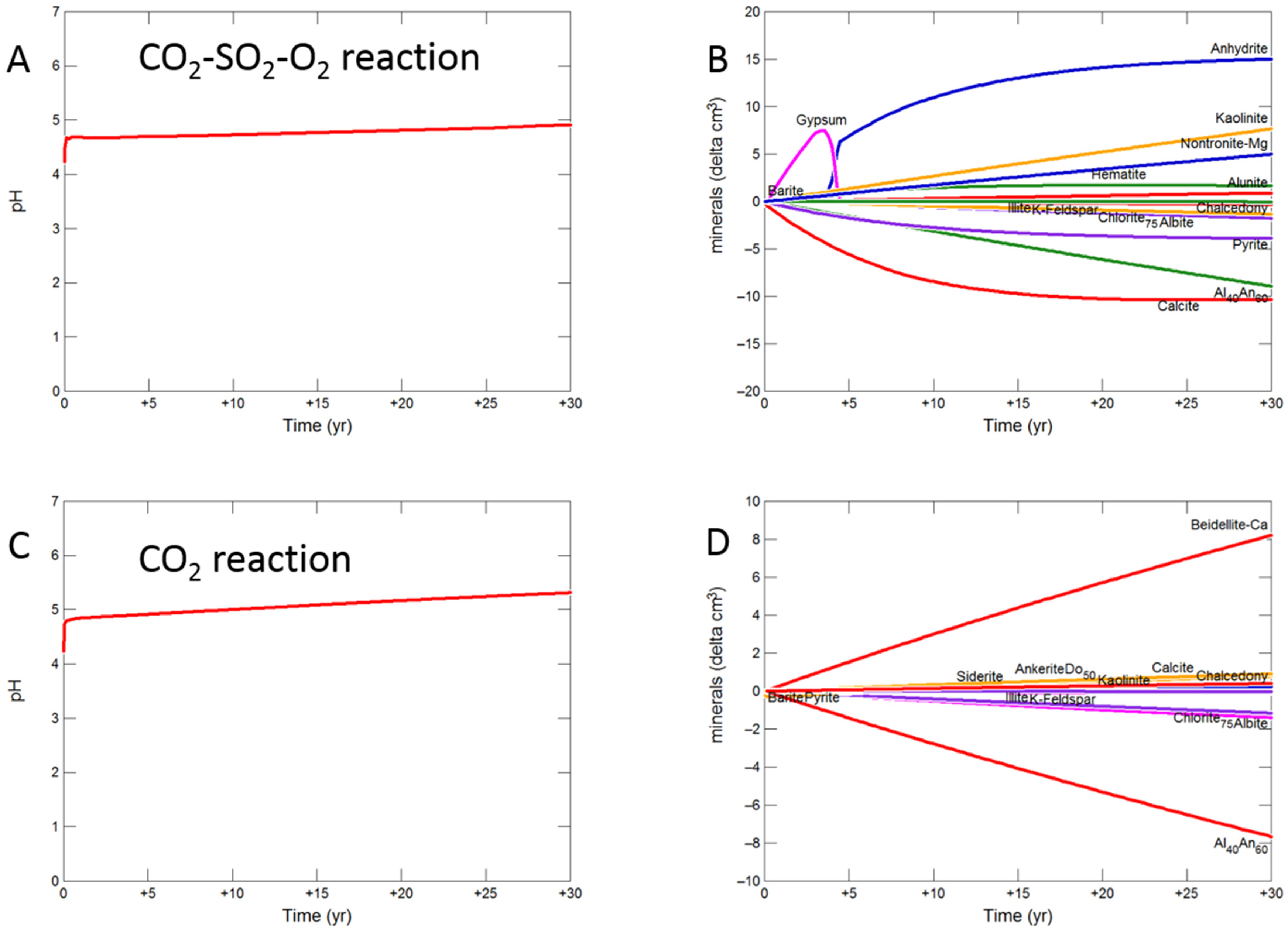
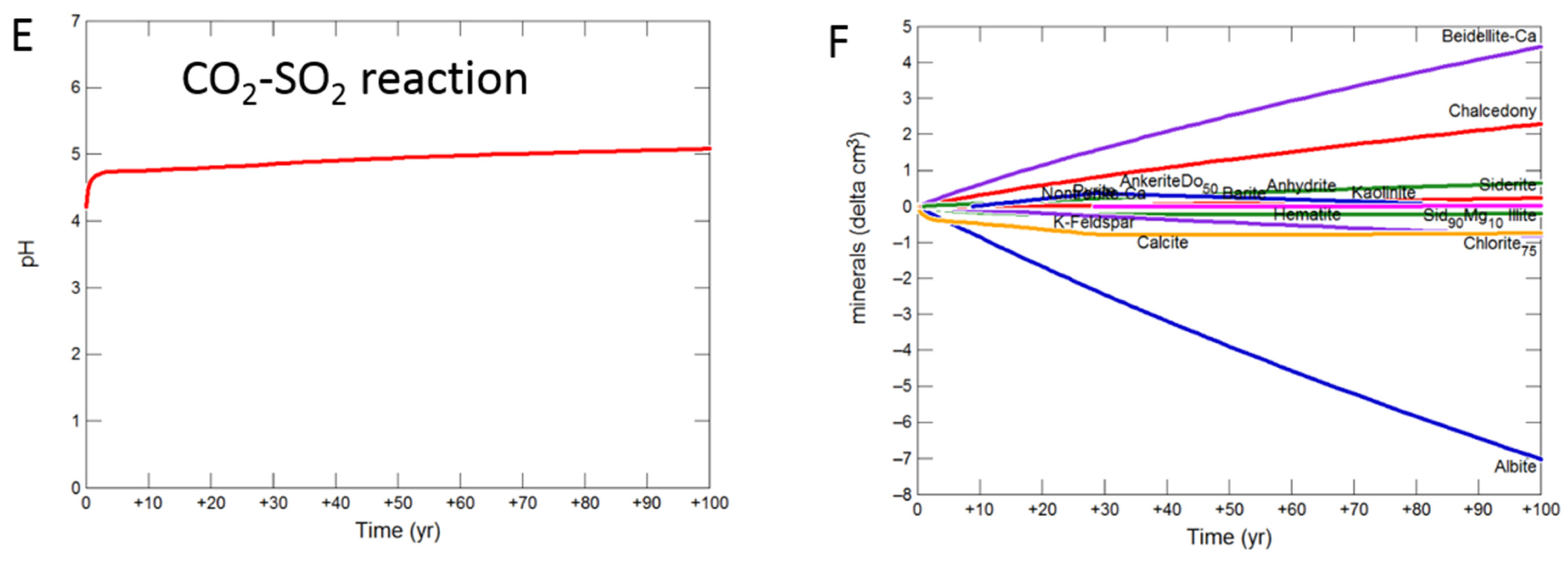
3.2.1. Calcite Cemented Cap-Rock
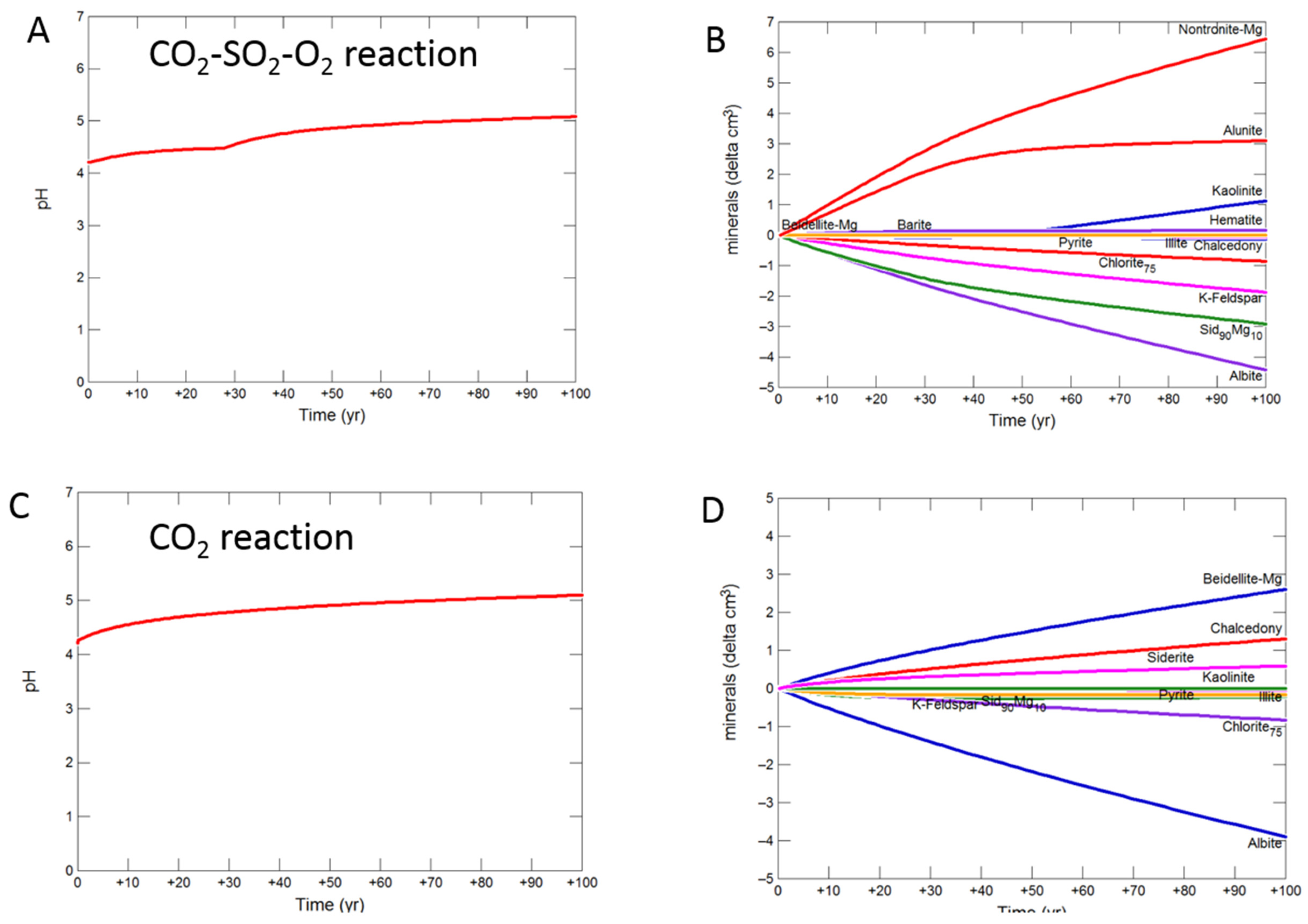
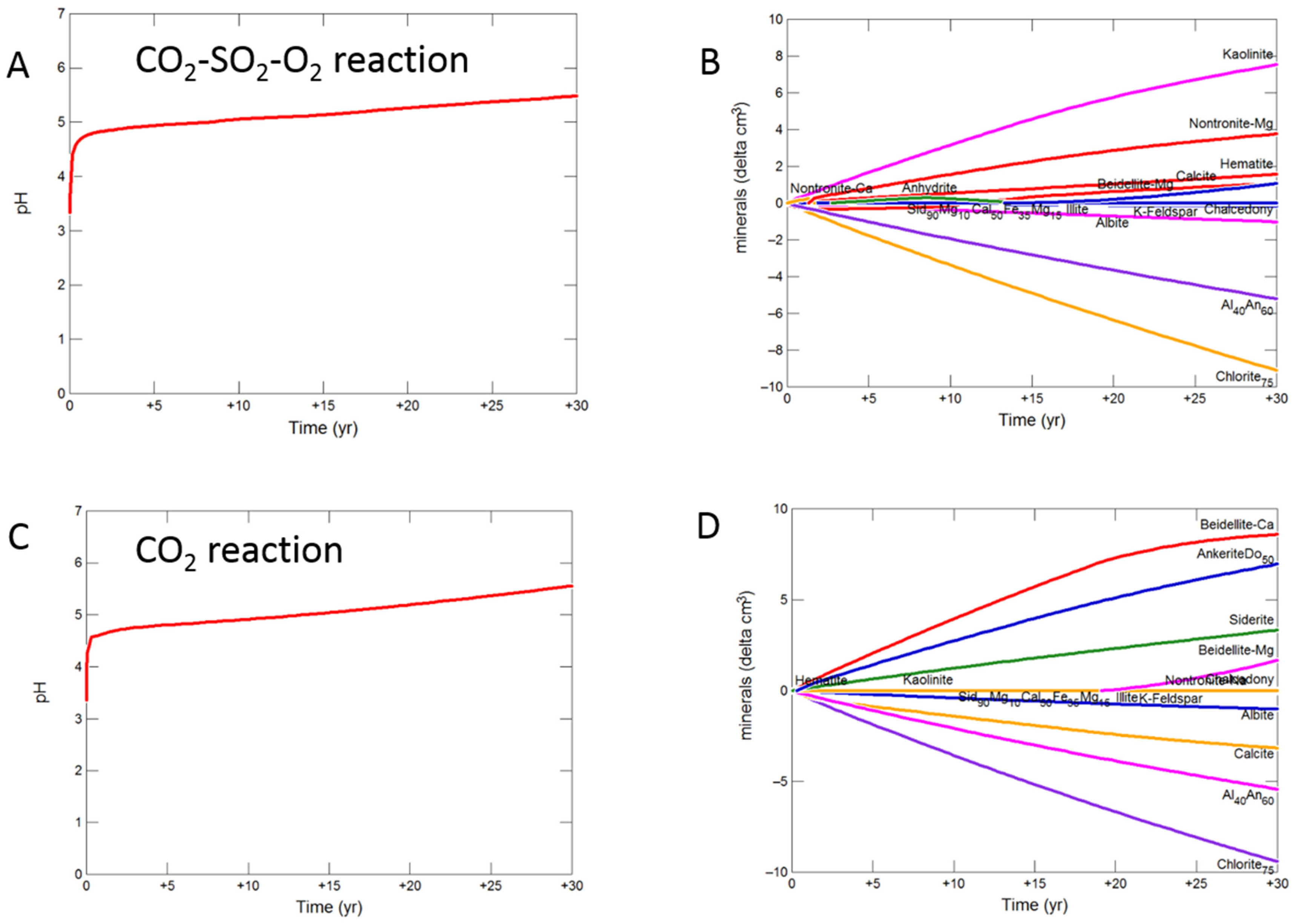
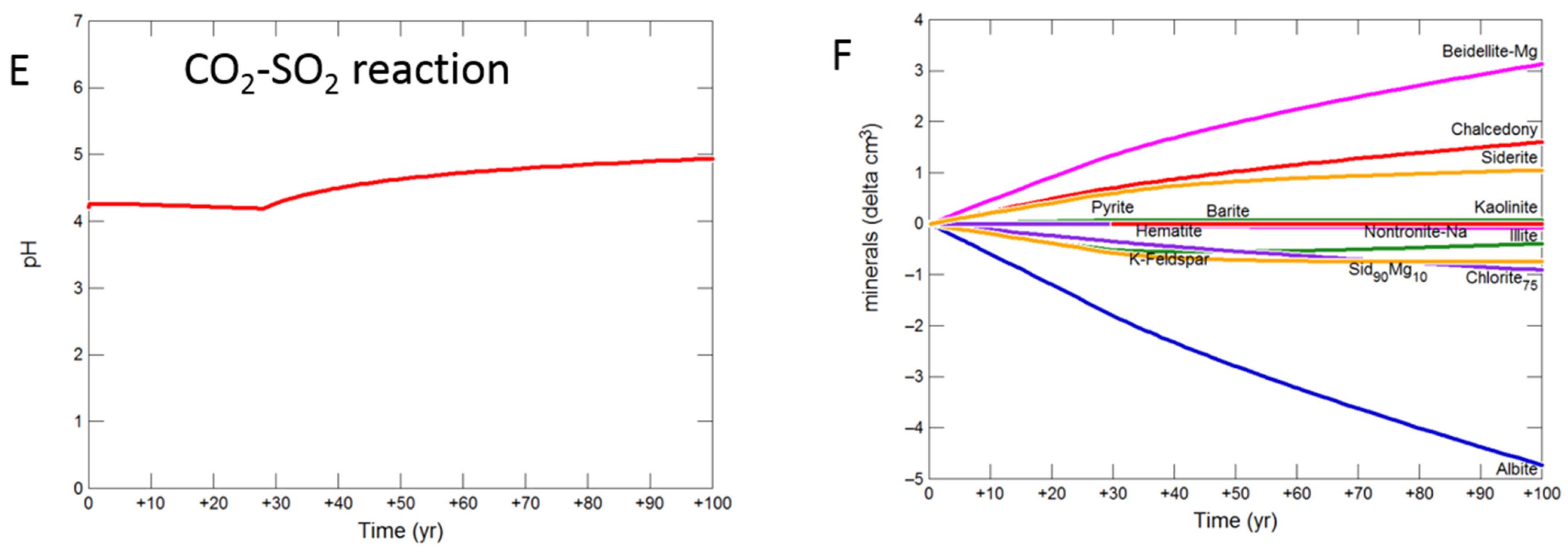
3.2.2. Mudstone Cap-Rock
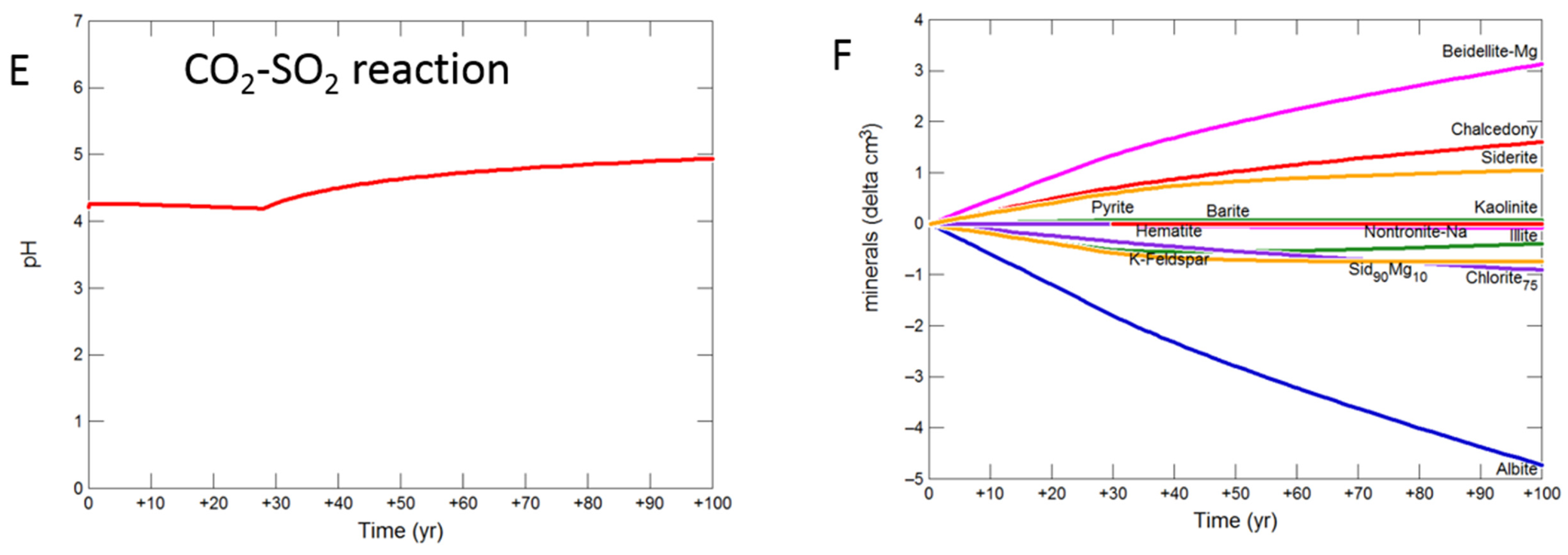
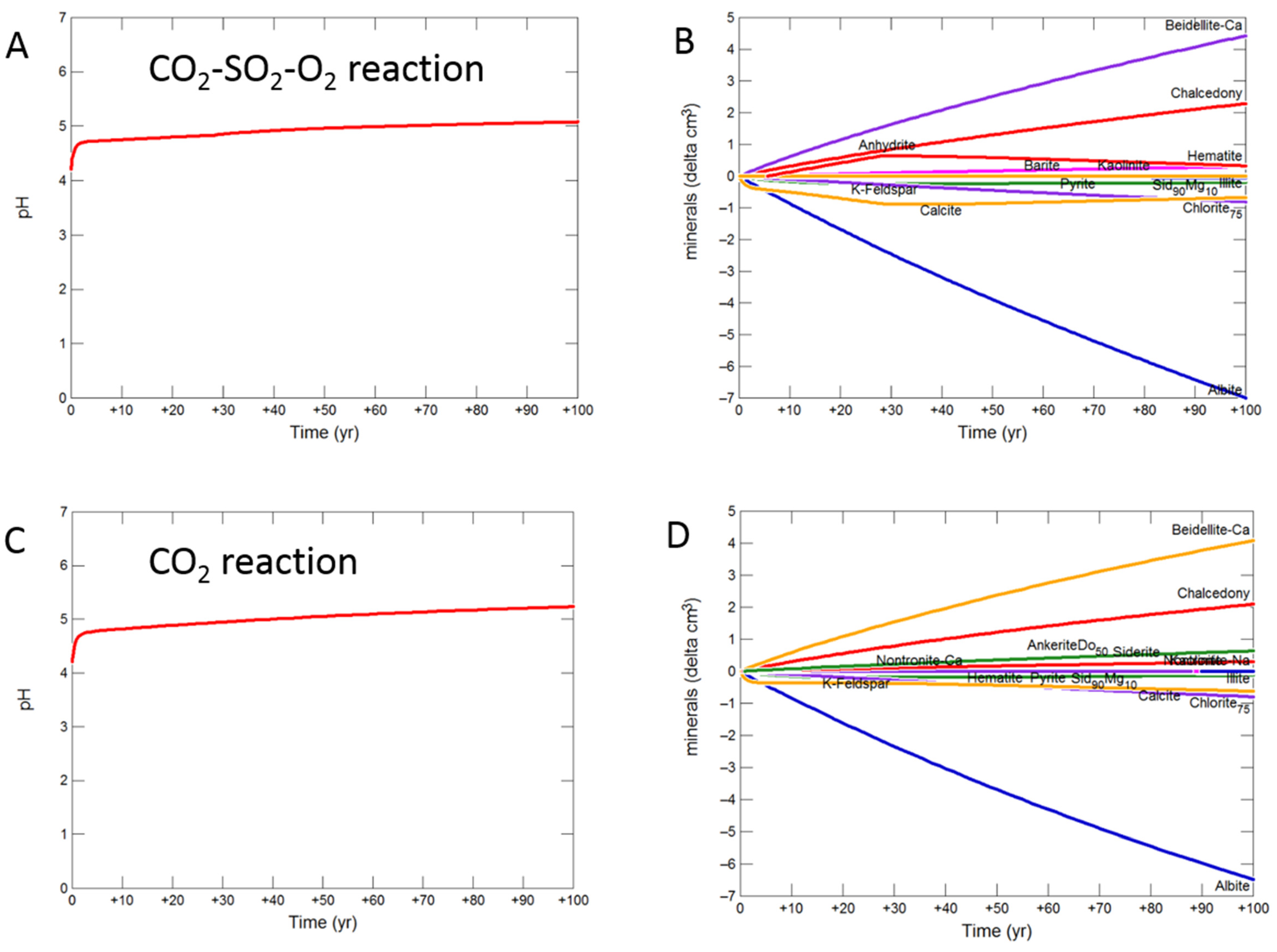
3.2.3. Siderite Cemented Cap-Rock
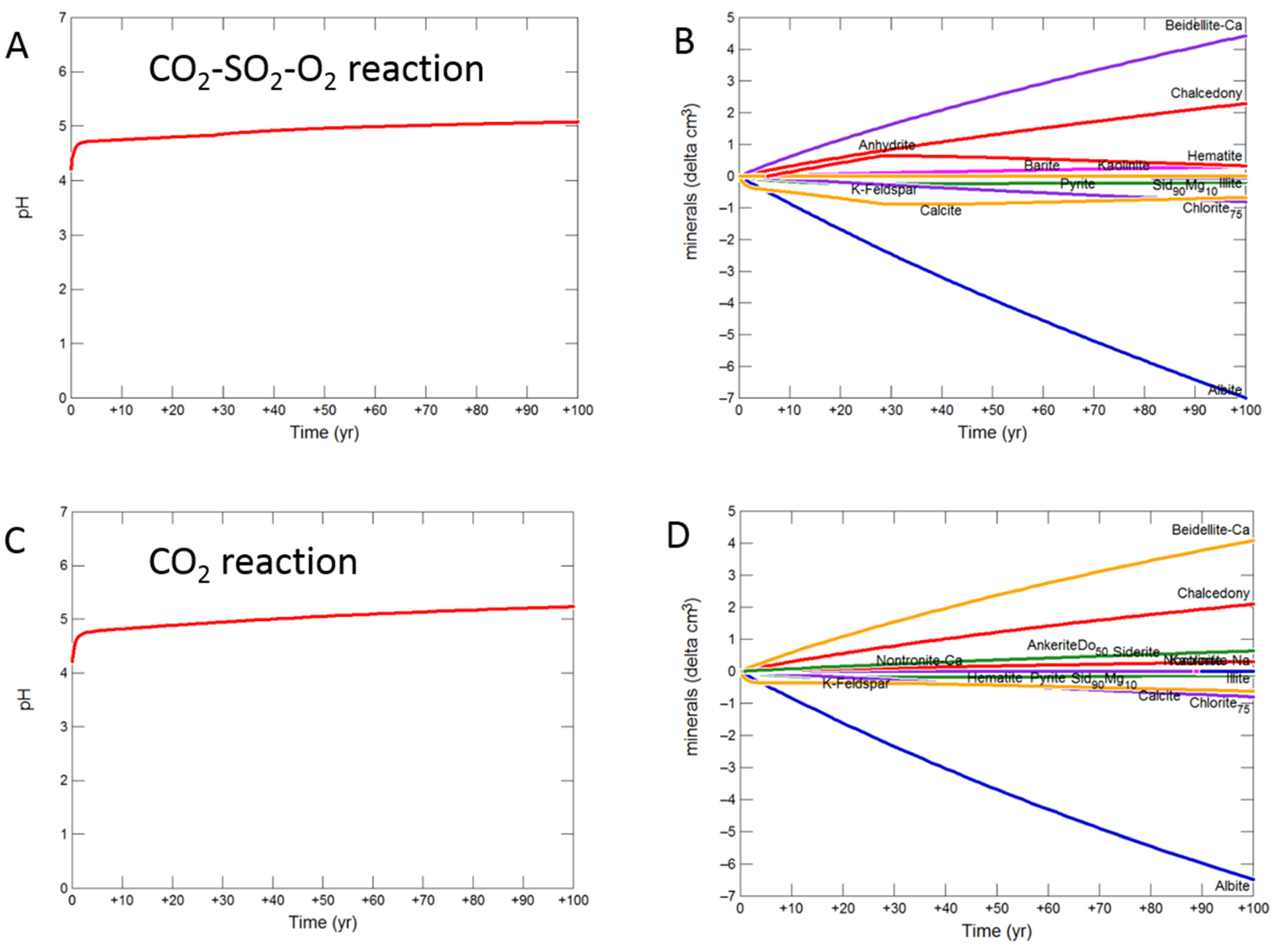
3.2.4. Shale Cap-Rock
4. Discussion
4.1. Experimental Results and Relevant Comparison Studies
4.2. Comparison of Modelling Outputs to Natural Analogue or Field Trial Observations
4.3. Significance
4.4. Potential Issues, Limitations, and Future Work
5. Conclusions
- Experimental CO2-SO2-O2 reaction of calcite cemented cap-rock resulted in calcite dissolution and chlorite corrosion, pH buffering and gypsum, barite, goethite and clay precipitation.
- To model the experimental data, reactive surface areas needed for calcite cement were low at 1 cm2/g, and high for silicates including plagioclase (300 cm2/g) and clays chlorite and illite (7000 cm2/g).
- Upscaled longer-term calcite cemented, siderite cemented, mudstone or shale cap-rock reactivity models predicted minimal net changes to porosity, favorably indicating cap-rock integrity was likely not significantly affected at these conditions.
- Smectite formation was predicted in all the long-term reactions, smectite has a high CO2 sorption capacity, favorable for trapping.
- Mineral trapping of CO2 as siderite and ankerite was only predicted to occur with pure CO2 or CO2-SO2. With O2 present, smectites, sulphate and oxide minerals were instead predicted to form. A limit on the O2 content co-injected may be needed to optimize CO2 mineral trapping, the most secure form of storage.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischer, S.; Liebscher, A.; De Lucia, M.; Hecht, L. Reactivity of sandstone and siltstone samples from the Ketzin pilot CO2 storage site-laboratory experiments and reactive geochemical modeling. Environ. Earth Sci. 2013, 70, 3687–3708. [Google Scholar] [CrossRef]
- Smith, M.M.; Sholokhova, Y.; Hao, Y.; Carroll, S.A. Evaporite Caprock integrity: An experimental study of reactive mineralogy and pore-scale heterogeneity during brine-CO2 exposure. Environ. Sci. Technol. 2013, 47, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Soong, Y.; Dilmore, R.M. Numerical investigation of lower Tuscaloosa sandstone and Selma chalk Caprock under geological CO2 sequestration conditions: Mineral precipitation and permeability evolution. Greenh. Gases Sci. Technol. 2017, 7, 988–1007. [Google Scholar] [CrossRef]
- Carroll, S.A.; McNab, W.W.; Dai, Z.; Torres, S.C. Reactivity of mount Simon sandstone and the eau Claire shale under CO2 storage conditions. Environ. Sci. Technol. 2013, 47, 252–261. [Google Scholar] [CrossRef] [PubMed]
- Kampman, N.; Bickle, M.J.; Maskell, A.; Chapman, H.J.; Evans, J.P.; Purser, G.; Zhou, Z.; Schaller, M.F.; Gattacceca, J.C.; Bertier, P.; et al. Drilling and sampling a natural CO2 reservoir: Implications for fluid flow and CO2-fluid–rock reactions during CO2 migration through the overburden. Chem. Geol. 2014, 369, 51–82. [Google Scholar] [CrossRef]
- Kampman, N.; Bertier, P.; Busch, A.; Snippe, J.; Harrington, J.; Pipich, V.; Maskell, A.; Bickle, M. Validating reactive transport models of CO2-brine-rock reactions in Caprocks using observations from a natural CO2 reservoir. Energy Procedia 2017, 114, 4902–4916. [Google Scholar] [CrossRef]
- Loring, J.S.; Schaef, H.T.; Turcu, R.V.F.; Thompson, C.J.; Miller, Q.R.S.; Martin, P.F.; Hu, J.; Hoyt, D.W.; Qafoku, O.; Ilton, E.S.; et al. In situ molecular spectroscopic evidence for CO2 intercalation into montmorillonite in supercritical carbon dioxide. Langmuir 2012, 28, 7125–7128. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Fitts, J.P.; Crandall, D.; McIntyre, D.; Peters, C.A. Alterations of fractures in carbonate rocks by CO2-acidified brines. Environ. Sci. Technol. 2015, 49, 10226–10234. [Google Scholar] [CrossRef] [PubMed]
- Dubacq, B.; Bickle, M.J.; Evans, K.A. An activity model for phase equilibria in the H2O–CO2–NaCl system. Geochim. Cosmochim. Acta 2013, 110, 229–252. [Google Scholar] [CrossRef]
- Ellis, B.; Fitts, J.; Bromhal, G.; McIntyre, D.; Tappero, R.; Peters, C. Dissolution-driven permeability reduction of a fractured carbonate Caprock. Environ. Eng. Sci. 2013, 30, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Wunsch, A.; Navarre-Sitchler, A.K.; Moore, J.; McCray, J.E. Metal release from limestones at high partial-pressures of CO2. Chem. Geol. 2014, 363, 40–55. [Google Scholar] [CrossRef]
- Bickle, M.; Kampman, N.; Chapman, H.; Ballentine, C.; Dubacq, B.; Galy, A.; Sirikitputtisak, T.; Warr, O.; Wigley, M.; Zhou, Z. Rapid reactions between CO2, brine and silicate minerals during geological carbon storage: Modelling based on a field CO2 injection experiment. Chem. Geol. 2017, 468, 17–31. [Google Scholar] [CrossRef]
- Wigley, M.; Dubacq, B.; Kampman, N.; Bickle, M. Controls of sluggish, CO2-promoted, hematite and k-feldspar dissolution kinetics in sandstones. Earth Planet. Sci. Lett. 2013, 362, 76–87. [Google Scholar] [CrossRef]
- Talman, S. Subsurface geochemical fate and effects of impurities contained in a CO2 stream injected into a deep saline aquifer: What is known? Int. J. Greenh. Gas Control 2015, 40, 267–291. [Google Scholar] [CrossRef]
- Porter, R.T.J.; Fairweather, M.; Pourkashanian, M.; Woolley, R.M. The range and level of impurities in CO2 streams from different carbon capture sources. Int. J. Greenh. Gas Control 2015, 36, 161–174. [Google Scholar] [CrossRef]
- Harkin, T.; Filby, I.; Sick, H.; Manderson, D.; Ashton, R. Development of a CO2 specification for a CCS hub network. Energy Procedia 2017, 114, 6708–6720. [Google Scholar] [CrossRef]
- Snæbjörnsdóttir, S.Ó.; Gislason, S.R.; Galeczka, I.M.; Oelkers, E.H. Reaction path modelling of in situ mineralisation of CO2 at the Carbfix site at Hellisheidi, SW-Iceland. Geochim. Cosmochim. Acta 2018, 220, 348–366. [Google Scholar] [CrossRef]
- Bachu, S.; Gunter, W.D. Overview of acid-gas injection operations in western Canada. In Greenhouse Gas Control Technologies 7; Rubin, E.S., Keith, D.W., Gilboy, C.F., Wilson, M., Morris, T., Gale, J., Thambimuthu, K., Eds.; Elsevier Science Ltd.: Oxford, UK, 2005; pp. 443–448. [Google Scholar]
- Hodgkinson, J.; Preda, M.; Hortle, A.; McKillop, M.; Dixon, O.; Foster, L. The Potential Impact of Carbon Dioxide Injection on Freshwater Aquifers: The Surat and Eromanga Basins in Queensland; Dept. of Employment, Economic Development and Innovation, Geological Survey of Queensland: Brisbane, Australia, 2010; p. 133. ISBN 9781921489570. [Google Scholar]
- Farquhar, S.M.; Pearce, J.K.; Dawson, G.K.W.; Golab, A.; Kirste, D.; Biddle, D.; Golding, S.D. A fresh approach to investigating CO2 storage: Experimental CO2-water-rock interactions in a freshwater reservoir system. Chem. Geol. 2015, 399, 98–122. [Google Scholar] [CrossRef]
- Hodgkinson, J.; Grigorescu, M. Background research for selection of potential geostorage targets—Case studies from the Surat basin, Queensland. Aust. J. Earth Sci. 2012, 60, 71–89. [Google Scholar] [CrossRef]
- Feitz, A.J.; Ransley, T.R.; Dunsmore, R.; Kuske, T.J.; Hodgkinson, J.; Preda, M.; Spulak, R.; Dixon, O.; Draper, J. Geoscience Australia and Geological Survey of Queensland Surat and Bowen Basins Groundwater Surveys Hydrochemistry Dataset (2009–2011); Geoscience Australia: Canberra, Australia, 2014. [Google Scholar]
- Farquhar, S.M.; Dawson, G.K.W.; Esterle, J.S.; Golding, S.D. Mineralogical characterisation of a potential reservoir system for CO2 sequestration in the Surat basin. Aust. J. Earth Sci. 2013, 60, 91–110. [Google Scholar] [CrossRef]
- Golab, A.; Arena, A.; Sommacal, S.; Goodwin, C.; Rajan, P.; Dodd, N.; Khor, J.; Deakin, L.; Zhang, J.; Young, B.; et al. Milestone 2.9: Final Report of Digital Core Analysis Results for Plug Samples from West Wandoan-1 Well; Report for ANLEC R&D; FEI-Lithicon: Hillsboro, OR, USA, 2015. [Google Scholar]
- Golab, A.; Arena, A.; Khor, J.; Goodwin, C.; Young, B.; Carnerup, A.; Hussain, F. Milestone 1.4 Final Report of RCA and SCAL Data on Plugs from West Wandoan-1 Well; Report for ANLEC R&D; FEI Lithicon: Hillsboro, OR, USA, 2015. [Google Scholar]
- Ellis, B.; Peters, C.; Fitts, J.; Bromhal, G.; McIntyre, D.; Warzinski, R.; Rosenbaum, E. Deterioration of a fractured carbonate Caprock exposed to CO2-acidified brine flow. Greenh. Gases Sci. Technol. 2011, 1, 248–260. [Google Scholar] [CrossRef]
- Kirste, D.; Pearce, J.; Golding, S. Parameterizing geochemical models: Do kinetics of calcite matter? Procedia Earth Planet. Sci. 2017, 17, 606–609. [Google Scholar] [CrossRef]
- Wigley, M.; Kampman, N.; Dubacq, B.; Bickle, M. Fluid-mineral reactions and trace metal mobilization in an exhumed natural CO2 reservoir, green river, Utah. Geology 2012, 40, 555–558. [Google Scholar] [CrossRef]
- Golab, A.; Knuefing, L.; Goodwin, C.; Sommacal, S.; Carnerup, A.; Dawson, G.; Pearce, J.K.; Golding, S.D. Milestone 5.7: Final Report on Geochemical Reactivity Studies of Core Material Using SCCO2; Report for ANLEC R&D; Lithicon FEI: Hillsboro, OR, USA, 2015. [Google Scholar]
- Dawson, G.K.W.; Biddle, D.; Farquhar, S.M.; Gao, J.; Golding, S.D.; Jiang, X.; Keck, R.; Khan, C.; Law, A.C.K.; Li, Q.; et al. Achieving Risk and Cost Reductions in CO2 Geosequestration through 4D Characterisation of Host Formations; ANLEC R&D; University of Queensland: Brisbane, Australia, 2015. [Google Scholar]
- Pearce, J.K.; Kirste, D.M.; Dawson, G.K.W.; Farquhar, S.M.; Biddle, D.; Golding, S.; Rudolph, V. SO2 impurity impacts on experimental and simulated CO2-water-reservoir rock reactions at carbon storage conditions. Chem. Geol. 2015, 399, 65–86. [Google Scholar] [CrossRef]
- Pearce, J.K.; Law, A.C.K.; Dawson, G.K.W.; Golding, S.D. SO2-CO2 and pure CO2 reactivity of ferroan carbonates at carbon storage conditions. Chem. Geol. 2015. [Google Scholar] [CrossRef]
- Bethke, C.M.; Yeakel, S. The Geochemist’s Workbench (Version 9.0): Reaction Modeling Guide; Aqueous Solutions LLC: Champaign, IL, USA, 2012; 96p. [Google Scholar]
- Delany, J.M.; Lundeen, S.R. The LLNL Thermodynamic Database; Lawrence Livermore National Laboratory Report UCRL-21658; Lawrence Livermore National Laboratory: Livermore, CA, USA, 1989. [Google Scholar]
- Duan, Z.; Sun, R. An improved model calculating CO2 solubility in pure water and aqueous NaCl solutions from 273 to 533 k and from 0 to 2000 bar. Chem. Geol. 2003, 193, 257–271. [Google Scholar] [CrossRef]
- Palandri, J.L.; Kharaka, Y.K. A Compilation of Rate Parameters of Water-Mineral Interaction Kinetics for Application to Geochemical Modeling; USGS Open File Report 2004-1068; USGS: Reston, VA, USA, 2004; p. 64. [Google Scholar]
- Lowson, R.T.; Brown, P.L.; Comarmond, M.C.J.; Rajaratnam, G. The kinetics of chlorite dissolution. Geochim. Cosmochim. Acta 2007, 71, 1431–1447. [Google Scholar] [CrossRef]
- Köhler, S.J.; Dufaud, F.; Oelkers, E.H. An experimental study of Illite dissolution kinetics as a function of pH from 1.4 to 12.4 and temperature from 5 to 50 °C. Geochim. Cosmochim. Acta 2003, 67, 3583–3594. [Google Scholar] [CrossRef]
- Pham, V.T.H.; Lu, P.; Aagaard, P.; Zhu, C.; Hellevang, H. On the potential of CO2–water–rock interactions for CO2 storage using a modified kinetic model. Int. J. Greenh. Gas Control 2011, 5, 1002–1015. [Google Scholar] [CrossRef]
- Steefel, C.I. Gimrt, Version 1.2: Software for Modeling Multicomponent, Multidimensional Reactive Transport. User’s Guide, UCRL-MA-143182; Lawrence Livermore National Laboratory: Livermore, CA, USA, 2001. [Google Scholar]
- White, A.F. Chemical weathering rates of silicate minerals in soils. Rev. Mineral. Geochem. 1995, 31, 407–461. [Google Scholar]
- Pearce, J.; Kirste, D.; Altaf, I.; Golding, S.; Undershultz, J. Geochemistry of storing CO2 and NOx in the deep precipice sandstone. In Proceedings of the Australasian Exploration Geoscience Conference, Sydney, Australia, 18–21 February 2018. [Google Scholar]
- Grigorescu, M. Mineralogy of the North-Eastern Bowen Basin and North-Eastern Surat Basin, Queensland; Geological Survey of Queensland: Queensland, Australian, 2011. [Google Scholar]
- Raza, A.; Hill, K.C.; Korsch, R.J. Mid-cretaceous uplift and denudation of the Bowen and Surat basins, Eastern Australia: Relationship to Tasman Sea rifting from apatite fission-track and vitrinite-reflectance data. Aust. J. Earth Sci. 2009, 56, 501–531. [Google Scholar] [CrossRef]
- Hu, Y.; Ray, J.R.; Jun, Y.-S. Biotite–brine interactions under acidic hydrothermal conditions: Fibrous illite, goethite, and kaolinite formation and biotite surface cracking. Environ. Sci. Technol. 2011, 45, 6175–6180. [Google Scholar] [CrossRef] [PubMed]
- Renard, S.; Sterpenich, J.; Pironon, J.; Chiquet, P.; Randi, A. Geochemical effects of an oxycombustion stream containing SO2 and O2 on carbonate rocks in the context of CO2 storage. Chem. Geol. 2014, 382, 140–152. [Google Scholar] [CrossRef]
- Pearce, J.K.; Dawson, G.K.W.; Blach, T.P.; Bahadur, J.; Melnichenko, Y.B.; Golding, S.D. Impure CO2 reaction of feldspar, clay, and organic matter rich cap-rocks: Decreases in the fraction of accessible mesopores measured by sans. Int. J. Coal Geol. 2018, 185, 79–90. [Google Scholar] [CrossRef]
- Chopping, C.; Kaszuba, J.P. Reactivity of supercritical sulfur dioxide and carbon dioxide in a carbonate reservoir: An experimental investigation of supercritical fluid-brine-rock interactions relevant to the Madison limestone of southwest Wyoming. Interpretation 2017, 5, SS43–SS58. [Google Scholar] [CrossRef]
- Smith, M.M.; Wolery, T.J.; Carroll, S.A. Kinetics of chlorite dissolution at elevated temperatures and CO2 conditions. Chem. Geol. 2013, 347, 1–8. [Google Scholar] [CrossRef]
- Bickle, M.J.; Kampman, N.; Wigley, M. Natural analogues. Geochemistry of Geologic Carbon Sequestration. Rev. Mineral. Geochem. 2013, 77, 15–71. [Google Scholar] [CrossRef] [Green Version]
- Watson, M.N.; Zwingmann, N.; Lemon, N.M. The ladbroke grove-katnook carbon dioxide natural laboratory: A recent CO2 accumulation in a lithic sandstone reservoir. Energy 2004, 29, 1457–1466. [Google Scholar] [CrossRef]
- Higgs, K.E.; Haese, R.R.; Golding, S.D.; Schacht, U.; Watson, M. The pretty hill formation as a natural analogue for CO2 storage; an investigation of mineralogical and isotopic changes associated with sandstones exposed to low, intermediate and high CO2 concentrations over geological time. Chem. Geol. 2015, 399, 36–64. [Google Scholar] [CrossRef]
- Higgs, K.E.; Funnell, R.H.; Reyes, A.G. Changes in reservoir heterogeneity and quality as a response to high partial pressures of CO2 in a gas reservoir, New Zealand. Mar. Pet. Geol. 2013, 48, 293–322. [Google Scholar] [CrossRef]
- Kaszuba, J.P.; Navarre-Sitchler, A.; Thyne, G.; Chopping, C.; Meuzelaar, T. Supercritical carbon dioxide and sulfur in the Madison limestone: A natural analog in southwest Wyoming for geologic carbon-sulfur co-sequestration. Earth Planet. Sci. Lett. 2011, 309, 131–140. [Google Scholar] [CrossRef]
- Ilgen, A.G.; Cygan, R.T. Mineral dissolution and precipitation during CO2 injection at the Frio-I brine pilot: Geochemical modeling and uncertainty analysis. Int. J. Greenh. Gas Control 2016, 44, 166–174. [Google Scholar] [CrossRef]
- Gaus, I.; Azaroual, M.; Czernichowski-Lauriol, I. Reactive transport modelling of the impact of CO2 injection on the clayey cap rock at Sleipner (North Sea). Chem. Geol. 2005, 217, 319–337. [Google Scholar] [CrossRef]
- Bickle, M.; Chadwick, A.; Huppert, H.E.; Hallworth, M.; Lyle, S. Modelling carbon dioxide accumulation at Sleipner: Implications for underground carbon storage. Earth Planet. Sci. Lett. 2007, 255, 164–176. [Google Scholar] [CrossRef]
- Xu, T.F.; Apps, J.A.; Pruess, K.; Yamamoto, H. Numerical modeling of injection and mineral trapping of CO2 with H2S and SO2 in a sandstone formation. Chem. Geol. 2007, 242, 319–346. [Google Scholar] [CrossRef]
- Hangx, S.; van der Linden, A.; Marcelis, F.; Bauer, A. The effect of CO2 on the mechanical properties of the captain sandstone: Geological storage of CO2 at the goldeneye field (UK). Int. J. Greenh. Gas Control 2013, 19, 609–619. [Google Scholar] [CrossRef]
- Aman, M.; Espinoza, D.N.; Ilgen, A.G.; Major, J.R.; Eichhubl, P.; Dewers, T.A. CO2 induced chemo mechanical alteration in reservoir rocks assessed via batch reaction experiments and scratch testing. Greenh. Gases Sci. Technol. 2018, 8, 133–149. [Google Scholar] [CrossRef]
- Golding, S.D.; Uysal, I.T.; Bolhar, R.; Boreham, C.J.; Dawson, G.K.W.; Baublys, K.A.; Esterle, J.S. Carbon dioxide-rich coals of the oaky creek area, central Bowen basin: A natural analogue for carbon sequestration in coal systems. Aust. J. Earth Sci. 2013, 60, 125–140. [Google Scholar] [CrossRef]
- Farquhar, S. CO2–Water–Rock Interactions in Low-Salinity Reservoir Systems. Ph.D. Thesis, University of Queensland, Brisbane, Australia, 2016. [Google Scholar]
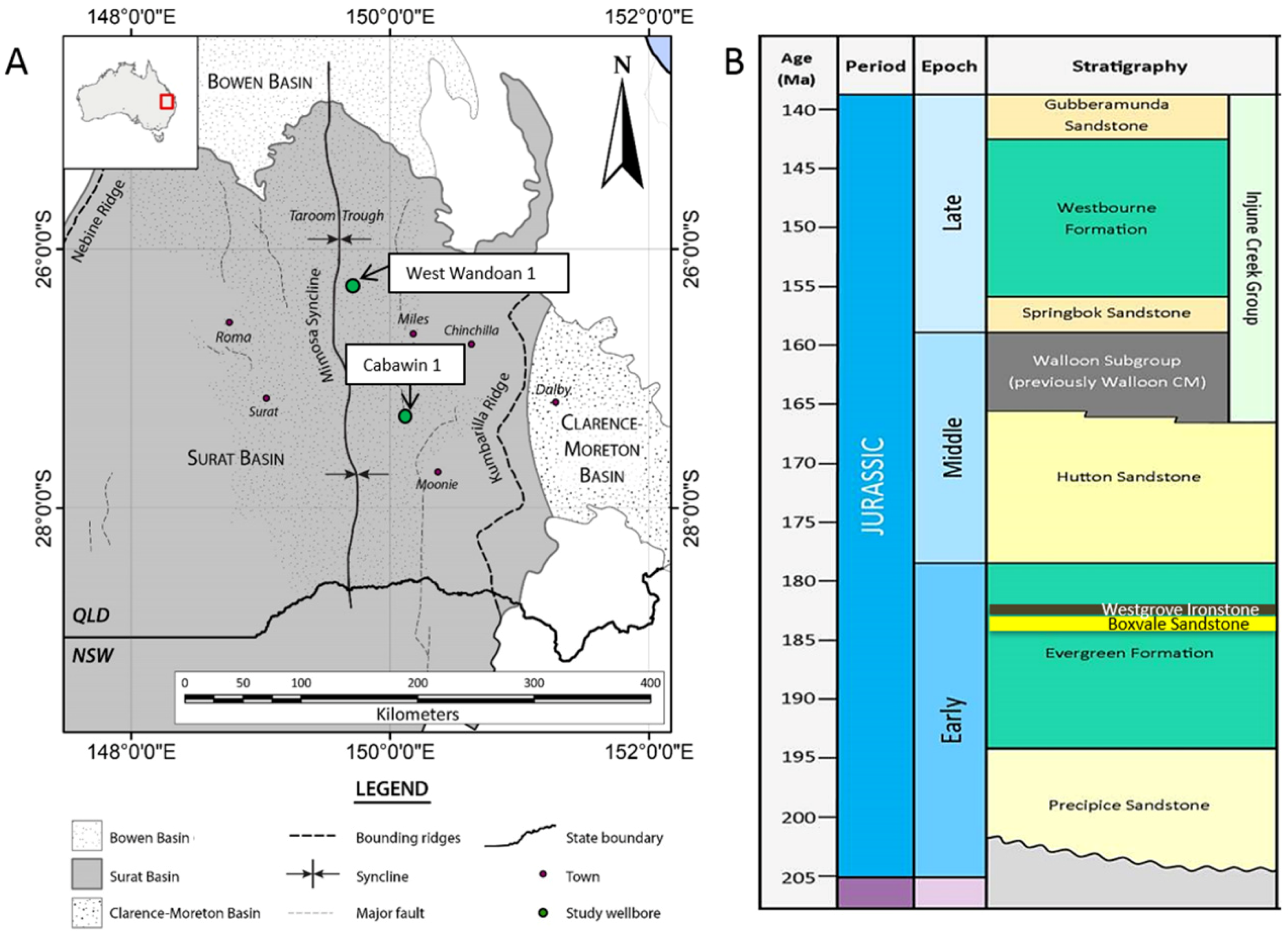
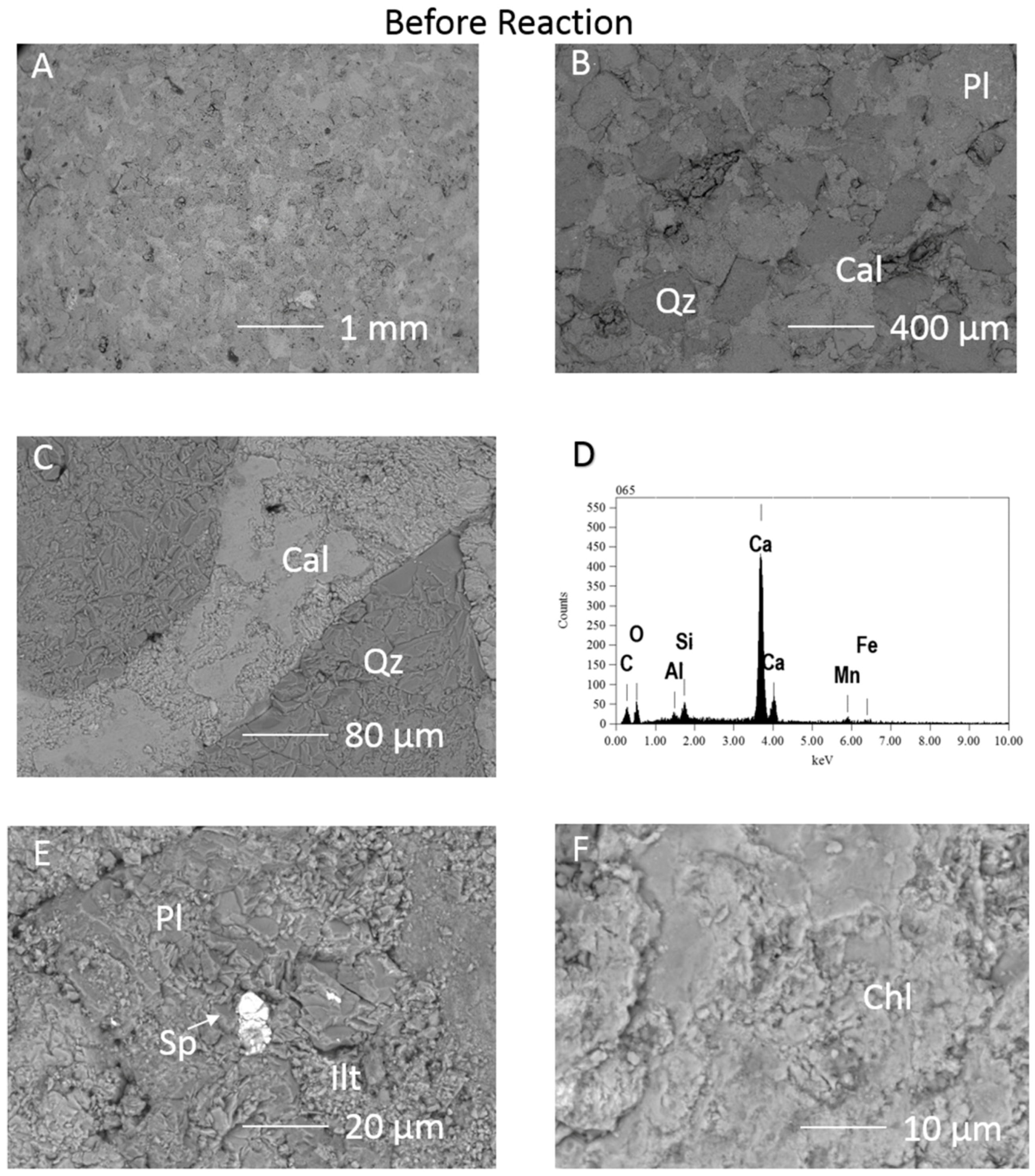




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mineral | QEMSCAN a | QEMSCAN b | Mineral | XRD | XRD Post |
|---|---|---|---|---|---|
| Depth/m | 1056.1 | 1056.1 | 1056.10–1056.18 | ||
| Quartz | 25 | 21 | Quartz | 27.3 | 37.6 |
| Alkali feldspar | 13 | 12 | Orthoclase | 6 | 12 |
| Plagioclase | 17.7 | 21.6 | Na(Ca)plagioclase | 14.7 | 12.5 |
| Musc/Illite | 0.4 | 0.2 | Illite/Musc M1 | 19 | 17.4 |
| Illite-Smec | 11.9 | 9.2 | Montmorillonite | 0 | 7.3 |
| Kaolinite | 0.9 | 0.6 | Kaolinite | 2 | 9 |
| Chlorite | 0.9 | 0.8 | Chlorite * | 1 | 4 |
| Calcite | 25.2 | 30.1 | Calcite | 16.3 | 0 |
| Dolomite/Mg-Cal | 0.5 | ||||
| Ca(Na)plagioclase | 13 | ||||
| Unclassified 1 | 4.9 | 4.7 | |||
| Total | 99.9 | 100.2 | 99.8 | 99.8 |
| Mineral | As * cm2/g | Asmod cm2/g | Asres cm2/g | ExpWW1 CalCem | WW1 CalCem | WW1 MudS | Cab1 SidCem | Cab1 Shale |
|---|---|---|---|---|---|---|---|---|
| Quartz | 10 | 10 | 1 | 24 | 27.3 | 43.0 | 39.0 | 40.1 |
| K-feld | 10 | 300 | 30 | 12.1 | 6.0 | 4.0 | 6.3 | 10.2 |
| Albite | 10 | 300 | 30 | 10.6 | 14.7 | 12.6 | 5.3 | 10.6 |
| Andesine | 10 | 300 | 30 | 6.4 | 13.0 | 9.0 | ||
| Kaolinite | 70 | 70 | 70 | 5.3 | 2.0 | 2.0 | 19.4 | 31.5 |
| Ill/Musc | 70 | 7000 | 70 | 13.2 | 19.0 | 23.0 | 6.7 | 4.4 |
| Smectite | 150 | 15 | ||||||
| Biotite | 70 | |||||||
| Chlorite | 70 | 7000 | 70 | 11.2 | 1.0 | 8.0 | 0.1 | 0.1 |
| Calcite | 10 | 1 | 0.01 | 16 | 16.8 | 1.0 | 0.7 | |
| Ankerite | 10 | 1 | 0.01 | 0.1 | ||||
| Siderite | 10 | 1 | 0.01 | 0.1 | 23.4 | 1.0 | ||
| Fe-oxide | 70 | 0.001 # | 10 | 1.5 | ||||
| Gyp/Anh | 10 | 0.05 # | 1 | |||||
| Pyrite | 10 | 10 | 10 | 0.8 | 0.2 | 0.05 | 0.05 | |
| Sphal | 10 | 10 | 10 | 0.5 |
| Time (h) | pH | Cond. | Al | Ba | Ca | Co | Cr | Fe | K | Li | Mg | Mn | Na | Ni | S | Si | Sr | Ti | Zn |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | 6.12 | 3 | 2.24 | 2.42 | 242.32 | 0.06 | 0.45 | 1.28 | 4.44 | <DL | 2.44 | 9.27 | 661.45 | 0.4 | 6.07 | 1.38 | 1.65 | 2.86 | 0.25 |
| 168 | 5.07 | 9.83 | 3.77 | 1068.89 | 0.33 | 8.39 | 98.96 | 19.63 | 0.1 | 15.37 | 49.08 | 1148.48 | 5.51 | 1398.85 | 45.94 | 9.36 | 0.08 | 0.97 | |
| 240 | 5.37 | 4.81 | 1.55 | 0.91 | 662.88 | 0.16 | 1.65 | 39.63 | 7.85 | 0.05 | 8.93 | 30.73 | 586.52 | 1.62 | 762.27 | 23.04 | 5.17 | 0.01 | 0.43 |
| 336 | 5.54 | 4.94 | 1.27 | 0.81 | 714.69 | 0.18 | 1.27 | 34.83 | 7.34 | 0.05 | 9.07 | 33.39 | 554.12 | 2.48 | 723.02 | 20.59 | 5.16 | 0.01 | 0.48 |
| 432 | 5.53 | 5.06 | 0.24 | 0.78 | 757.6 | 0.17 | 0.3 | 28 | 13.54 | 0.06 | 9.38 | 35.23 | 772.98 | 0.95 | 679.4 | 13.92 | 5.22 | 0.01 | 0.52 |
| 528 | 5.51 | 5.08 | <DL | 0.72 | 764.97 | 0.16 | 0.16 | 34.47 | 12.63 | 0.06 | 9.79 | 35.29 | 737.24 | 0.67 | 654.3 | 14.11 | 5.14 | 0 | 0.38 |
| 600 | 5.59 | 5.41 | <DL | 0.74 | 816.87 | 0.17 | 0.1 | 29.49 | 13.33 | 0.06 | 10.03 | 37.51 | 752.06 | 0.51 | 653.44 | 14.83 | 5.34 | 0.01 | 0.54 |
| 696 | 5.64 | 4.86 | 0.08 | 0.73 | 841.76 | 0.16 | 0.06 | 34.92 | 14.62 | 0.06 | 10.78 | 39.04 | 800.88 | 0.48 | 670.81 | 16.19 | 5.76 | 0 | 0.44 |
| 720 | 5.61 | 5.24 | <DL | 0.74 | 839.74 | 0.2 | 0.04 | 42.22 | 12.73 | 0.07 | 11.07 | 38.44 | 765.62 | 0.53 | 672.82 | 16.09 | 5.44 | 0.02 | 0.45 |
| quench | <DL | 0.68 | 768.49 | 0.15 | 0.02 | 25.17 | 11.75 | 0.07 | 10.19 | 35.75 | 710.06 | 0.2 | 618.41 | 15.06 | 5.01 | <DL | 0.3 | ||
| DL | 0.001 | 0.0001 | 0.0051 | 0.0007 | 0.0034 | 0.0009 | 0.27 | 0.0001 | 0.021 | 0.0002 | 0.27 | 0.0019 | 0.0018 | 0.03 | 0 | 0.0002 | 0.001 | ||
| DL*DF | 0.01 | 0.0011 | 0.051 | 0.0067 | 0.034 | 0.0088 | 2.7 | 0.0007 | 0.21 | 0.0015 | 2.7 | 0.019 | 0.018 | 0.3 | 0.0004 | 0.0024 | 0.01 |
| Time (h) | P | V | Cu | Ga | Ge | Rb | Nb | Sn | Sb | Th | U |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 0 | <DL | 0.81 | 52.24 | 0.30 | 0.45 | 31.19 | 0.07 | 3.63 | 0.81 | 0.06 | 0.63 |
| 168 | <DL | 34.54 | 151.85 | 2.10 | 2.57 | 38.63 | 1.03 | 5.02 | 1.57 | 0.66 | 1.28 |
| 240 | <DL | 10.82 | 101.38 | 1.89 | 2.33 | 41.23 | 0.29 | 5.61 | 3.95 | 0.31 | 1.41 |
| 336 | <DL | 4.32 | 68.50 | 1.47 | 1.73 | 43.94 | 0.15 | 4.25 | 1.00 | 0.16 | 1.38 |
| 432 | <DL | 0.33 | 0.33 | 0.42 | 43.02 | 0.04 | 4.46 | 0.99 | 0.07 | 1.06 | |
| 528 | <DL | 3.47 | 56.67 | 1.32 | 0.84 | 34.02 | 0.10 | 6.52 | 1.59 | 0.17 | 1.30 |
| 600 | <DL | 3.00 | 50.29 | 1.09 | 1.41 | 46.29 | 0.17 | 4.93 | 1.54 | 0.13 | 1.71 |
| 696 | <DL | 3.51 | 46.72 | 1.86 | 1.87 | 50.50 | 0.13 | 8.35 | 9.61 | 0.15 | 2.05 |
| 720 | <DL | 2.59 | 24.17 | 1.33 | 1.67 | 48.45 | 0.11 | 5.87 | 9.96 | 0.14 | 1.92 |
| Quench | <DL | 4.27 | 68.56 | 2.14 | 1.77 | 43.32 | 0.22 | 4.41 | 1.02 | 0.14 | 1.36 |
| DL | 3.8200 | 0.0076 | 0.0487 | 0.0021 | 0.0048 | 0.0062 | 0.0018 | 0.0385 | 0.0076 | 0.0015 | 0.0040 |
| DL*DF | 38.200 | 0.0762 | 0.4870 | 0.0208 | 0.0483 | 0.0615 | 0.0183 | 0.3850 | 0.0763 | 0.0146 | 0.0398 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pearce, J.K.; Dawson, G.K.W. Experimental Determination of Impure CO2 Alteration of Calcite Cemented Cap-Rock, and Long Term Predictions of Cap-Rock Reactivity. Geosciences 2018, 8, 241. https://doi.org/10.3390/geosciences8070241
Pearce JK, Dawson GKW. Experimental Determination of Impure CO2 Alteration of Calcite Cemented Cap-Rock, and Long Term Predictions of Cap-Rock Reactivity. Geosciences. 2018; 8(7):241. https://doi.org/10.3390/geosciences8070241
Chicago/Turabian StylePearce, Julie K., and Grant K. W. Dawson. 2018. "Experimental Determination of Impure CO2 Alteration of Calcite Cemented Cap-Rock, and Long Term Predictions of Cap-Rock Reactivity" Geosciences 8, no. 7: 241. https://doi.org/10.3390/geosciences8070241
APA StylePearce, J. K., & Dawson, G. K. W. (2018). Experimental Determination of Impure CO2 Alteration of Calcite Cemented Cap-Rock, and Long Term Predictions of Cap-Rock Reactivity. Geosciences, 8(7), 241. https://doi.org/10.3390/geosciences8070241