
Closed-System Magma Degassing and Disproportionation of SO2 Revealed by Changes in the Concentration and δ34S Value of H2S(g) in the Solfatara Fluids (Campi Flegrei, Italy)




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
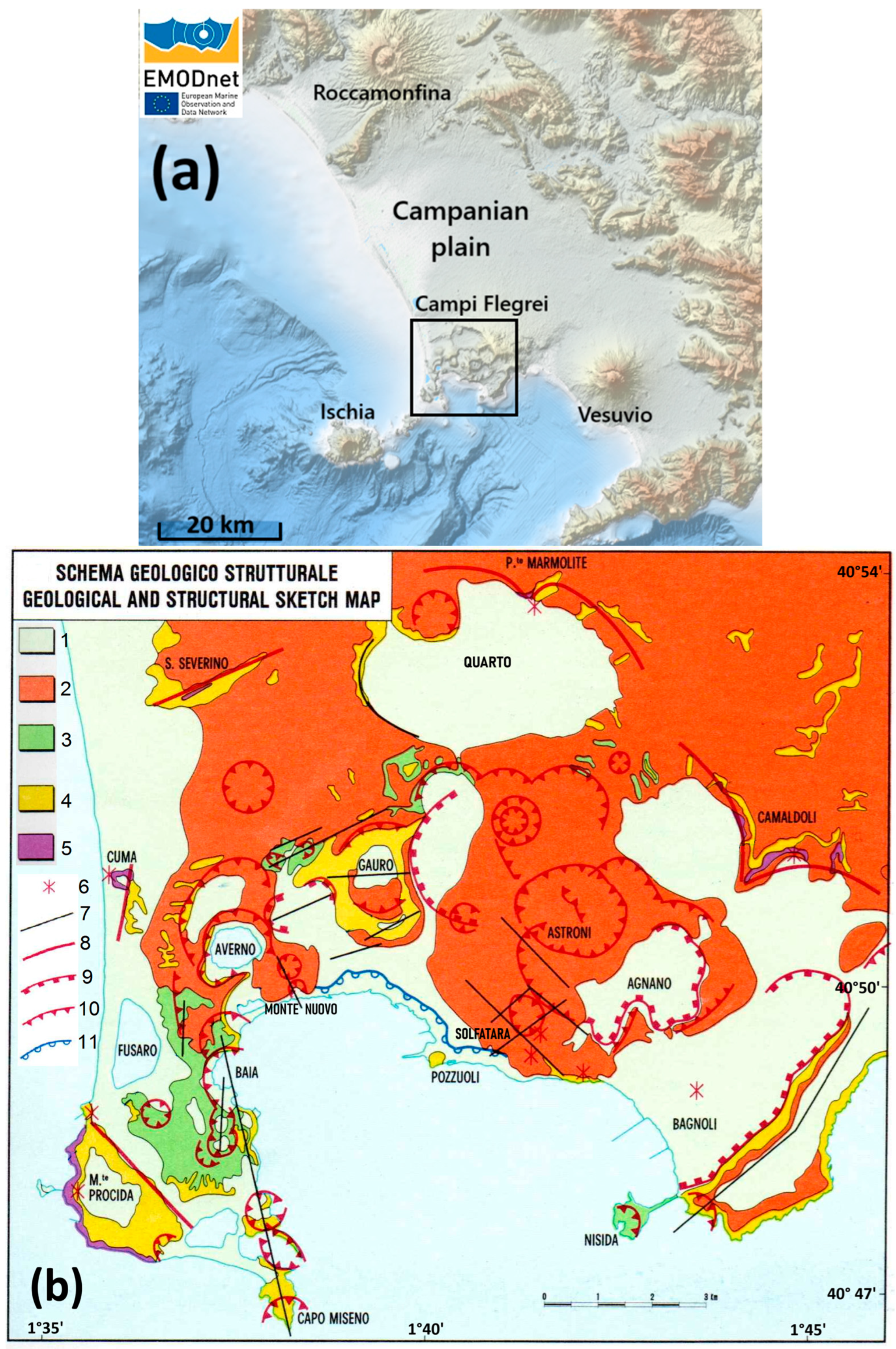
2. Geological Background
3. Materials and Methods
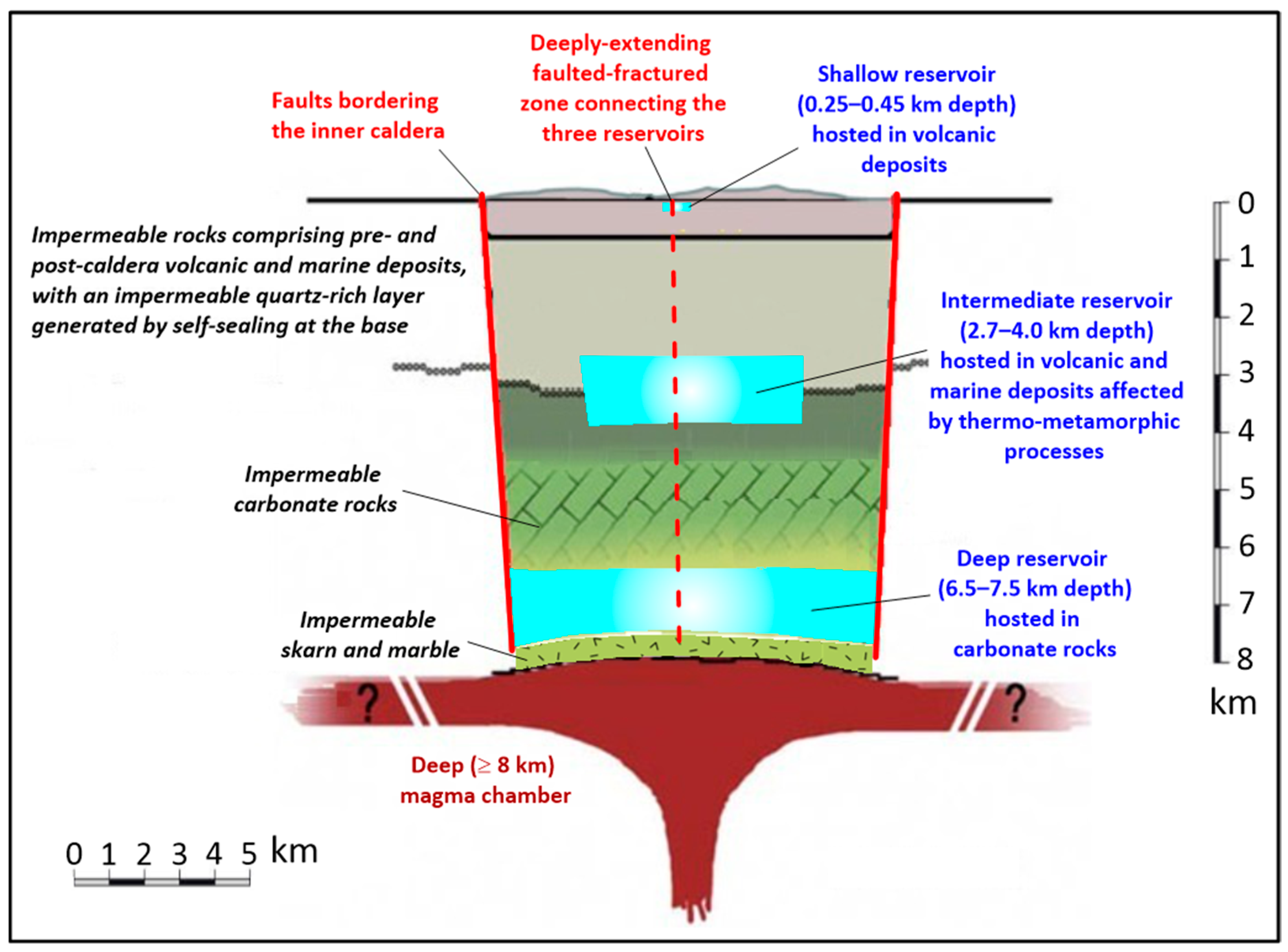
3.1. The Conceptual Model of the Solfatara Magmatic–Hydrothermal System
- (1)
- The cover of the shallow hydrothermal reservoir (0–0.25 km depth), made up of volcanic deposits influenced by advanced argillic hydrothermal alteration close to the surface and by argillic hydrothermal alteration away from it [18,45]. Its area is around 1 km2, as indicated by the Solfatara diffuse degassing structure [46,47].
- (2)
- The shallow hydrothermal reservoir (0.25–0.45 km depth), a steam and gas pocket hosted in volcanic deposits. It has the same area as its cover and a small volume, ~0.2 km3.
- (3)
- An impermeable unit (0.45–2.7 km depth), including volcanic and marine deposits, influenced by phyllitic hydrothermal alteration in the upper portion and by propylitic hydrothermal alteration in the lower part, with an impermeable quartz-rich layer generated by self-sealing at the bottom [18,45]. The area of this impermeable unit corresponds to that of the inner caldera block of Barberi et al. [20].
- (4)
- The intermediate hydrothermal reservoir (2.7–4.0 km depth) hosted in volcanic and marine deposits modified by thermometamorphic hydrothermal alteration. According to several studies, e.g., [21,48,49,50,51,52,53], over-pressurized supercritical fluids are stored in the intermediate hydrothermal reservoir, which, consequently, is the engine of the ground uplift and the associated shallow seismicity. Again, the area of the intermediate hydrothermal reservoir matches that of the inner caldera block of Barberi et al. [20]. Nevertheless, the intermediate hydrothermal reservoir might be constituted by distinct compartments rather than a unique aquifer, as indicated by the piecemeal collapse of the inner caldera [54] and the distribution of seismic events during the last years of the ongoing unrest (https://terremoti.ov.ingv.it/gossip/flegrei/index.html, last accessed 14 February 2025).
- (5)
- A thick carbonate sequence (4.0–6.5 km depth), behaving as aquiclude evidently due to nil to negligible dissolution and fracturing, similar to what was found by the deep geothermal well Nisyros-1 [55], which intersected an 830 m thick pile of impermeable carbonate rocks overlying a diorite intrusion and the associated thermometamorphic rocks.
- (6)
- The deep hydrothermal reservoir (6.5–7.5 km depth) hosted in fractured carbonate rocks, also affected by dissolution–precipitation processes and/or gas–solid reactions governed by acidic magmatic fluids and/or gases. Its area is the same as that of the underlying units.
- (7)
- An impermeable unit made up of skarn and marble (7.5–8.0 km depth) generated by thermometamorphic and metasomatic processes and behaving as an aquiclude due to their nil porosity [56].
- (8)
3.2. Temperature and Pressure Conditions
- (i)
- CO equilibrated in the shallow hydrothermal reservoir at nearly constant temperature, 217 ± 9 °C, and total fluid pressure, 24.5 ± 3.9 bar.
- (ii)
- CH4 attained thermochemical equilibrium in the intermediate hydrothermal reservoir at temperature and total fluid pressure increasing with time, from 235 °C and 31.1 bar in March 1984 to peak values of 609 °C and 1340 bar in September 2023.
- (iii)
- H2S achieved the equilibrium condition in the deep hydrothermal reservoir at temperature and total fluid pressure incrementing with time, from 618 °C and 1120 bar in July 1984 to maximum values of 1040 °C and 3280 bar in October 2019. These temperatures are in satisfactory agreement with those of 880–1020 °C obtained by extrapolating the geothermal gradient of ca. 134 °C/km measured in the deepest levels of the San Vito 1 well [58].
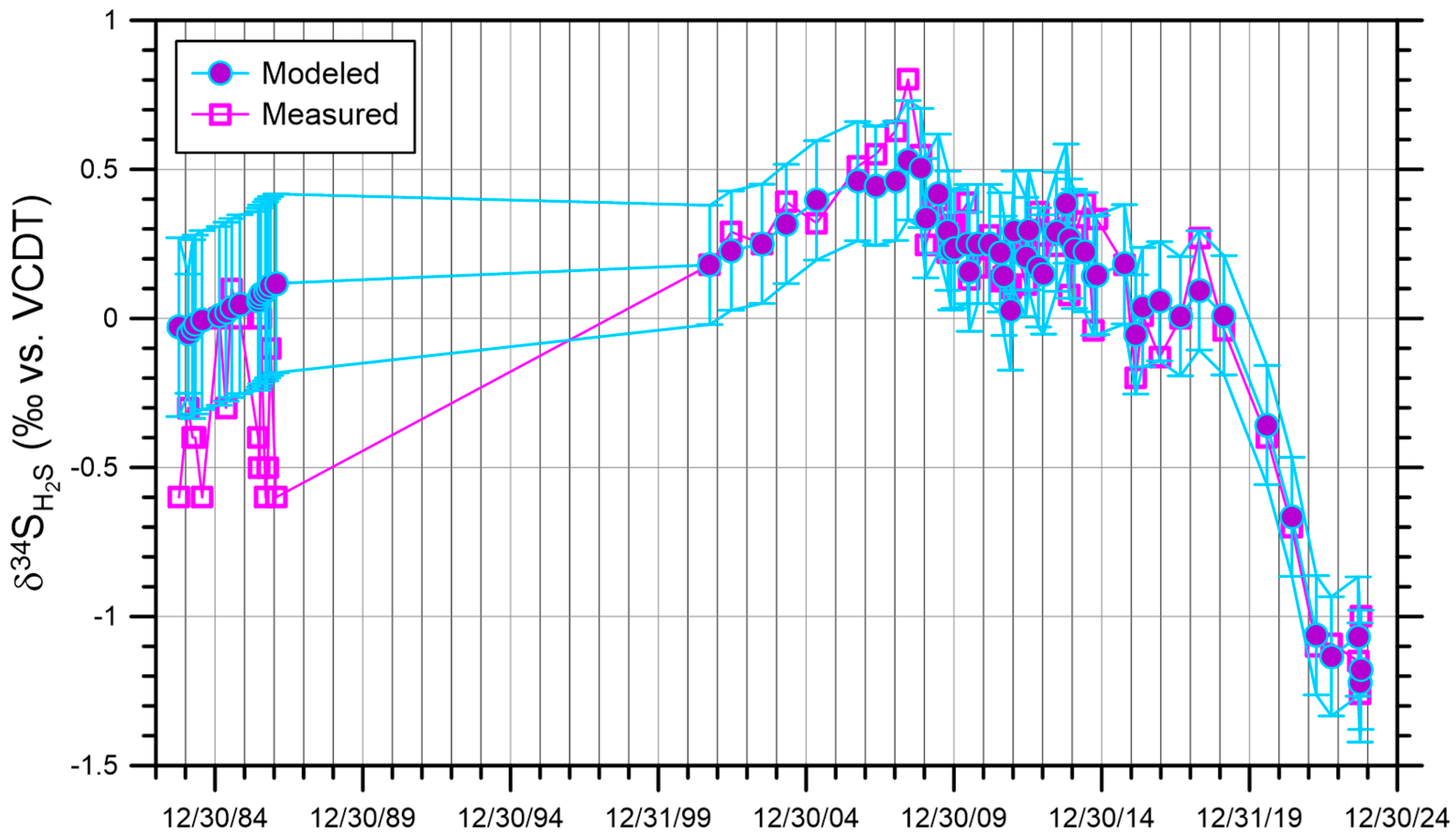
3.3. Processes Controlling the Concentration and the δ34S of H2S(g)
- (1)
- The separation of SO2(g)- and H2S(g)-bearing magmatic gases from the melt, occurring in the melt zone, at depths ≥ 8 km, under closed-system conditions;
- (2)
- The SO2 disproportionation reaction occurring in the deep hydrothermal reservoir and the related production of anhydrite.
3.4. Modeling of Magma Degassing
3.5. Modeling the SO2 Disproportionation Reaction in the Deep Hydrothermal Reservoir
3.6. Calculation Strategy
4. Results and Discussion
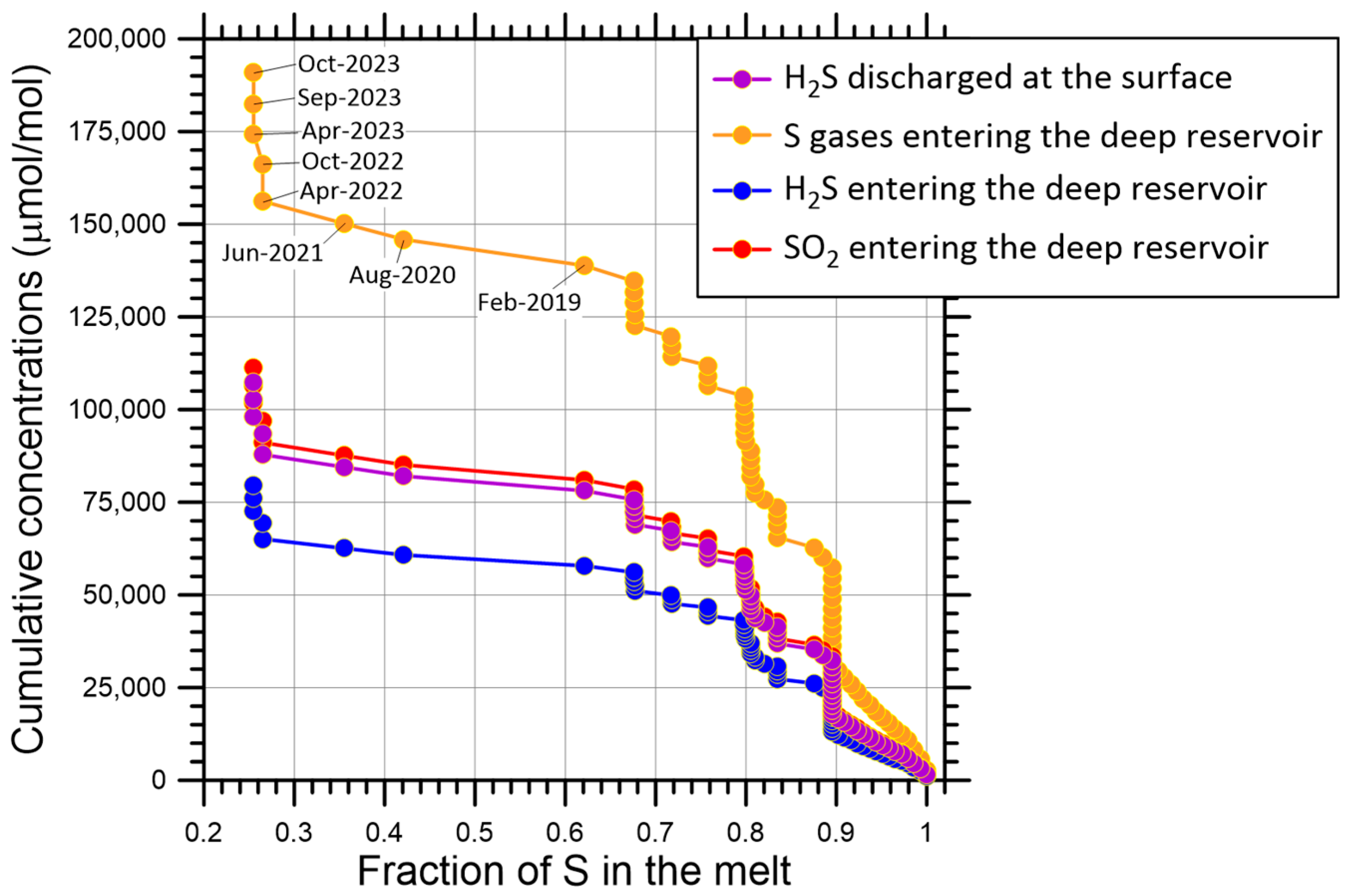
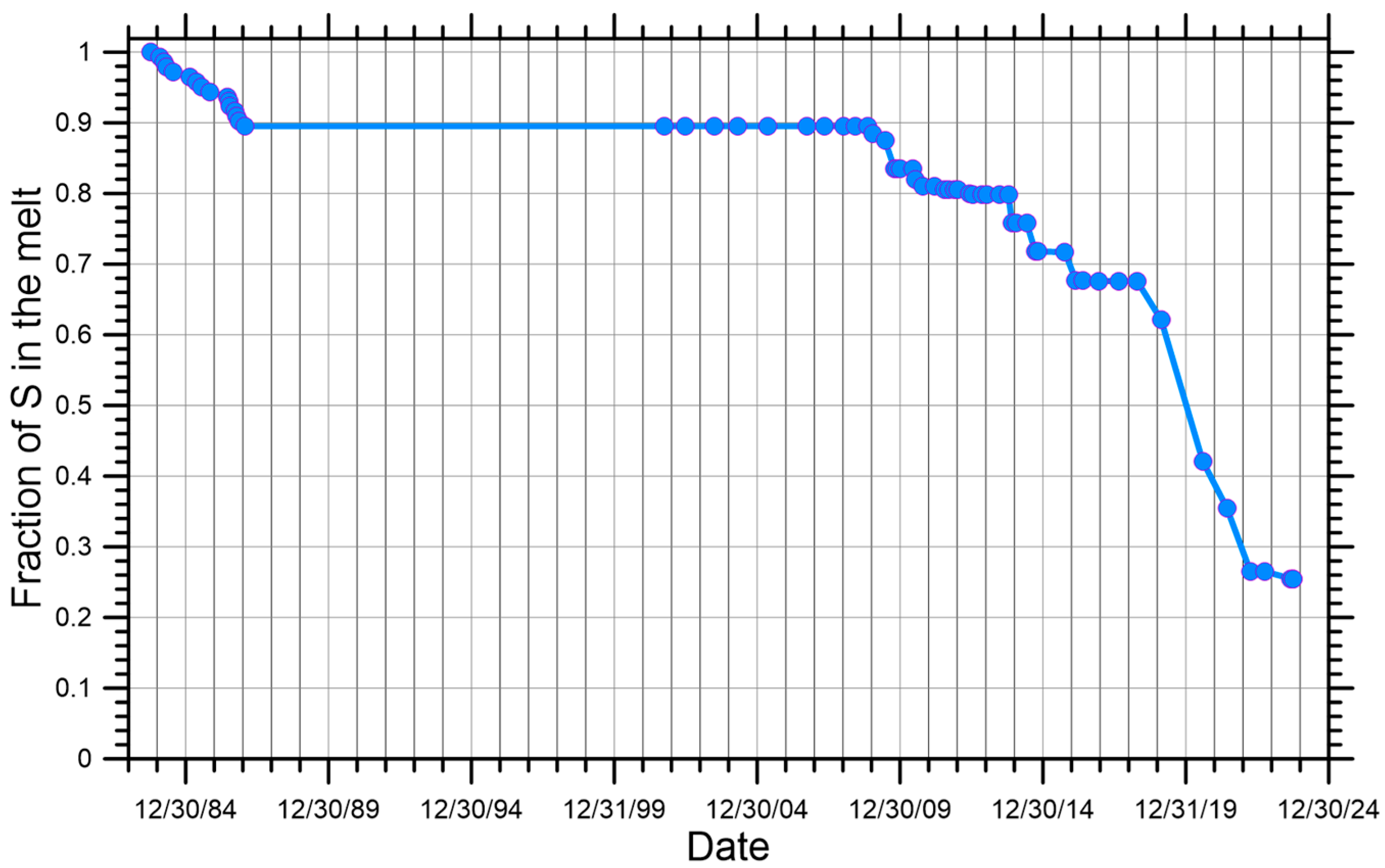
- (i)
- Total ionic S dissolved in the melt, which decreases from 0.00 to −2.40‰ and has a perfect linear relationship with F, as dictated by Equation (9).
- (ii)
- Total gaseous S in the gas mixture separated from the melt, which declines from +3.22 to +0.82‰ and, again, has an exact linear relationship with F, constrained by Equations (9) and (16).
- (iii)
- Sulfur dioxide in the gas mixture separated from the melt, which diminishes from +4.02 to +1.62‰ and, again, has a perfect linear relationship with F, defined by Equations (9) and (16)–(18).
- (iv)
- Hydrogen sulfide in the gas mixture separated from the melt, which decreases from +2.10 to −0.30‰ and has an exact linear relationship with F, constrained by Equations (9), (16) and (17).
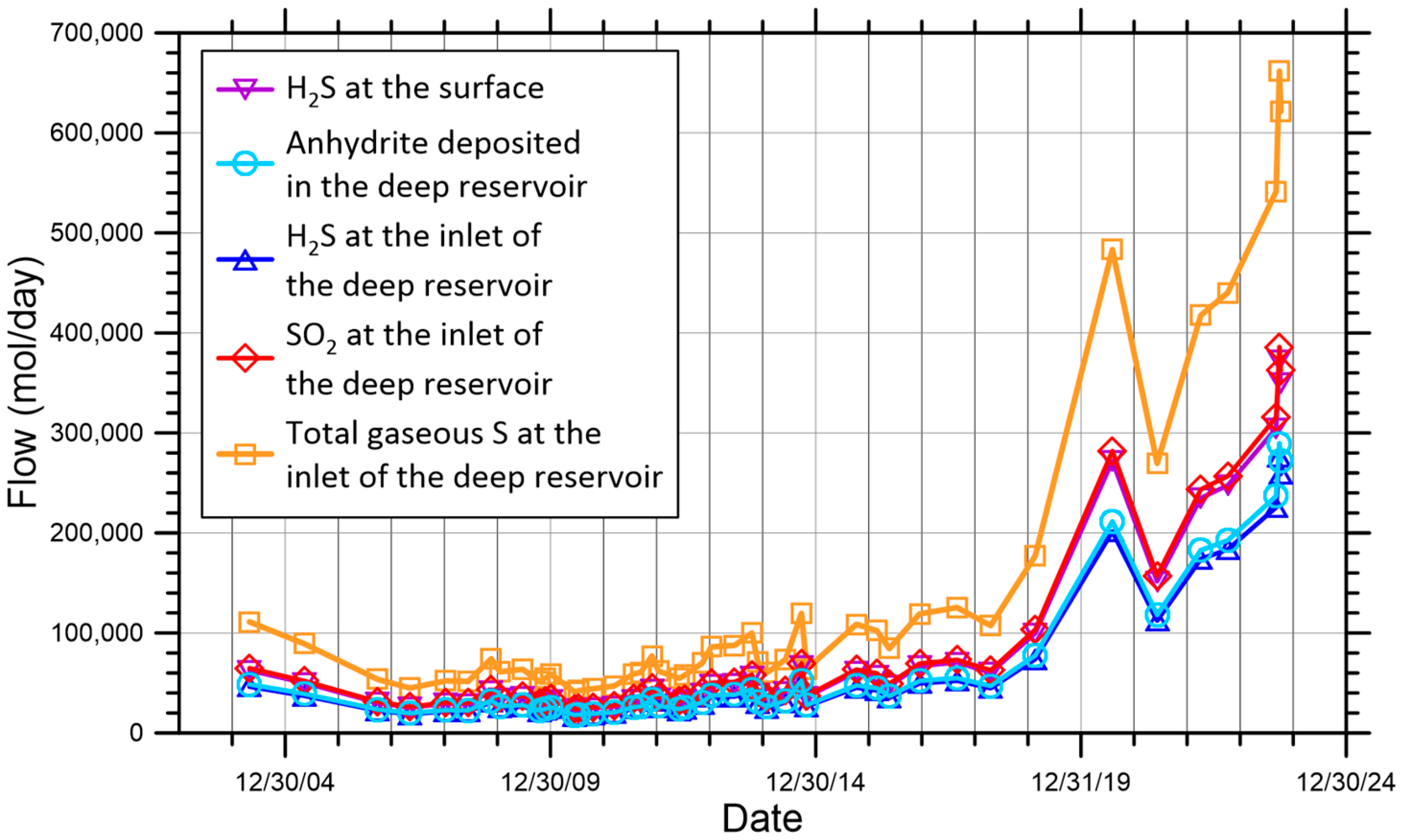
- (v)
- Sulfur dioxide in the gas mixture at the inlet of the deep hydrothermal reservoir, which declines from +5.21 to +1.90‰, describing a non-linear trend with a weak upward curvature and several small oscillations. These are due to the joint effects of magma degassing and SO2 disproportionation. It is worth noting that magma degassing, in the absence of other processes, would determine a linear trend, as in previous points. Moreover, SO2 disproportionation is responsible for the deviations from linearity because of the temperature changes in the deep hydrothermal reservoir from 671 to 1022 °C.
- (vi)
- Hydrogen sulfide in the gas mixture at the inlet of the deep hydrothermal reservoir, which oscillates from +0.94 to −0.83‰, delineating a generally decreasing, non-linear trend with a weak downward curvature and numerous little fluctuations due to the combined influences of magma degassing and SO2 disproportionation, similar to the previous point.
- (vii)
- Hydrogen sulfide in the gas mixture at the outlet of the deep hydrothermal reservoir, which varies from +0.53 to −1.22‰, outlining a trend similar to that of the previous point but shifted downward by 0.46–0.37‰ units, corresponding to the difference .
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A. Relevant Reactions Involving Anhydrite and Pyrite
Appendix A.1. SO2 Disproportionation and Anhydrite Formation
Appendix A.2. Reactions Involving Pyrite
Appendix A.3. The Distribution and Role of Pyrite and Anhydrite in the Solfatara Magmatic–Hydrothermal System
- (i)
- The temperature and pressure conditions are probably constrained by the coexistence, at equilibrium, of a vapor phase and a high-salinity sodium-chloride brine, because the other possible conditions are highly unlikely [16]. In fact, the occurrence of a single liquid phase at relatively low temperatures and high pressures is at variance with the large heat flow released from the deep melt zone and transferred to the overlying hydrothermal portion of the system, whereas the occurrence of a single vapor phase coexisting with solid NaCl (which is typical of depressurized vapor-cored magmatic systems [117]) is at variance with the current pressurization of the intermediate hydrothermal reservoir of the Solfatara hydrothermal–magmatic system and related ground uplift, shallow seismicity, and increasing emission of fluids from the Solfatara-Pisciarelli fumaroles [14]. The related implication is that SO2 disproportionation occurring in the deep hydrothermal reservoir follows the wet path described by reaction (A1).
- (ii)
- Accepting that the deep hydrothermal reservoir is hosted in Ca-rich, Fe-poor carbonate rocks, it is likely that the H2SO4 generated by reaction (A1) is neutralized by reaction (A6), causing the conversion of calcite into anhydrite, whereas it is unlikely that H2SO4 neutralization is controlled by reaction (A10). The occurrence of reaction (A6) is supported by the abundance of calcite and anhydrite, as vein minerals, in the carbonate–evaporite geothermal systems of Central Italy [118], such as the Latera geothermal system, where both anhydrite and calcite are very abundant secondary minerals, even in the contact-metasomatism assemblage close to the magma chamber [119], which was penetrated for >350 m by deep geothermal drilling, being positioned at ~2 km depth [120].
- (iii)
- Furthermore, anhydrite is stable at the temperatures of 618–1040 °C estimated for the deep hydrothermal reservoir using the H2S-CO2 gas geothermometer [16,58], whereas pyrite is expected to decompose rapidly at these temperatures (see Appendix A.2). Calcite and anhydrite are the two solid phases controlling the H2S-CO2 gas geothermometer, which is based on the following heterogeneous equilibrium reaction:occurring in the deep hydrothermal reservoir. In this gas geothermometer, calibrated by Marini et al. [58] and applied by Marini et al. [16], Equation (A14) is treated as an equilibrium reaction. Therefore, anhydrite was not considered to be a potential source of H2S by Marini et al. [16], as erroneously reported in Caliro et al. [15]. Indeed, as discussed in Appendix A.1, anhydrite is a sink of H2S and is not a source of H2S.
- (iv)
- The hydrothermal S-bearing mineral most commonly encountered in drilled geothermal systems is pyrite [121,122,123,124]. Not surprisingly, pyrite is stable at almost all depths and temperatures, up to 350 °C, in wells Mofete 1 and 2 [125]. In general, pyrrhotite is much less abundant than pyrite in drilled geothermal systems, and in wells Mofete 1 and 2, pyrrhotite is present only in the propylitic and thermometamorphic zones, at temperatures of 250–350 °C [125]. Based on this evidence, Marini et al. [58] calibrated the gas geothermometers pyrite-pyrrhotite, pyrite-fayalite-quartz, pyrite-magnetite, and pyrite-hematite, considering deviations from ideality, but the equilibrium temperatures computed by these pyrite-bearing gas geothermometers resulted in being at variance with the CH4 equilibrium temperature for the Solfatara fumarolic fluids, suggesting that the H2S concentration of Solfatara fluids is not controlled by reactions involving pyrite. Therefore, pyrite and reactions involving it were disregarded in this work.
References
- Sakai, H.; Casadevall, T.J.; Moore, J.G. Chemistry and isotope ratios of sulfur in basalts and volcanic gases at Kilauea volcano, Hawaii. Geochim. Cosmochim. Acta 1982, 46, 729–738. [Google Scholar] [CrossRef]
- Ueda, A.; Sakai, H. Sulfur isotope study of Quaternary volcanic rocks from the Japanese Islands Arc. Geochim. Cosmochim. Acta 1984, 48, 1837–1848. [Google Scholar] [CrossRef]
- Taylor, B.E. Magmatic volatiles—Isotopic variation of C, H, and S. Rev. Mineral. 1986, 16, 185–225. [Google Scholar]
- Marini, L.; Paiotti, A.; Principe, C.; Ferrara, G.; Cioni, R. Isotopic ratio and concentration of sulfur in the undersaturated alkaline magmas of Vulture Volcano (Italy). Bull. Volcanol. 1994, 56, 487–492. [Google Scholar] [CrossRef]
- Marini, L.; Chiappini, V.; Cioni, R.; Cortecci, G.; Dinelli, E.; Principe, C.; Ferrara, G. Effect of degassing on sulfur contents and δ34S values in Somma-Vesuvius magmas. Bull. Volcanol. 1998, 60, 187–194. [Google Scholar] [CrossRef]
- Marini, L.; Moretti, R.; Accornero, M. Sulfur isotopes in magmatic-hydrothermal systems, melts, and magmas. Rev. Mineral. Geochem. 2011, 73, 423–492. [Google Scholar] [CrossRef]
- Carroll, M.R.; Rutherford, M.J. Sulfur speciation in hydrous experimental glasses of varying oxidation-state—Results from measured wavelength shifts of sulfur X-rays. Am. Mineral. 1988, 73, 845–849. [Google Scholar]
- Wallace, P.; Carmichael, I.S.E. Sulfur in basaltic magmas. Geochim. Cosmochim. Acta 1992, 56, 1863–1874. [Google Scholar] [CrossRef]
- Jugo, P.J.; Wilke, M.; Botcharnikov, R.E. Sulfur K-edge XANES analysis of natural and synthetic basaltic glasses: Implications for S speciation and S content as function of oxygen fugacity. Geochim. Cosmochim. Acta 2010, 74, 5926–5938. [Google Scholar] [CrossRef]
- Baker, D.R.; Moretti, R. Modeling the solubility of sulfur in magmas: A 50-year old geochemical challenge. Rev. Mineral. Geochem. 2011, 73, 167–213. [Google Scholar] [CrossRef]
- Mandeville, C.W.; Sasaki, A.; Saito, G.; Faure, K.; King, R.; Hauri, E. Open-system degassing of sulfur from Krakatau, magma. Earth Planet. Sci. Lett. 1998, 160, 709–722. [Google Scholar] [CrossRef]
- Burgisser, A.; Scaillet, B. Redox evolution of a degassing magma rising to the surface. Nature 2007, 445, 194–197. [Google Scholar] [CrossRef] [PubMed]
- Rye, R.O. A review of the stable-isotope geochemistry of sulfate minerals in selected igneous environments and related hydrothermal systems. Chem. Geol. 2005, 215, 5–36. [Google Scholar] [CrossRef]
- Giudicepietro, F.; Avino, R.; Bellucci Sessa, E.; Bevilacqua, A.; Bonano, M.; Caliro, S.; Francesco Casu, F.; De Cesare, W.; De Luca, C.; De Martino, P.; et al. Burst-like swarms in the Campi Flegrei caldera accelerating unrest from 2021 to 2024. Nature Commun. 2025, 16, 1548. [Google Scholar] [CrossRef] [PubMed]
- Caliro, S.; Chiodini, G.; Avino, R.; Carandente, A.; Cuoco, E.; Di Vito, M.A.; Minopoli, C.; Rufino, F.; Santi, A.; Lages, J.; et al. Escalation of caldera unrest indicated by increasing emission of isotopically light sulfur. Nat. Geosci. 2025, 18, 167–174. [Google Scholar] [CrossRef]
- Marini, L.; Principe, C.; Lelli, M. Time changes during the last 40 years in the Solfatara magmatic-hydrothermal system (Campi Flegrei, Italy): New conceptual model and future scenarios. EGUsphere 2024, 1–25. [Google Scholar] [CrossRef]
- Rosi, M.; Sbrana, A.; Principe, C. The Phlegraean Fields: Structural evolution, volcanic history and eruptive mechanisms. J. Volcanol. Geotherm. Res. 1983, 17, 273–288. [Google Scholar] [CrossRef]
- Chelini, W.; Sbrana, A. Subsurface Geology. In Phlegrean Fields, Progetto Finalizzato ‘Geodinamica’, Monografie Finali; Quaderni de “La Ricerca Scientifica”; Rosi, M., Sbrana, A., Eds.; Consiglio Nazionale delle Ricerche: Roma, Italy, 1987; Volume 9, pp. 94–103. [Google Scholar]
- Cassano, E.; La Torre, P. Geophysics. In Phlegrean Fields, Progetto Finalizzato ‘Geodinamica’, Monografie Finali; Quaderni de “La Ricerca Scientifica”; Rosi, M., Sbrana, A., Eds.; Consiglio Nazionale delle Ricerche: Roma, Italy, 1987; Volume 9, pp. 103–131. [Google Scholar]
- Barberi, F.; Cassano, E.; La Torre, P.; Sbrana, A. Structural evolution of Campi Flegrei caldera in light of volcanological and geophysical data. J. Volcanol. Geotherm. Res. 1991, 48, 33–49. [Google Scholar] [CrossRef]
- Zollo, A.; Maercklin, N.; Vassallo, M.; Dello Iacono, D.; Virieux, J.; Gasparini, P. Seismic reflections reveal a massive melt layer feeding Campi Flegrei caldera. Geophys. Res. Lett. 2008, 35, L12306. [Google Scholar] [CrossRef]
- EMODnet Consortium. Available online: https://www.emodnet-bathymetry.eu/ (accessed on 12 April 2025).
- Principe, C.; Rosi, M.; Sbrana, A.; Zan, L. Geological and gravimetric map of Phlegrean Fields, scale 1:15,000. In Phlegrean Fields, Progetto Finalizzato ‘Geodinamica’, Monografie Finali; Quaderni de “La Ricerca Scientifica”; Rosi, M., Sbrana, A., Eds.; Consiglio Nazionale delle Ricerche: Roma, Italy, 1987; Volume 9. [Google Scholar]
- Giaccio, B.; Hajdas, I.; Isaia, R.; Deino, A.; Nomade, S. High-precision 14C and 40Ar/39Ar dating of the Campanian Ignimbrite (Y-5) reconciles the timescales of climatic-cultural processes at 40 ka. Sci. Rep. 2017, 7, 45940. [Google Scholar] [CrossRef]
- Acocella, V. Activating and reactivating pairs of nested collapses during caldera-forming eruptions: Campi Flegrei (Italy). Geophys. Res. Lett. 2008, 35, L17304. [Google Scholar] [CrossRef]
- Deino, A.L.; Orsi, G.; de Vita, S.; Piochi, M. The age of the Neapolitan Yellow Tuff caldera-forming eruption (Campi Flegrei caldera-Italy) assessed by 40Ar/39Ar dating method. J. Volcanol. Geotherm. Res. 2004, 133, 157–170. [Google Scholar] [CrossRef]
- Lirer, L.; Luongo, G.; Scandone, R. On the volcanological evolution of Campi Flegrei. EOS Trans. Am. Geophys. Union. 1987, 68, 226–234. [Google Scholar] [CrossRef]
- Rosi, M.; Sbrana, A. Petrography. In Phlegrean Fields, Progetto Finalizzato ‘Geodinamica’, Monografie Finali; Quaderni de “La Ricerca, Scientifica”; Rosi, M., Sbrana, A., Eds.; Consiglio Nazionale delle Ricerche: Roma, Italy, 1987; Volume 9, pp. 60–80. [Google Scholar]
- Armienti, P.; Barberi, F.; Bizojard, H.; Clocchiatti, R.; Innocenti, F.; Metrich, N.; Rosi, M.; Sbrana, A. The Phlegraean Fields: Magma evolution within a shallow chamber. J. Volcanol. Geotherm. Res. 1983, 17, 289–311. [Google Scholar] [CrossRef]
- Rosi, M.; Sbrana, A. Description of mapped products. In Phlegrean Fields, Progetto Finalizzato ‘Geodinamica’, Monografie Finali; Quaderni de “La Ricerca, Scientifica”; Rosi, M., Sbrana, A., Eds.; Consiglio Nazionale delle Ricerche: Roma, Italy, 1987; Volume 9, pp. 39–60. [Google Scholar]
- Cipriani, F.; Marianelli, P.; Sbrana, A. Studio di una sequenza piroclastica del vulcano della Solfatara (Campi Flegrei). Considerazioni vulcanologiche e sul sistema di alimentazione. Atti Soc. Tosc. Sci. Nat. Mem. Serie A 2008, 113, 39–48. [Google Scholar]
- Isaia, R.; Vitale, S.; Di Giuseppe, M.G.; Iannuzzi, E.; D’Assisi, T.F.; Troiano, A. Stratigraphy, structure, and volcano-tectonic evolution of Solfatara maar-diatreme (Campi Flegrei, Italy). Geol. Soc. Am. Bull. 2015, 127, 1485–1504. [Google Scholar] [CrossRef]
- Principe, C. Managing different eruptive scenarios at Phlegraean Fields and Vesuvius. In Proceedings of the EGU General Assembly, EGU24-22302. Vienna, Austria, 14–19 April 2024. EGU24-22302. [Google Scholar] [CrossRef]
- Smith, V.C.; Isaia, R.; Pearce, N.J.G. Tephrostratigraphy and glass compositions of post-15 kyr Campi Flegrei eruptions: Implications for eruption history and chronostratigraphic markers. Quat. Sci. Rev. 2011, 30, 3638–3660. [Google Scholar] [CrossRef]
- Guidoboni, E.; Ciuccarelli, C. The Campi Flegrei caldera: Historical revision and new data on seismic crises, bradyseisms, the Monte Nuovo eruption and ensuing earthquakes (twelfth century 1582 AD). Bull. Volcanol. 2011, 73, 655–677. [Google Scholar] [CrossRef]
- Parascandola, A. I fenomeni bradisismici del Serapeo di Pozzuoli; Genovese, G., Ed.; Guida Editori: Napoli, Italy, 1947; pp. 1–156. [Google Scholar]
- Sbrana, A.; Marianelli, P.; Pasquini, G. The Phlegrean Fields volcanological evolution. J. Maps 2021, 17, 545–558. [Google Scholar] [CrossRef]
- Lyell, C. Principles of Geology; Murray, J., Ed.; Penguin Classics: London, UK, 1830; Volume 1, pp. 489–498. [Google Scholar]
- Del Gaudio, C.; Aquino, I.; Ricciardi, G.P.; Ricco, C.; Scandone, R. Unrest episodes at Campi Flegrei: A reconstruction of vertical ground movements during 1905–2009. J. Volcanol. Geotherm. Res. 2010, 195, 48–56. [Google Scholar] [CrossRef]
- De Martino, P.; Tammaro, U.; Obrizzo, F. GPS time series at Campi Flegrei caldera (2000–2013). Ann. Geophys. 2014, 57, 0213. [Google Scholar] [CrossRef]
- Tramelli, A.; Godano, C.; Ricciolino, P.; Giudicepietro, F.; Caliro, S.; Orazi, M.; De Martino, P.; Chiodini, G. Statistics of seismicity to investigate the Campi Flegrei caldera unrest. Sci. Rep. 2021, 11, 7211. [Google Scholar] [CrossRef] [PubMed]
- INGV—Osservatorio Vesuviano, Bollettino di Sorveglianza, Campi Flegrei, Marzo 2025. Available online: https://www.ov.ingv.it/index.php/monitoraggio-e-infrastrutture/bollettini-tutti/bollett-mensili-cf/anno-2025-3/1754-bollettino-mensile-campi-flegrei-2025-03/file (accessed on 12 April 2025).
- D’Auria, L.; Pepe, S.; Castaldo, R.; Giudicepietro, F.; Macedonio, G.; Ricciolino, P.; Tizzani, P.; Casu, F.; Lanari, R.; Manzo, M.; et al. Magma injection beneath the urban area of Naples: A new mechanism for the 2012–2013 volcanic unrest at Campi Flegrei caldera. Sci. Rep. 2015, 5, 13100. [Google Scholar] [CrossRef]
- Todesco, M. Signals from the Campi Flegrei hydrothermal system: Role of a “magmatic” source of fluids. J. Geophys. Res. Solid. Earth 2009, 114, B05201. [Google Scholar] [CrossRef]
- Piochi, M.; Mormone, A.; Balassone, G.; Strauss, H.; Troise, C.; De Natale, G. Native sulfur, sulfates and sulfides from the active Campi Flegrei volcano (southern Italy): Genetic environments and degassing dynamics revealed by mineralogy and isotope geochemistry. J. Volcanol. Geotherm. Res. 2015, 304, 180–193. [Google Scholar] [CrossRef]
- Chiodini, G.; Frondini, F.; Cardellini, C.; Granieri, D.; Marini, L.; Ventura, G. CO2 degassing and energy release at Solfatara volcano, Campi Flegrei, Italy. J. Geophys. Res. 2001, 106, 16213–16222. [Google Scholar] [CrossRef]
- Cardellini, C.; Chiodini, G.; Frondini, F.; Avino, R.; Bagnato, E.; Caliro, S.; Lelli, M.; Rosiello, A. Monitoring diffuse volcanic degassing during volcanic unrests: The case of Campi Flegrei (Italy). Sci. Rep. 2017, 7, 6757. [Google Scholar] [CrossRef]
- Zollo, A.; Judenherc, S.; Auger, E.; D’Auria, L.; Virieux, J.; Capuano, P.; Chiarabba, C.; de Franco, R.; Makris, J.; Michelini, A.; et al. Evidence for the buried rim of Campi Flegrei caldera from 3-d active seismic imaging. Geophys. Res. Lett. 2003, 30. [Google Scholar] [CrossRef]
- Judenherc, S.; Zollo, A. The Bay of Naples (southern Italy): Constraints on the volcanic structures inferred from a dense seismic survey. J. Geophys. Res. Solid. Earth 2004, 109, B10312. [Google Scholar] [CrossRef]
- Vanorio, T.; Virieux, J.; Capuano, P.; Russo, G. Three-dimensional seismic tomography from P wave and S wave microearthquake travel times and rock physics characterization of the Campi Flegrei Caldera. J. Geophys. Res. Solid. Earth 2005, 110. [Google Scholar] [CrossRef]
- Chiarabba, C.; Moretti, M. An insight into the unrest phenomena at the Campi Flegrei caldera from Vp and Vp/Vs tomography. Terra Nova 2006, 18, 373–379. [Google Scholar] [CrossRef]
- Battaglia, J.; Zollo, A.; Virieux, J.; Dello Iacono, D. Merging active and passive data sets in traveltime tomography: The case study of Campi Flegrei caldera southern Italy. Geophys. Prospect. 2008, 56, 555–573. [Google Scholar] [CrossRef]
- De Siena, L.; Chiodini, G.; Vilardo, G.; Del Pezzo, E.; Castellano, M.; Colombelli, S.; Tisato, N.; Ventura, G. Source and dynamics of a volcanic caldera unrest: Campi Flegrei, 1983–1984. Sci. Rep. 2017, 7, 8099. [Google Scholar] [CrossRef]
- Capuano, P.; Russo, G.; Civetta, L.; Orsi, G.; D’Antonio, M.; Moretti, R. The active portion of the Campi Flegrei caldera structure imaged by 3-D inversion of gravity data. Geochem. Geophys. Geosyst. 2013, 14, 4681–4697. [Google Scholar] [CrossRef]
- Ambrosio, M.; Doveri, M.; Fagioli, M.T.; Marini, L.; Principe, C.; Raco, B. Water–rock interaction in the magmatic-hydrothermal system of Nisyros Island (Greece). J. Volcanol. Geotherm. Res. 2010, 192, 7–68. [Google Scholar] [CrossRef]
- Kerrick, D.M. The genesis of zoned skarns in the Sierra Nevada, California. J. Petrol. 1977, 18, 144–181. [Google Scholar] [CrossRef]
- Caliro, S.; Chiodini, G.; Paonita, A. Geochemical evidences of magma dynamics at Campi Flegrei (Italy). Geochim. Cosmochim. Acta 2014, 132, 1–15. [Google Scholar] [CrossRef]
- Marini, L.; Principe, C.; Lelli, M. The Solfatara Magmatic-Hydrothermal System: Geochemistry, Geothermometry and Geobarometry of Fumarolic Fluids; Springer Nature: Cham, Switzerland, 2022; p. 375. [Google Scholar]
- Cioni, R.; Corazza, E.; Marini, L. The gas/steam ratio as indicator of heat transfer at the Solfatara fumaroles, Phlegraean Fields (Italy). Bull. Volcanol. 1984, 47, 295–302. [Google Scholar] [CrossRef]
- Cioni, R.; Corazza, E.; Fratta, M.; Guidi, M.; Magro, G.; Marini, L. Geochemical precursors at Solfatara Volcano, Pozzuoli (Italy). In Volcanic Hazards; Latter, J.H., Ed.; Springer: Berlin, Germany, 1989; Volume 1, pp. 384–398. [Google Scholar]
- Chiodini, G.; Marini, L. Hydrothermal gas equilibria: The H2O-H2-CO2-CO-CH4 system. Geochim. Cosmochim. Acta 1998, 62, 2673–2687. [Google Scholar] [CrossRef]
- Caliro, S.; Chiodini, G.; Moretti, R.; Avino, R.; Granieri, D.; Russo, M.; Fiebig, J. The origin of the fumaroles of La Solfatara (Campi Flegrei, south Italy). Geochim. Cosmochim. Acta 2007, 71, 3040–3055. [Google Scholar] [CrossRef]
- Chiodini, G. CO2/CH4 ratio in fumaroles a powerful tool to detect magma degassing episodes atn quiescent volcanoes. Geophys. Res. Lett. 2009, 36, L02302. [Google Scholar] [CrossRef]
- Chiodini, G.; Caliro, S.; Cardellini, C.; Granieri, D.; Avino, R.; Baldini, A.; Donnini, M.; Minopoli, C. Long-term variations of the Campi Flegrei, Italy, volcanic system as revealed by the monitoring of hydrothermal activity. J. Geophys. Res. Solid. Earth 2010, 115, B03205. [Google Scholar] [CrossRef]
- Chiodini, G.; Avino, R.; Caliro, S.; Minopoli, C. Temperature and pressure gas geoindicators at the Solfatara fumaroles (Campi Flegrei). Ann. Geophys. 2011, 54, 151–160. [Google Scholar] [CrossRef]
- Chiodini, G.; Caliro, S.; De Martino, P.; Avino, R.; Gherardi, F. Early signals of new volcanic unrest at Campi Flegrei caldera? Insights from geochemical data and physical simulations. Geology 2012, 40, 943–946. [Google Scholar] [CrossRef]
- Chiodini, G.; Vandemeulebrouck, J.; Caliro, S.; D’Auria, L.; De Martino, P.; Mangiacapra, A.; Petrillo, Z. Evidence of thermal-driven processes triggering the 2005–2014 unrest at Campi Flegrei caldera. Earth Planet. Sci. Lett. 2015, 414, 58–67. [Google Scholar] [CrossRef]
- Chiodini, G.; Paonita, A.; Aiuppa, A.; Costa, A.; Caliro, S.; De Martino, P.; Acocella, V.; Vandemeulebrouck, J. Magmas near the critical degassing pressure drive volcanic unrest towards a critical state. Nat. Commun. 2016, 7, 13712. [Google Scholar] [CrossRef]
- Chiodini, G.; Selva, J.; Del Pezzo, E.; Marsan, D.; De Siena, L.; D’Auria, L.; Bianco, F.; Caliro, S.; De Martino, P.; Ricciolino, P.; et al. Clues on the origin of post-2000 earthquakes at Campi Flegrei caldera (Italy). Sci. Rep. 2017, 7, 4472. [Google Scholar] [CrossRef]
- Chiodini, G.; Giudicepietro, F.; Vandemeulebrouck, J.; Aiuppa, A.; Caliro, S.; De Cesare, W.; Tamburello, G.; Avino, R.; Orazi, M.; D’Auria, L. Fumarolic tremor and geochemical signals during a volcanic unrest. Geology 2017, 45, 1131–1134. [Google Scholar] [CrossRef]
- Chiodini, G.; Caliro, S.; Avino, R.; Bini, G.; Giudicepietro, F.; De Cesare, W.; Ricciolino, P.; Aiuppa, A.; Cardellini, C.; Petrillo, Z.; et al. Hydrothermal pressure-temperature control on CO2 emissions and seismicity at Campi Flegrei (Italy). J. Volcanol. Geotherm. Res. 2021, 414, 107245. [Google Scholar] [CrossRef]
- Buono, G.; Caliro, S.; Paonita, A.; Pappalardo, L.; Chiodini, G. Discriminating carbon dioxide sources during volcanic unrest: The case of Campi Flegrei caldera (Italy). Geology 2023, 51, 397–401. [Google Scholar] [CrossRef]
- Lelli, M.; Caliro, S.; Dallara, E.; Chiodini, G.; Marini, L. Sulphur trace components in La Solfatara and Vulcano volcanic gases: Searching for suitable new geo-indicators. In Proceedings of the EGU General Assembly, Vienna, Austria, 23–28 April 2023. EGU-4492. [Google Scholar] [CrossRef]
- Symonds, R.B.; Gerlach, T.M.; Reed, M.H. Magmatic gas scrubbing: Implications for volcano monitoring. J. Volcanol. Geotherm. Res. 2001, 108, 303–341. [Google Scholar] [CrossRef]
- Marini, L.; Gambardella, B. Geochemical modeling of magmatic gas scrubbing. Ann. Geophys. 2005, 48, 739–753. [Google Scholar] [CrossRef]
- Piochi, M.; Cantucci, B.; Montegrossi, G.; Currenti, G. Hydrothermal alteration at the San Vito area of the Campi Flegrei geothermal system in Italy: Mineral review and geochemical modeling. Minerals 2021, 11, 810. [Google Scholar] [CrossRef]
- Giggenbach, W.F. Redox processes governing the chemistry of fumarolic gas discharges from White Island, New Zeland. Appl. Geochem. 1987, 2, 143–161. [Google Scholar] [CrossRef]
- Allard, P.; Maiorani, A.; Tedesco, D.; Cortecci, G.; Turi, B. Isotopic study of the origin of sulfur and carbon in Solfatara fumaroles, Campi Flegrei caldera. J. Volcanol. Geotherm. Res. 1991, 48, 139–159. [Google Scholar] [CrossRef]
- Buono, G.; Paonita, A.; Pappalardo, L.; Caliro, S.; Tramelli, A.; Chiodini, G. New insights into the recent magma dynamics under Campi Flegrei caldera (Italy) from petrological and geochemical evidence. J. Geophys. Res. Solid. Earth 2022, 127, e2021JB023773. [Google Scholar] [CrossRef]
- Huebner, J.S.; Sato, M. The oxygen fugacity-temperature relationships of manganese oxide and nickel oxide buffers. Am. Mineral. 1970, 55, 934–952. [Google Scholar]
- Myers, J.T.; Eugster, H.P. The system Fe-Si-O: Oxygen buffer calibrations to 1,500 K. Contrib. Mineral. Petrol. 1983, 82, 75–90. [Google Scholar] [CrossRef]
- Stull, D.R.; Westrum, E.F.; Sinke, G.C. The Chemical Thermodynamics of Organic Compounds; Wiley: New York, NY, USA, 1969; p. 882. [Google Scholar]
- Ding, S.; Plank, T.; Wallace, P.J.; Rasmussen, D.J. Sulfur_X: A model of sulfur degassing during magma ascent. Geochem. Geophys. Geosystems 2023, 24, e2022GC010552. [Google Scholar] [CrossRef]
- Hutchison, W.; Finch, A.A.; Boyce, A.J. The sulfur isotope evolution of magmatic-hydrothermal fluids: Insights into ore-forming processes. Geochim. Cosmochim. Acta 2020, 288, 176–198. [Google Scholar] [CrossRef]
- Fiege, A.; Holtz, F.; Shimizu, N.; Mandeville, C.W.; Behrens, H.; Knipping, J.L. Sulfur isotope fractionation between fluid and andesitic melt: An experimental study. Geochim. Cosmochim. Acta 2014, 142, 501–521. [Google Scholar] [CrossRef]
- Miyoshi, T.; Sakai, H.; Chiba, H. Experimental-study of sulfur isotope fractionation factors between sulfate and sulfide in high-temperature melts. Geochem. J. 1984, 18, 75–84. [Google Scholar] [CrossRef]
- Ohmoto, H.; Rye, R.O. Isotopes of sulfur and carbon. In Geochemistry of Hydrothermal Ore Deposits, 2nd ed.; Barnes, H.L., Ed.; Wiley: New York, NY, USA, 1979; pp. 509–567. [Google Scholar]
- Eldridge, D.L.; Guo, W.; Farquhar, J. Theoretical estimates of equilibrium sulfur isotope effects in aqueous sulfur systems: Highlighting the role of isomers in the sulfite and sulfoxylate systems. Geochim. Cosmochim. Acta 2016, 195, 171–200. [Google Scholar] [CrossRef]
- Giggenbach, W.F. Chemical composition of volcanic gases. In Monitoring and Mitigation of Volcano Hazards; Scarpa, R., Tilling, R.I., Eds.; Springer: Berlin, Germany, 1996; pp. 221–256. [Google Scholar]
- Giggenbach, W.F. The origin and evolution of fluids in magmatic-hydrothermal systems. In Geochemistry of Hydrothermal Ore Deposits, 3rd ed.; Barnes, H.L., Ed.; Wiley: New York, NY, USA, 1997; pp. 737–796. [Google Scholar]
- Moretti, R.; Ottonello, G. Polymerization and disproportionation of iron and sulfur in silicate melts: Insights from an optical basicity-based approach. J. Non-Cryst. Solids 2003, 323, 111–119. [Google Scholar] [CrossRef]
- Moretti, R.; Ottonello, G. A polymeric approach to the sulfide capacity of silicate slags and melts. Metall. Mater. Trans. 2003, 34, 399–410. [Google Scholar] [CrossRef]
- Moretti, R.; Ottonello, G. Solubility and speciation of sulfur in silicate melts: The Conjugated Toop-Samis-Flood-Grjotheim (CTSFG) model. Geochim. Cosmochim. Acta 2005, 69, 801–823. [Google Scholar] [CrossRef]
- Holland, H.D. Some applications of thermochemical data to problems of ore deposits. II. Mineral assemblages and the composition of ore forming fluids. Econ. Geol. 1965, 60, 1101–1166. [Google Scholar] [CrossRef]
- Ármannsson, H.; Gíslason, G.; Hauksson, T. Magmatic gases in well fluids aid the mapping of the flow pattern in a geothermal system. Geochim. Cosmochim. Acta 1982, 46, 167–177. [Google Scholar] [CrossRef]
- Sturchio, N.C.; Williams, S.N.; Garcia, N.P.; Londono, A.C. The hydrothermal system of Nevado del Ruiz volcano, Colombia. Bull. Volcanol. 1988, 50, 399–412. [Google Scholar] [CrossRef]
- Williams, S.N.; Sturchio, N.C.; Mendez, R.; Londoño, A.; García, N. Sulfur dioxide from Nevado del Ruiz volcano, Colombia: Total flux and isotopic constraints on its origin. J. Volcanol. Geotherm. Res. 1990, 42, 53–68. [Google Scholar] [CrossRef]
- Reed, M.H. Hydrothermal alteration and its relationship to ore fluid composition. In Geochemistry of Hydrothermal Ore Deposits, 3rd ed.; Barnes, H.L., Ed.; Wiley: New York, NY, USA, 1997; pp. 303–366. [Google Scholar]
- Marini, L.; Gambardella, B.; Principe, C.; Arias, A.; Brombach, T.; Hunziker, J.C. Characterization of magmatic sulfur in the Aegean Island arc by means of the δ34S values of fumarolic H2S, elemental S, and hydrothermal gypsum from Nisyros and Milos Islands. Earth Planet. Sci. Lett. 2002, 200, 15–31. [Google Scholar] [CrossRef]
- Marini, L.; Yock Fung, A.; Sanchez, E. Use of reaction path modeling to identify the processes governing the generation of neutral Na-Cl and acidic Na-Cl-SO4 deep geothermal liquids at Miravalles geothermal system, Costa Rica. J. Volcanol. Geotherm. Res. 2003, 128, 363–387. [Google Scholar] [CrossRef]
- Ayris, P.M.; Lee, A.F.; Wilson, K.; Kueppers, U.; Dingwell, D.B.; Delmelle, P. SO2 sequestration in large volcanic eruptions: High-temperature scavenging by tephra. Geochim. Cosmochim. Acta 2013, 110, 58–69. [Google Scholar] [CrossRef]
- Henley, R.W.; King, P.L.; Wykes, J.L.; Renggli, C.J.; Brink, F.J.; Clark, D.A.; Troitzsch, U. Porphyry copper deposit formation by sub-volcanic sulphur dioxide flux and chemisorption. Nat. Geosci. 2015, 8, 210–215. [Google Scholar] [CrossRef]
- Renggli, C.J.; King, P.L.; Henley, R.W.; Guagliardo, P.; McMorrow, L.; Middleton, J.P.; Turner, M. An experimental study of SO2 reactions with silicate glasses and supercooled melts in the system anorthite–diopside–albite at high temperature. Contrib. Mineral. Petrol. 2019, 174, 3. [Google Scholar] [CrossRef]
- Delmelle, P.; Wadsworth, F.B.; Maters, E.C.; Ayris, P.M. High temperature uptake of volcanic gases on ash in eruption plumes. Rev. Mineral. Geochem. 2018, 84, 285–308. [Google Scholar] [CrossRef]
- Maters, E.C.; Delmelle, P.; Rossi, M.J.; Ayris, P.M. Reactive uptake of sulfur dioxide and ozone on volcanic glass and ash at ambient temperature. J. Geophys. Res. Atmos. 2017, 122, 10077–10088. [Google Scholar] [CrossRef]
- Renggli, C.J.; King, P.L. SO2 gas reactions with silicate glasses. Rev. Mineral. Geochem. 2018, 84, 229–256. [Google Scholar] [CrossRef]
- Henley, R.W.; Brink, F.J.; King, P.L.; Leys, C.; Ganguly, J.; Mernagh, T.; Middleton, J.; Renggli, C.J.; Sieber, M.; Troitzsch, U.; et al. High temperature gas–solid reactions in calc–silicate Cu–Au skarn formation; Ertsberg, Papua Province, Indonesia. Contrib. Mineral. Petrol. 2017, 172, 106. [Google Scholar] [CrossRef]
- Henley, R.W.; Seward, T.M. Gas–solid reactions in arc volcanoes: Ancient and modern. Rev. Mineral. Geochem. 2018, 84, 309–350. [Google Scholar] [CrossRef]
- Henley, R.W.; Fischer, T.P. Sulfur sequestration and redox equilibria in volcanic gases. J. Volcanol. Geotherm. Res. 2021, 414, 107181. [Google Scholar] [CrossRef]
- Saadatfar, M.; Brink, F.; Latham, S.; King, P.; Middleton, J.; Troitzsch, U.; Turner, M.; Henley, R.W. High resolution 3D mapping of grain kinematics during high temperature sequestration of SO2 from flue gas by carbonate aggregates. Sci. Rep. 2020, 10, 2201. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.S. Behavior of calcium sulfate at high temperatures. J. Res. Nat. Bur. Stand. 1941, 27, 191–196. [Google Scholar] [CrossRef]
- Krejsová, J.; Kužel, R.; Keppert, M.; Scheinherrová, L.; Vimmrová, A. New insight into the phase changes of gypsum. Mater. Struct. 2024, 57, 128. [Google Scholar] [CrossRef]
- Clark, I.D. Isotope fractionation. In Encyclopedia of Geochemistry. Marshall, C.P., Fairbridge, R.W., Eds; Springer: Dordrecht, The Netherlands, 1998; pp. 354–357. [Google Scholar] [CrossRef]
- Hong, Y.; Fegley, B., Jr. The kinetics and mechanism of pyrite thermal decomposition. Berichte Bunsenges. Phys. Chem. 1997, 101, 1870–1881. [Google Scholar] [CrossRef]
- Schwab, G.M.; Philinis, J. Reactions of iron pyrite: Its thermal decomposition, reduction by hydrogen and air oxidation. J. Am. Chem. Soc. 1947, 69, 2588–2596. [Google Scholar] [CrossRef]
- Fournier, R.O. Hydrothermal processes related to movement of fluid from plastic into brittle rock in the magmatic-epithermal environment. Econ. Geol. 1999, 94, 1193–1211. [Google Scholar] [CrossRef]
- Reyes, A.G.; Giggenbach, W.F.; Saleras, J.R.; Salonga, N.D.; Vergara, M.C. Petrology and geochemistry of Alto Peak, a vapor-cored hydrothermal system, Leyte Province, Philippines. Geothermics 1993, 22, 479–519. [Google Scholar] [CrossRef]
- Marini, L.; Chiodini, G. The role of carbon dioxide in the carbonate-evaporite geothermal systems of Tuscany and Latium (Italy). Acta Vulcanol. 1994, 5, 95–104. [Google Scholar]
- Cavarretta, G.; Gianelli, G.; Scandiffio, G.; Tecce, F. Evolution of the Latera geothermal system II: Metamorphic, hydrothermal mineral assemblages and fluid chemistry. J. Volcanol. Geotherm. Res. 1985, 26, 337–364. [Google Scholar] [CrossRef]
- Turbeville, B.N. Sidewall differentiation in an alkalic magma chamber: Evidence from syenite xenoliths in tuffs of the Latera caldera, Italy. Geol. Mag. 1993, 130, 453–470. [Google Scholar] [CrossRef]
- Browne, P.R.L.; Ellis, A.J. The Ohaaki-Broadlands hydrothermal area, New Zealand: Mineralogy and related geochemistry. Am. J. Sci. 1970, 269, 97–131. [Google Scholar] [CrossRef]
- Kristmannsdóttir, H. Alteration of basaltic rocks by hydrothermal activity at 100–300 °C. In International Clay Conference 1978, Proceedings of the VI International Clay Conference 1978, Oxford, UK, 10–14 July, 1978; Mortland, M.M., Farmer, V.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1979; Volume 27, pp. 359–367. [Google Scholar]
- Browne, P.R.L. Hydrothermal alteration in active geothermal fields. Annu. Rev. Earth. Pl. Sc. 1978, 6, 229–248. [Google Scholar] [CrossRef]
- Reyes, A.G. Petrology of Philippine geothermal systems and the application of alteration mineralogy to their assessment. J. Volcanol. Geotherm. Res. 1990, 43, 279–309. [Google Scholar] [CrossRef]
- Balducci, S.; Chelini, W. Hydrothermal equilibria in the active Mofete geothermal system, Phlegraean Fields, Naples, Italy. Acta Vulcanol. 1992, 2, 17–34. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marini, L.; Principe, C.; Lelli, M. Closed-System Magma Degassing and Disproportionation of SO2 Revealed by Changes in the Concentration and δ34S Value of H2S(g) in the Solfatara Fluids (Campi Flegrei, Italy). Geosciences 2025, 15, 162. https://doi.org/10.3390/geosciences15050162
Marini L, Principe C, Lelli M. Closed-System Magma Degassing and Disproportionation of SO2 Revealed by Changes in the Concentration and δ34S Value of H2S(g) in the Solfatara Fluids (Campi Flegrei, Italy). Geosciences. 2025; 15(5):162. https://doi.org/10.3390/geosciences15050162
Chicago/Turabian StyleMarini, Luigi, Claudia Principe, and Matteo Lelli. 2025. "Closed-System Magma Degassing and Disproportionation of SO2 Revealed by Changes in the Concentration and δ34S Value of H2S(g) in the Solfatara Fluids (Campi Flegrei, Italy)" Geosciences 15, no. 5: 162. https://doi.org/10.3390/geosciences15050162
APA StyleMarini, L., Principe, C., & Lelli, M. (2025). Closed-System Magma Degassing and Disproportionation of SO2 Revealed by Changes in the Concentration and δ34S Value of H2S(g) in the Solfatara Fluids (Campi Flegrei, Italy). Geosciences, 15(5), 162. https://doi.org/10.3390/geosciences15050162