
An Observation Related to the Pressure Dependence of Ionic Radii


Abstract
1. Introduction
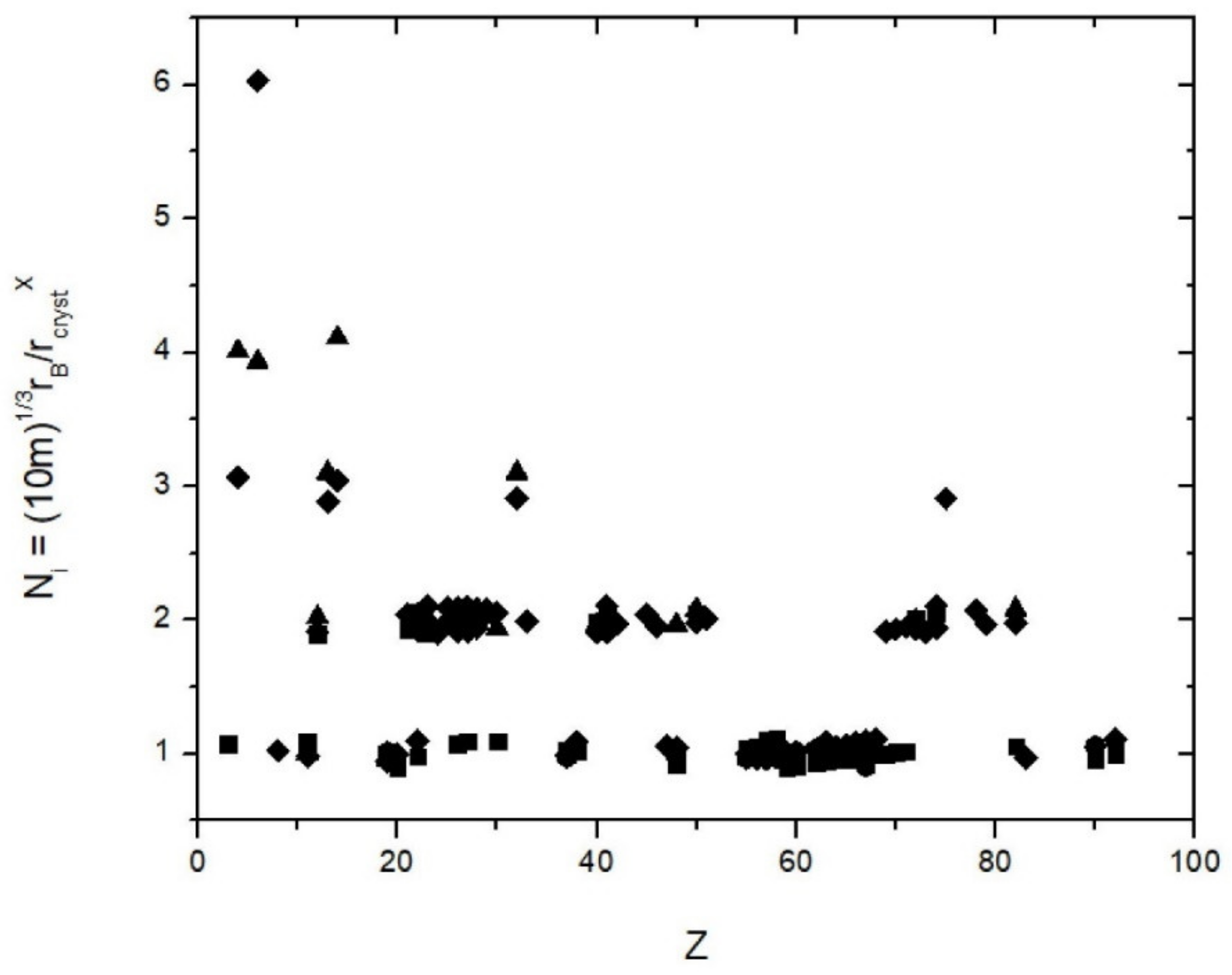
2. Method
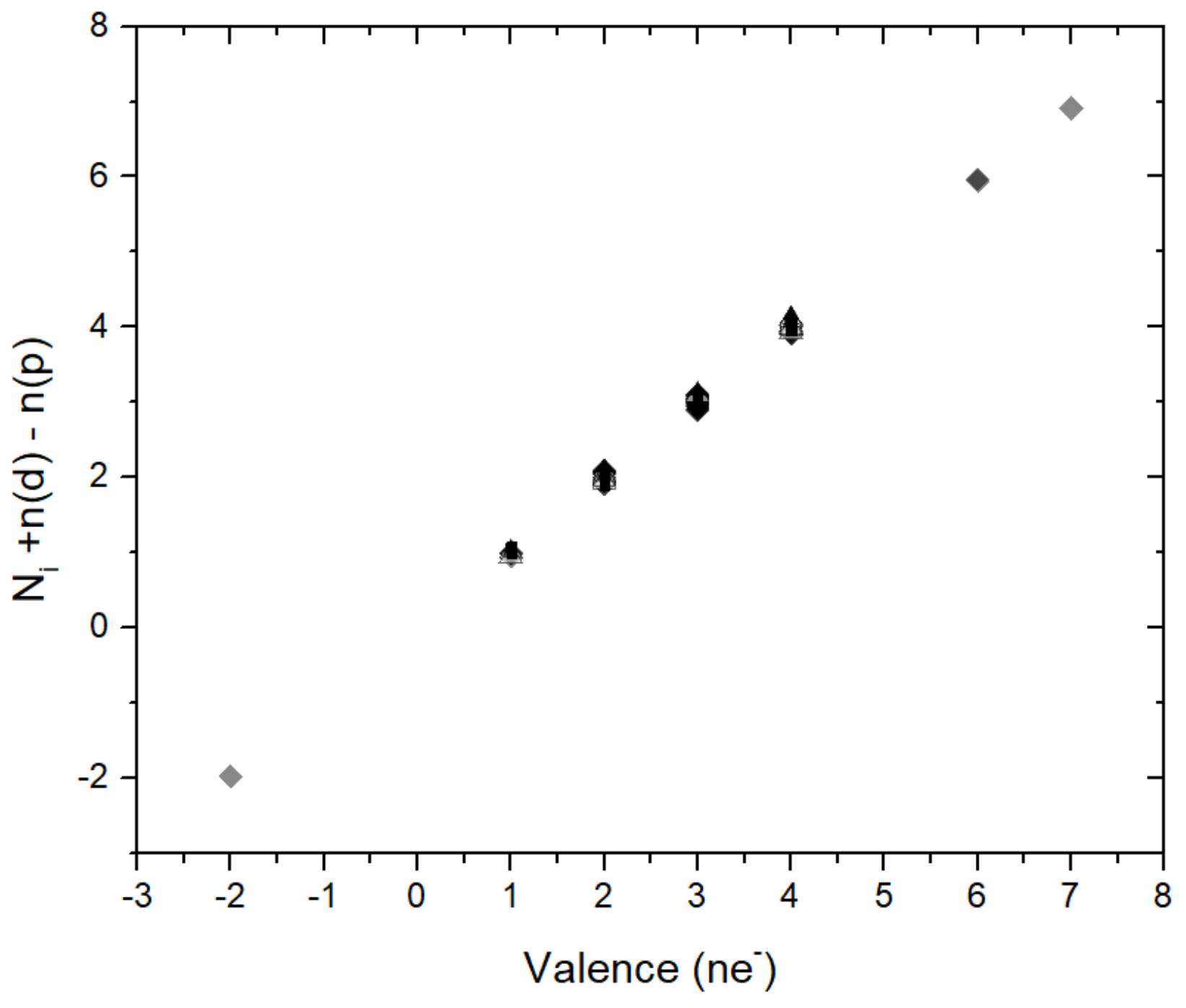
3. Results
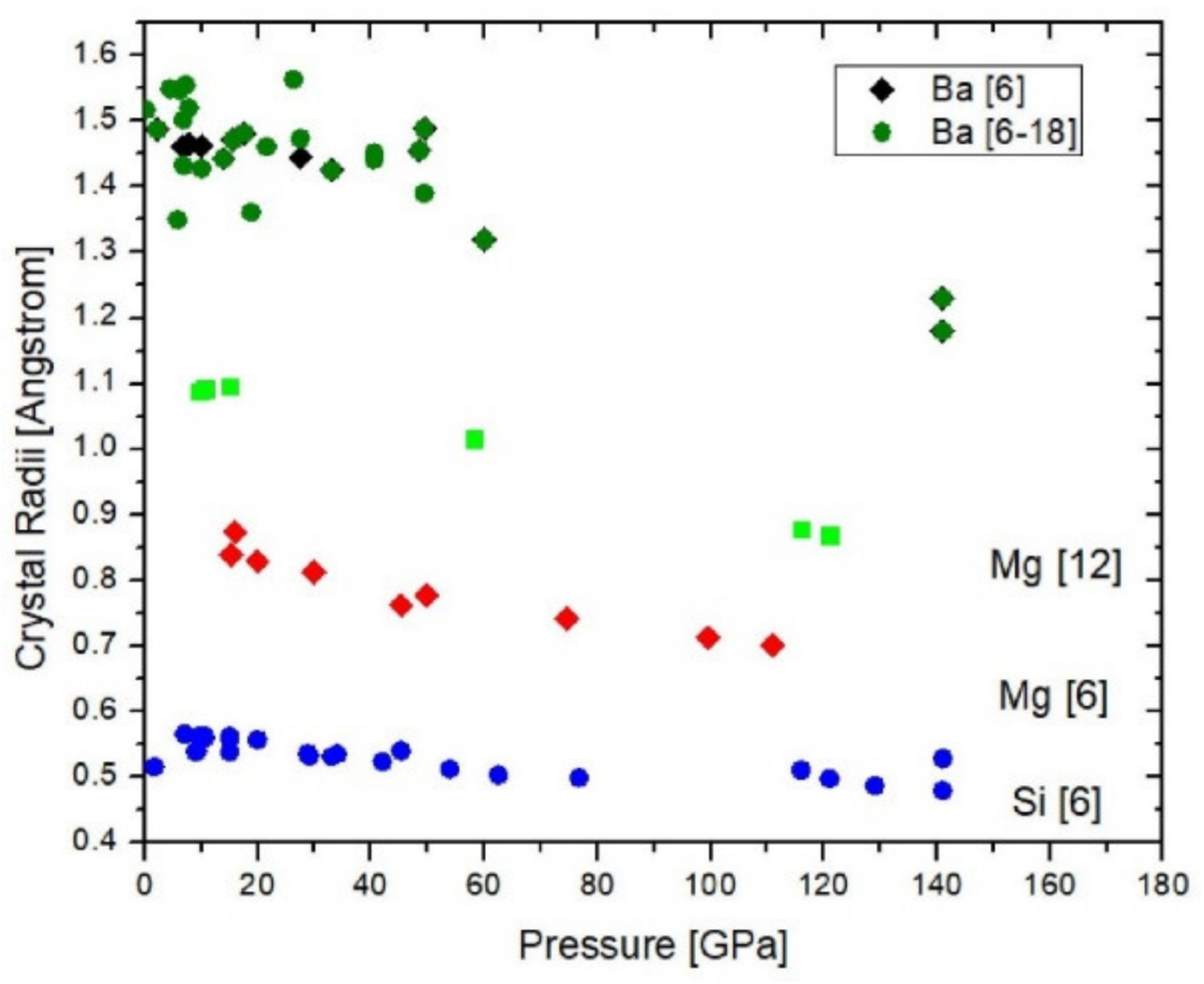
4. Effect of Pressure on Ionic Radii
5. Summary
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldschmidt, V.M. The principles of distribution of chemical elements in minerals and rocks. The seventh Hugo Muller Lecture, delivered before the Chemical Society on March 17th, 1937. J. Chem. Soc. 1937, 655–673. [Google Scholar] [CrossRef]
- Bukowinski, M.S.T. Quantum Geophysics. Annu. Rev. Earth Planet. Sci. 1994, 22, 167–205. [Google Scholar] [CrossRef]
- Mao, H.K.; Chen, X.J.; Ding, Y.; Li, B.; Wang, L. Solids, liquids, and gases under high pressure. Rev. Mod. Phys. 2018, 90, 015007. [Google Scholar] [CrossRef]
- Sturhahn, W.; Jackson, J.M.; Lin, J.F. The spin state of iron in minerals of Earth’s lower mantle. Geophys. Res. Lett. 2005, 32, L12307. [Google Scholar] [CrossRef]
- Frost, D.J.; McCammon, C.A. The redox state of Earth’s mantle. Annu. Rev. Earth Planet. Sci. 2008, 36, 389–420. [Google Scholar] [CrossRef]
- Iota, V.; Klepeis, J.-H.P.; Yoo, C.-S.; Lang, J.; Haskel, D.; Srajer, G. Electronic structure and magnetism in compressed 3d transition metals. Appl. Phys. Lett. 2007, 90, 042505. [Google Scholar] [CrossRef]
- Fabbris, G.; Lim, J.; Veiga, L.; Haskel, D.; Schilling, J.S. Electronic and structural ground state of heavy alkali metals at high pressure. Phys. Rev. B 2015, 91, 085111. [Google Scholar] [CrossRef]
- Eriksson, O.; Brooks, M.S.S.; Johansson, B. Orbital polarization in narrow-band systems: Application to volume collapses in light lanthanides. Phys. Rev. B 1990, 41, 7311–7314. [Google Scholar] [CrossRef]
- Royce, E.B. Stability of the Electronic Configuration and Compressibility of Electron Orbitals in Metals under Shock-Wave Compression. Phys. Rev. Ser. I 1967, 164, 929–943. [Google Scholar] [CrossRef]
- Batsanov, S.S. Cationic radii from structures of extremely compressed solids. Acta Cryst. 2013, B69, 563–569. [Google Scholar] [CrossRef]
- Cammi, R.; Rahm, M.; Hoffmann, R.; Ashcroft, N.W. Varying Electronic Configurations in Compressed Atoms: From the Role of the Spatial Extension of Atomic Orbitals to the Change of Electronic Configuration as an Isobaric Transformation. J. Chem. Theory Comput. 2020, 16, 5047–5056. [Google Scholar] [CrossRef] [PubMed]
- Rahm, M.; Ångqvist, M.; Rahm, J.M.; Erhart, P.; Cammi, R. Non-Bonded Radii of the Atoms Under Compression. ChemPhysChem 2020, 21, 2441–2453. [Google Scholar] [CrossRef] [PubMed]
- Shannon, R.D. Revised effective ionicradii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Du, X.; Tse, J.S. Oxygen Packing Fraction and the Structure of Silicon and Germanium Oxide Glasses. J. Phys. Chem. B 2017, 121, 10726–10732. [Google Scholar] [CrossRef]
- Jacobsen, S.D.; Holl, C.M.; Adams, K.A.; Fischer, R.A.; Martin, E.S.; Bina, C.R.; Lin, J.-F.; Prakapenka, V.B.; Kubo, A.; Dera, P. Compression of single-crystal magnesium oxide to 118 GPa and a ruby pressure gauge for helium pressure media. Am. Miner. 2008, 93, 1823–1828. [Google Scholar] [CrossRef]
- Weir, S.T.; Vohra, Y.K.; Ruoff, A.L. High-pressure phase transitions and the equations of state of BaS and BaO. Phys. Rev. B 1986, 33, 4221–4226. [Google Scholar] [CrossRef] [PubMed]
- Liu, L. A Dense Modification of BaO and Its Crystal Structure. J. Appl. Phys. 1971, 42, 3702–3704. [Google Scholar] [CrossRef]
- Crichton, W.A.; Merlini, M.; Hanfland, M.; Müller, H. The crystal structure of barite, BaSO4, at high pressure. Am. Miner. 2011, 96, 364–367. [Google Scholar] [CrossRef]
- Santamaría-Pérez, D.; Chulia-Jordan, R. Compression of mineral barite, BaSO4: A structural study. High Press. Res. 2012, 32, 81–88. [Google Scholar] [CrossRef]
- Errandonea, D.; Pellicer-Porres, J.; Manjón, F.J.; Segura, A.; Ferrer-Roca, C.; Kumar, R.S.; Tschauner, O.; López-Solano, J.; Rodríguez-Hernández, P.; Radescu, S.; et al. Determination of the high-presure crystal structure of BaWO4 and PbWO4. Phys. Rev. B Condens. Matter Mater. Phys. 2006, 73, 224103. [Google Scholar] [CrossRef]
- Tan, D.; Xiao, W.; Zhou, W.; Chen, M.; Zhou, W.; Li, X.; Li, Y.; Liu, J. High pressure X-ray diffraction study on BaWO4-II. High Press. Res. 2012, 32, 262–269. [Google Scholar] [CrossRef]
- Yusa, H.; Sata, N.; Ohishi, Y. Rhombohedral (9R) and hexagonal (6H) perovskites in barium silicates under high pressure. Am. Miner. 2007, 92, 648–654. [Google Scholar] [CrossRef]
- Efthimiopoulos, I.; Kunc, K.; Karmakar, S.; Syassen, K.; Hanfland, M.; Vajenine, G. Structural transformation and vibrational properties of BaO2 at high pressures. Phys. Rev. B 2010, 82, 134125. [Google Scholar] [CrossRef]
- Friese, K.; Kanke, Y.; Grzechnik, A. Characterization of the pressure-induced second-order phase transition in the mixed-valence vanadate BaV6O11. Acta Crystallogr. Sect. B Struct. Sci. 2009, 65, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Curetti, N.; Benna, P.; Bruno, E. High-pressure structural configuration and phase transition in celsian, BaAl2Si2O8. Phys. Chem. Miner. 2016, 44, 181–192. [Google Scholar] [CrossRef]
- Panchal, V.; Garg, N.; Sharma, S.M. Raman and X-ray diffraction investigations on BaMoO4 under high pressures. J. Phys. Condens. Matter 2006, 18, 3917–3929. [Google Scholar] [CrossRef] [PubMed]
- Hazen, R.M.; Finger, L.W. High-pressure and high-temperature crystallographic study of the gillespite I–II phase transition. Am. Mineral. 1983, 68, 595–603. [Google Scholar]
- Andrault, D.; Angel, R.J.; Mosenfelder, J.L.; Le Bihan, T. Equation of state of stishovite to lower mantle pressures. Am. Miner. 2003, 88, 301–307. [Google Scholar] [CrossRef]
- Zhang, L.; Popov, D.; Meng, Y.; Wang, J.; Ji, C.; Li, B.; Mao, H.-K. In-situ crystal structure determination of seifertite SiO2 at 129 GPa: Studying a minor phase near Earth’s core–mantle boundary. Am. Miner. 2016, 101, 231–234. [Google Scholar] [CrossRef][Green Version]
- Ross, N.L.; Shu, J.-F.; Hazen, R.M.; Gasparik, T. High-pressure crystal chemistry of stishovite. Am. Mineral. 1990, 75, 739–747. [Google Scholar]
- Yamanaka, T.; Fukuda, T.; Mimaki, J. Bonding character of SiO2 stishovite under high pressures up to 30 Gpa. Phys. Chem. Miner. 2002, 29, 633–641. [Google Scholar] [CrossRef]
- Oganov, A.R.; Ono, S. Theoretical and experimental evidence for a post-perovskite phase of MgSiO3 in Earth’s D″ layer. Nature 2004, 430, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.; Hirose, K.; Kawamura, K.; Sata, N.; Ohishi, Y. Post-Perovskite Phase Transition in MgSiO3. Science 2004, 304, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Sugahara, M.; Yoshiasa, A.; Komatsu, Y.; Yamanaka, T.; Bolfan-Casanova, N.; Nakatsuka, A.; Sasaki, S.; Tanaka, M. Reinvestigation of the MgSiO3 perovskite structure at high pressure. Am. Miner. 2006, 91, 533–536. [Google Scholar] [CrossRef]
- Yamanaka, T.; Komatsu, Y.; Sugahara, M.; Nagai, T. Structure change of MgSiO3, MgGeO3, and MgTiO3 ilmenites under compression. Am. Miner. 2005, 90, 1301–1307. [Google Scholar] [CrossRef]
- Ross, N.; Hazen, R.M. High-pressure crystal chemistry of MgSiO3 perovskite. Phys. Chem. Miner. 1990, 17, 228–237. [Google Scholar] [CrossRef]
- Kudoh, Y.; Ito, E.; Takeda, H. Effect of pressure on the crystal structure of perovskite-type MgSiO3. Phys. Chem. Miner. 1987, 14, 350–354. [Google Scholar] [CrossRef]
- Finkelstein, G.J.; Dera, P.K.; Jahn, S.; Oganov, A.R.; Holl, C.M.; Meng, Y.; Duffy, T.S. Phase transitions and equation of state of forsterite to 90 GPa from single-crystal X-ray diffraction and molecular modeling. Am. Miner. 2014, 99, 35–43. [Google Scholar] [CrossRef]
- Jacobsen, S.D.; Demouchy, S.; Frost, D.J.; Ballaran, T.B.; Kung, J. A systematic study of OH in hydrous wadsleyite from polarized FTIR spectroscopy and single-crystal X-ray diffraction: Oxygen sites for hydrogen storage in Earth’s interior. Am. Miner. 2005, 90, 61–70. [Google Scholar] [CrossRef]
- Clementi, E.; Raimondi, D.L.; Reinhardt, W.P. Atomic Screening Constants from SCF Functions. II. Atoms with 37 to 86 Electrons. J. Chem. Phys. 1967, 47, 1300–1307. [Google Scholar] [CrossRef]
- Spruch, L. Pedagogic notes on Thomas-Fermi theory (and on some improvements)—Atoms, stars and the stability of bulk matter. Rev. Mod. Phys. 1991, 63, 151–209. [Google Scholar] [CrossRef]
- Shannon, R.D.; Prewitt, C.T. Coordination and volume changes accompanying high-pressure phase trans-formations of oxides. Mater. Res. Bull. 1969, 4, 57–59. [Google Scholar] [CrossRef]
- Tschauner, O. High-pressure minerals. Am. Miner. 2019, 104, 1701–1731. [Google Scholar] [CrossRef]
- Neaton, J.B.; Ashcroft, N.W. On the Constitution of Sodium at Higher Densities. Phys. Rev. Lett. 2001, 86, 2830–2833. [Google Scholar] [CrossRef] [PubMed]
- Miao, M.-S.; Hoffmann, R. High Pressure Electrides: A Predictive Chemical and Physical Theory. Acc. Chem. Res. 2014, 47, 1311–1317. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Kim, D.Y.; Yang, L.; Li, N.; Tang, L.; Amine, K.; Mao, H.K. Oxygen-Rich Lithium Oxide Phases Formed at High Pressure for Potential Lithium-Air Battery Electrode. Adv. Sci. 2017, 4, 1600453. [Google Scholar] [CrossRef]
- Zhang, W.; Oganov, A.R.; Goncharov, A.F.; Zhu, Q.; Boulfelfel, S.E.; Lyakhov, A.O.; Stavrou, E.; Somayazulu, M.; Prakapenka, V.B.; Konôpková, Z. Unexpected Stable Stoichiometries of Sodium Chlorides. Science 2013, 342, 1502–1505. [Google Scholar] [CrossRef]
- Zhu, Q.; Oganov, A.R.; Lyakhov, A.O. Novel stable compounds in the Mg–O system under high pressure. Phys. Chem. Chem. Phys. 2013, 15, 7696–7700. [Google Scholar] [CrossRef]
- Hirose, K.; Shimizu, N.; van Westrenen, W.; Fei, Y.W. Trace element partitioning in Earth’s lower mantle and implications for geochemical consequences of partial melting at the core-mantle boundary. Phys. Earth Planet. Inter. 2004, 146, 249–260. [Google Scholar] [CrossRef]
- Tschauner, O.; Huang, S.; Yang, S.; Humayun, M.; Liu, W.; Corder, S.N.G.; Bechtel, H.A.; Tischler, J.; Rossman, G.R. Discovery of davemaoite, CaSiO3 -perovskite, as a mineral from the lower mantle. Science 2021, 374, 891–894. [Google Scholar] [CrossRef]
- Hirose, K.; Sinmyo, R.; Hernlund, J. Perovskite in Earth’s deep interior. Science 2017, 358, 734–738. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| a. Barium | |||||||
| Compound | Coordination | Pressure (GPa) | Ba-O(Å) | r (Å) | References | ||
| BaO_III | 8 | 18.8 | 2.566 | 1.361 | [16] | ||
| BaO_III | 8 | 60 | 2.50 | 1.32 | [16] | ||
| BaO, NiAs-type | 6 | 13.9 | 2.654 | 1.443 | [17] | ||
| Baryte | 8 | 17.5 | 2.760 | 1.5 | [18] | ||
| Baryte | 6 | 17.5 | 2.688 | 1.482 | [18] | ||
| Baryte | 8 | 2.15 | 2.815 | 1.479 | [18] | ||
| Baryte | 6 | 2.15 | 2.739 | 1.487 | [18] | ||
| Baryte | 8 | 0.22 | 2.82 | 1.563 | [18] | ||
| Baryte-II | 8 | 0 | 2.85 | 1.59 | [19] | ||
| Baryte-II | 8 | 40.5 | 2.63 | 1.441 | [19] | ||
| Baryte-II | 12 | 40.5 | 2.653 | 1.46 | [19] | ||
| BaWO4, fergusonite-type | 12 | 15.6 | 2.708 | 1.52 | [20,21] | ||
| BaSiO3, BaFeO3-type | 6 | 48.5 | 2.64 | 1.455 | [22] | ||
| BaSiO3, BaRuO3-type | 18 | 27.9 | 2.766 | 1.569 | [22] | ||
| BaSiO3, BaTiO3-type | 6 | 141 | 2.393 | 1.23 | [22] | ||
| BaO2 | 8 | 49.4 | 2.57 | 1.39 | [23] | ||
| BaAlSi3O8 -II | 14 | 7.81 | 2.75 | 1.52 | [24] | ||
| BaV6O11 | 12 | 5.82 | 1.52 | [25] | |||
| BaMoO4 | 12 | 7.2 | 1.41 * | [26] | |||
| Gillespite, BaFeSi4O10 | 8 | 4.5 | 2.784 | 1.549 | [27] | ||
| BaO2_CuCl2-typ | 12 | 6.8 | 2.729 | 1.50 | [23] | ||
| b. Mg-Silicates, Silica, Periclase | |||||||
| Compound | P [GPa] | Mg-O | Si-O | rSi (Å) {6,7} | rMg (Å) {6,12} | ||
| Periclase, MgO | 15.3 | 2.0485 | 0.839 | [15] | |||
| 29.9 | 2.0085 | 0.813 | [15] | ||||
| 49.7 | 1.962 | 0.778 | [15] | ||||
| 74.6 | 1.918 | 0.742 | [15] | ||||
| 99.6 | 1.8825 | 0.712 | [15] | ||||
| 111 | 1.869 | 0.701 | [15] | ||||
| Stishovite, SiO2 | 7 | 1.792 | 0.566 | [28] | |||
| 10.5 | 1.781 | 0.564 | [28] | ||||
| 15 | 1.772 | 0.562 | [28] | ||||
| 34 | 1.728 | 0.536 | [28] | ||||
| 76.75 | 1.675 | 0.500 | [28] | ||||
| 1.7 | 1.773 | 0.516 | [29] | ||||
| 9 | 1.76 | 0.539 | [29] | ||||
| 15 | 1.749 | 0.539 | [29] | ||||
| 9.26 | 1.7603 | 0.540 | [30] | ||||
| 29.1 | 1.7287 | 0.533 | [30] | ||||
| Seifertite, SiO2 | 129 | 1.652 | 0.487 | [31] | |||
| Akimotoite, MgSiO3 | 19.91 | 2.033 | 1.761 | 0.557 | 0.829 | [32] | |
| Bridgmanite, MgSiO3 | 10.6 | 2.31 | 1.777 | 0.560 | 1.093 | [33] | |
| 9.6 | 2.3075 | 1.782 | 0.563 | 1.088 | [34] | ||
| 15 | 2.3057 | 1.769 | 0.559 | 1.096 | [35] | ||
| postperovskite, MgSiO3 | 121 | 2.035 | 1.664 | 0.498 | 0.869 | [36] | |
| 116 | 2.045 | 1.678 | 0.511 | 0.878 | [37] | ||
| Forsterite-II, Mg2SiO4 | 45.3 | 1.949 | 1.727 | 0.541 | 0.763 | [38] | |
| 45.3 | 1.949 | (1.866) | (0.68) | 0.763 | [38] | ||
| Forsterite-III, Mg2SiO4 | 58.2 | 2.198 | 1.815 | 0.634 | 1.017 | [38] | |
| Wadsleyite, Mg2SiO4 | 16 | 2.082 | ** | ** | 0.874 | [39] | |
| a | ||||||||
| Ion | n(4) = rB 3√(10 m)/r(4)obs | m | n(6) = rB 3√(10 m)/r(6)obs | m | n(8) = rB 3√(10 m)/r(8)obs | m | n(12) = rB 3√(10 m)/r(12)obs | M |
| Li+ | 1.07547 | 1 | ||||||
| Na+ | 1.00885 | 1 | 0.98276 | 1 | 1.08808 | 2 | 1.07457 | 3 |
| K+ | 0.95117 | 2 | 0.94492 | 2 | 0.99642 | 3 | 1.0166 | 4 |
| Rb+ | 0.99042 | 3 | 1.03403 | 4 | 0.97288 | 4 | ||
| Cs+ | 0.99975 | 4 | 1.03684 | 5 | 0.96498 | 5 | ||
| Be2+ | 4.01 | 3 | 3.06703 | 4 | ||||
| Mg2+ | 2.02292 | 2 | 1.91174 | 3 | 1.89249 | 5 | ||
| Ca2+ | 1 | 1 | 0.90476 | 1 | 0.97045 | 2 | ||
| Sr2+ | 1.08808 | 2 | 1.02591 | 2 | 1.04057 | 3 | ||
| Ba2+ | 0.96394 | 2 | 1.05391 | 3 | 1.03403 | 4 | ||
| Al3+ | 3.10207 | 3 | ||||||
| Si4+ | 4.11025 | 3 | 3.04463 | 3 | ||||
| Sn4+ | 2.08155 | 2 | 1.98084 | 3 | 2.05185 | 5 | ||
| Pb4+ | 2.08114 | 3 | 1.97765 | 4 | 1.05555 | 1 | ||
| Ge4+ | 3.10207 | 3 | 2.90935 | 5 | ||||
| C4+ | 3.93102 | 1 | 6.03183 | 4 | ||||
| Sc3+ | 2.04469 | 4 | 1.92996 | 5 | ||||
| Y3+ | 1.09615 | 1 | 0.9836 | 1 | ||||
| La3+ | 0.97269 | 1 | 1.10482 | 2 | 0.95751 | 2 | ||
| Ce3+ | 0.9913 | 1 | 1.11946 | 2 | 0.97045 | 2 | ||
| Pr3+ | 1.00885 | 1 | 0.90047 | 1 | ||||
| Nd3+ | 1.01513 | 1 | 0.91273 | 1 | 1.01863 | 1 | ||
| Sm3+ | 1.03825 | 1 | 0.93519 | 1 | 1.04078 | 2 | ||
| Eu3+ | 1.04875 | 1 | 0.94527 | 1 | ||||
| Eu2+ | 1.09639 | 2 | 1.03329 | 2 | ||||
| Gd3+ | 1.05751 | 1 | 0.95557 | 1 | ||||
| Tb3+ | 1.07243 | 1 | 0.9661 | 1 | ||||
| Dy3+ | 1.08365 | 1 | 0.97686 | 1 | ||||
| Ho3+ | 1.0951 | 1 | 0.98701 | 1 | 0.90476 | 1 | ||
| Er3+ | 1.10679 | 1 | 0.9965 | 1 | ||||
| Tm3+ | 1.91104 | 5 | 1.00529 | 1 | ||||
| Yb3+ | 1.93379 | 5 | 1.01333 | 1 | ||||
| Lu3+ | 1.94731 | 5 | 1.02059 | 1 | ||||
| Th4+ | 1.05555 | 1 | 0.95798 | 1 | 1.06391 | 2 | ||
| U4+ | 1.10679 | 1 | 1 | 1 | 1.09639 | 2 | ||
| Ti2+ | 1.94926 | 5 | ||||||
| Ti3+ | 2.02975 | 3 | ||||||
| Ti4+ | 2.03571 | 1 | 1.92788 | 2 | 2.0563 | 4 | ||
| Zr4+ | 1.9675 | 2 | 1.91174 | 3 | 1.98904 | 5 | ||
| Hf4+ | 1.99482 | 2 | 1.93423 | 3 | 2.00955 | 5 | ||
| V2+ | 1.94575 | 4 | ||||||
| V3+ | 2.10782 | 3 | ||||||
| V4+ | 1.99482 | 2 | 1.91174 | 3 | ||||
| Nb3+ | 1.91174 | 3 | ||||||
| Nb5+ | 2.10782 | 3 | 2.0563 | 4 | ||||
| Ta3+ | 1.91174 | 3 | ||||||
| Ta5+ | 2.10782 | 3 | 2.0563 | 4 | ||||
| Cr3+ | 1.90235 | 2 | ||||||
| Mo3+ | 1.98084 | 3 | ||||||
| Mo6+ | 1.9675 | 2 | ||||||
| W6+ | 1.94091 | 2 | ||||||
| Mn3+HS | 2.09439 | 3 | ||||||
| Mn3+LS | 1.99482 | 2 | ||||||
| Re7+ | 2.90935 | 5 | ||||||
| Fe2+HS | 1.9669 | 4 | 1.07547 | 1 | ||||
| Fe2+LS | 1.91503 | 2 | ||||||
| Fe3+HS | 2.09439 | 3 | 1.9669 | 4 | ||||
| Fe3+LS | 2.08155 | 2 | ||||||
| Ru4+ | ||||||||
| Os4+ | ||||||||
| Co2+HS | 1.99482 | 2 | 2.04469 | 4 | 1.09615 | 1 | ||
| Co2+LS | 2.08114 | 3 | ||||||
| Co3+HS | 1.91503 | 2 | ||||||
| Co3+LS | 2.09675 | 2 | ||||||
| Rh3+ | 2.04236 | 3 | ||||||
| Ir4+ | ||||||||
| Ni2+ | 2.08155 | 2 | 1.98084 | 3 | ||||
| Ni3+HS | 1.94091 | 2 | ||||||
| Ni3+LS | 2.05182 | 2 | ||||||
| Pd2+ | 1.94926 | 5 | ||||||
| Pt2+ | 2.07368 | 5 | ||||||
| Cu2+ | 2.02292 | 2 | 2.07994 | 4 | ||||
| Ag2+ | 1.05555 | 1 | ||||||
| Au3+ | 1.96895 | 5 | ||||||
| Zn2+ | 1.94091 | 2 | 2.0563 | 4 | 1.09615 | 1 | ||
| Cd2+ | 1.9669 | 4 | 1.04587 | 1 | 0.91935 | 1 | 0.99053 | 2 |
| As3+ | 1.99482 | 2 | ||||||
| Sb3+ | 2.01061 | 4 | ||||||
| Bi3+ | 0.97436 | 1 | ||||||
| O2− | 1.02756 | 3 | ||||||
| b | ||||||||
| Ion | r (4)obs (Å) | r (4)clc (Å) | r (6)obs (Å) | r (6)clc (Å) | r (8)obs (Å) | r (8)clc (Å) | r (12)obs (Å) | r (12)clc (Å) |
| Li+ | 0.73 | 0.72 | 0.9 | 0.900 | 1.06 | 1.060 | ||
| Na+ | 1.13 | 1.145 | 1.16 | 1.160 | 1.32 | 1.380 | 1.53 | 1.527 |
| K+ | 1.51 | 1.523 | 1.52 | 1.527 | 1.65 | 1.647 | 1.78 | 1.785 |
| Rb+ | 1.66 | 1.706 | 1.75 | 1.785 | 1.86 | 1.837 | ||
| Cs+ | 1.81 | 1.834 | 1.88 | 1.837 | 2.02 | 2.132 | ||
| Be2+ | 0.41 | 0.411 | 0.59 | 0.595 | ||||
| Mg2+ | 0.71 | 0.711 | 0.86 | 0.853 | 1.03 | 1.020 | ||
| Ca2+ | 1.14 | 1.145 | 1.26 | 1.219 | 1.48 | 1.482 | ||
| Sr2+ | 1.32 | 1.377 | 1.4 | 1.378 | 1.58 | 1.527 | ||
| Ba2+ | 1.49 | 1.485 | 1.56 | 1.482 | 1.75 | 1.785 | ||
| Al3+ | 0.53 | 0.533 | 0.675 | 0.689 | ||||
| Si4+ | 0.4 | 0.411 | 0.54 | 0.551 | ||||
| Sn4+ | 0.69 | 0.689 | 0.83 | 0.853 | 0.95 | 1.020 | ||
| Pb4+ | 0.79 | 0.793 | 0.915 | 0.916 | 1.08 | 1.102 | ||
| Ge4+ | 0.53 | 0.533 | 0.67 | 0.689 | ||||
| C4+ | 0.29 | 0.288 | 0.3 | 0.296 | ||||
| Sc3+ | 0.885 | 0.892 | 1.01 | 1.020 | ||||
| Y3+ | 1.04 | 0.96 | 1.159 | 1.145 | ||||
| La3+ | 1.172 | 1.19 | 1.3 | 1.377 | 1.5 | 1.482 | ||
| Ce3+ | 1.15 | 1.14 | 1.283 | 1.219 | 1.48 | 1.482 | ||
| Pr3+ | 1.13 | 1.14 | 1.266 | 1.219 | ||||
| Nd3+ | 1.123 | 1.19 | 1.249 | 1.219 | 1.41 | 1.422 | ||
| Sm3+ | 1.098 | 1.14 | 1.219 | 1.219 | 1.38 | 1.378 | ||
| Eu3+ | 1.087 | 1.14 | 1.206 | 1.219 | ||||
| Eu2+ | 1.31 | 1.43 | 1.39 | 1.377 | ||||
| Gd3+ | 1.078 | 1.14 | 1.193 | 1.218 | ||||
| Tb3+ | 1.063 | 1.14 | 1.18 | 1.22 | ||||
| Dy3+ | 1.052 | 1.14 | 1.167 | 1.22 | ||||
| Ho3+ | 1.041 | 1.14 | 1.155 | 1.14 | 1.26 | 1.219 | ||
| Er3+ | 1.03 | 0.98 | 1.144 | 1.14 | ||||
| Tm3+ | 1.02 | 0.98 | 1.134 | 1.14 | ||||
| Yb3+ | 1.008 | 0.98 | 1.125 | 1.14 | ||||
| Lu3+ | 1.001 | 0.96 | 1.117 | 1.14 | ||||
| Th4+ | 1.08 | 1.190 | 1.19 | 1.218 | 1.35 | 1.377 | ||
| U4+ | 1.03 | 1.190 | 1.14 | 1.145 | 1.31 | 1.377 | ||
| Ti2+ | 1 | 0.960 | ||||||
| Ti3+ | 0.81 | 0.793 | ||||||
| Ti4+ | 0.56 | 0.552 | 0.745 | 0.740 | 0.88 | 0.893 | ||
| Zr4+ | 0.73 | 0.720 | 0.86 | 0.853 | 0.98 | 0.960 | ||
| Hf4+ | 0.72 | 0.720 | 0.85 | 0.853 | 0.97 | 0.960 | ||
| V2+ | 0.93 | 0.918 | ||||||
| V3+ | 0.78 | 0.787 | ||||||
| V4+ | 0.72 | 0.720 | 0.86 | 0.853 | ||||
| Nb3+ | 0.86 | 0.853 | ||||||
| Nb5+ | 0.78 | 0.775 | 0.88 | 0.892 | ||||
| Ta3+ | 0.86 | 0.853 | ||||||
| Ta5+ | 0.78 | 0.787 | 0.88 | 0.892 | ||||
| Cr3+ | 0.755 | 0.763 | ||||||
| Mo3+ | 0.83 | 0.793 | ||||||
| Mo6+ | 0.73 | 0.720 | ||||||
| W6+ | 0.74 | 0.740 | ||||||
| Mn3+HS | 0.785 | 0.787 | ||||||
| Mn3+LS | 0.72 | 0.720 | ||||||
| Re7+ | 0.67 | 0.689 | ||||||
| Fe2+HS | 0.77 | 0.775 | 0.92 | 0.916 | 1.06 | 1.066 | ||
| Fe2+LS | 0.75 | 0.740 | ||||||
| Fe3+HS | 0.785 | 0.763 | 0.92 | 0.918 | ||||
| Fe3+LS | 0.69 | 0.689 | ||||||
| Ru4+ | 0.76 | 0.763 | ||||||
| Os4+ | 0.77 | 0.775 | ||||||
| Co2+HS | 0.72 | 0.720 | 0.885 | 0.892 | 1.04 | 1.020 | ||
| Co2+LS | 0.79 | 0.793 | ||||||
| Co3+HS | 0.75 | 0.740 | ||||||
| Co3+LS | 0.685 | 0.689 | ||||||
| Rh3+ | 0.805 | 0.763 | ||||||
| Ir4+ | 0.765 | 0.763 | ||||||
| Ni2+ | 0.69 | 0.720 | 0.83 | 0.793 | ||||
| Ni3+HS | 0.74 | 0.74 | ||||||
| Ni3+LS | 0.7 | 0.689 | ||||||
| Pd2+ | 1 | 0.960 | ||||||
| Pt2+ | 0.94 | 0.960 | ||||||
| Cu2+ | 0.71 | 0.720 | 0.87 | 0.916 | ||||
| Ag2+ | 1.08 | 1.102 | ||||||
| Au3+ | 0.99 | 0.916 | ||||||
| Zn2+ | 0.74 | 0.720 | 0.88 | 0.916 | ||||
| Cd2+ | 0.92 | 1.102 | 1.09 | 1.102 | 1.24 | 1.219 | 1.45 | 1.440 |
| As3+ | 0.72 | 0.720 | ||||||
| Sb3+ | 0.9 | 0.893 | ||||||
| Bi3+ | 1.17 | 1.19 | ||||||
| O2− | 1.60 | 1.64 | ||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tschauner, O. An Observation Related to the Pressure Dependence of Ionic Radii. Geosciences 2022, 12, 246. https://doi.org/10.3390/geosciences12060246
Tschauner O. An Observation Related to the Pressure Dependence of Ionic Radii. Geosciences. 2022; 12(6):246. https://doi.org/10.3390/geosciences12060246
Chicago/Turabian StyleTschauner, Oliver. 2022. "An Observation Related to the Pressure Dependence of Ionic Radii" Geosciences 12, no. 6: 246. https://doi.org/10.3390/geosciences12060246
APA StyleTschauner, O. (2022). An Observation Related to the Pressure Dependence of Ionic Radii. Geosciences, 12(6), 246. https://doi.org/10.3390/geosciences12060246