Rumen and Fecal Microbial Community Structure of Holstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake

 , ,
, ,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
3. Results
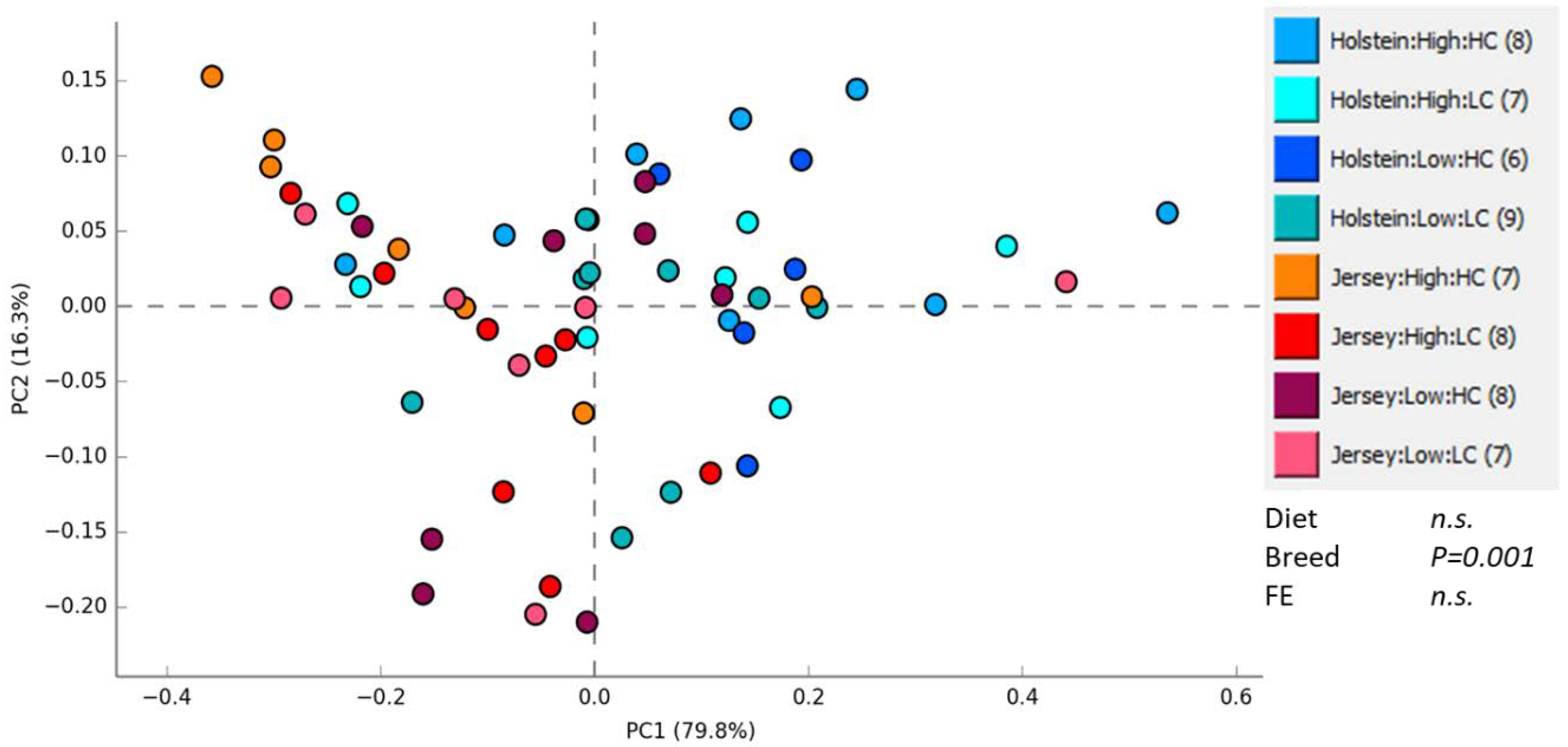
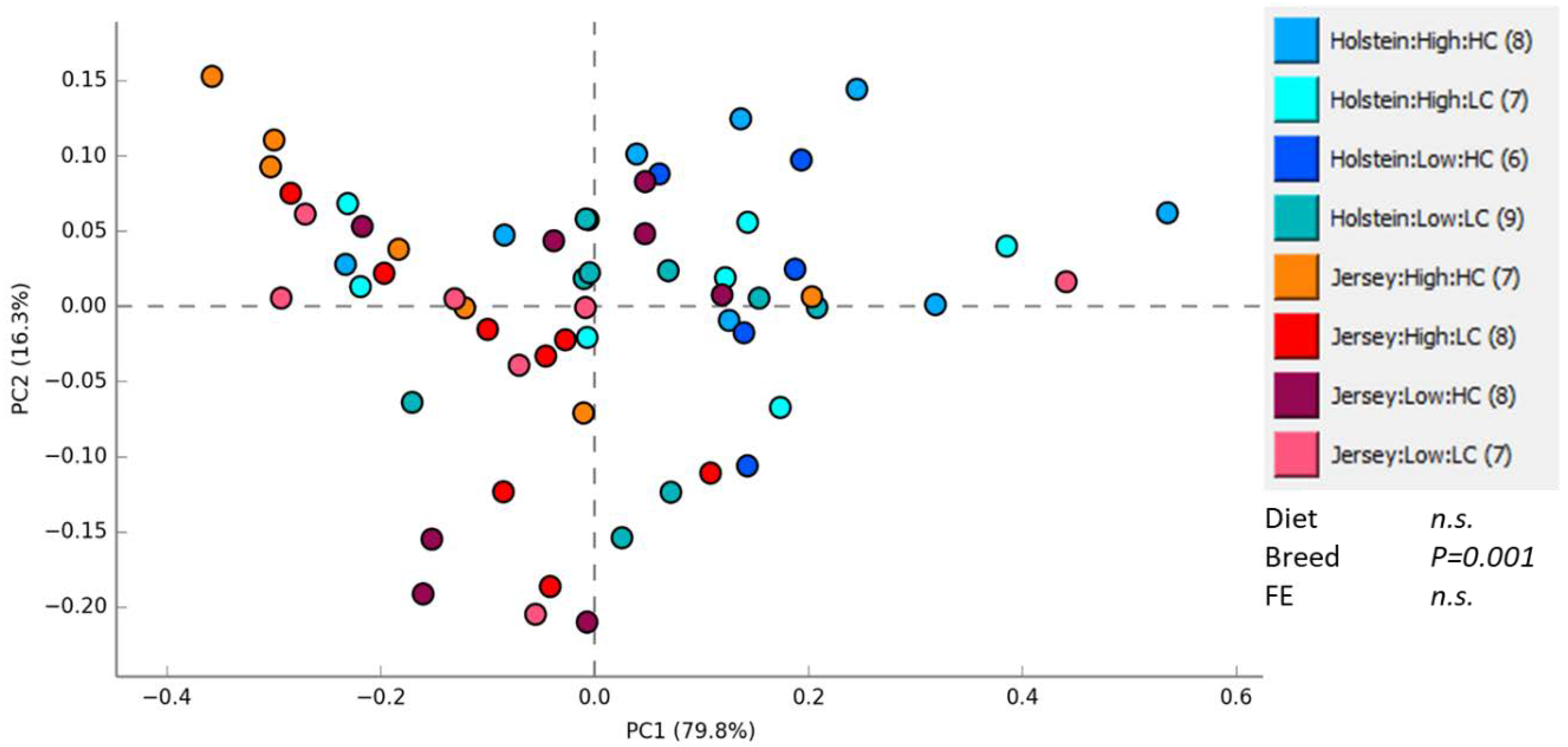
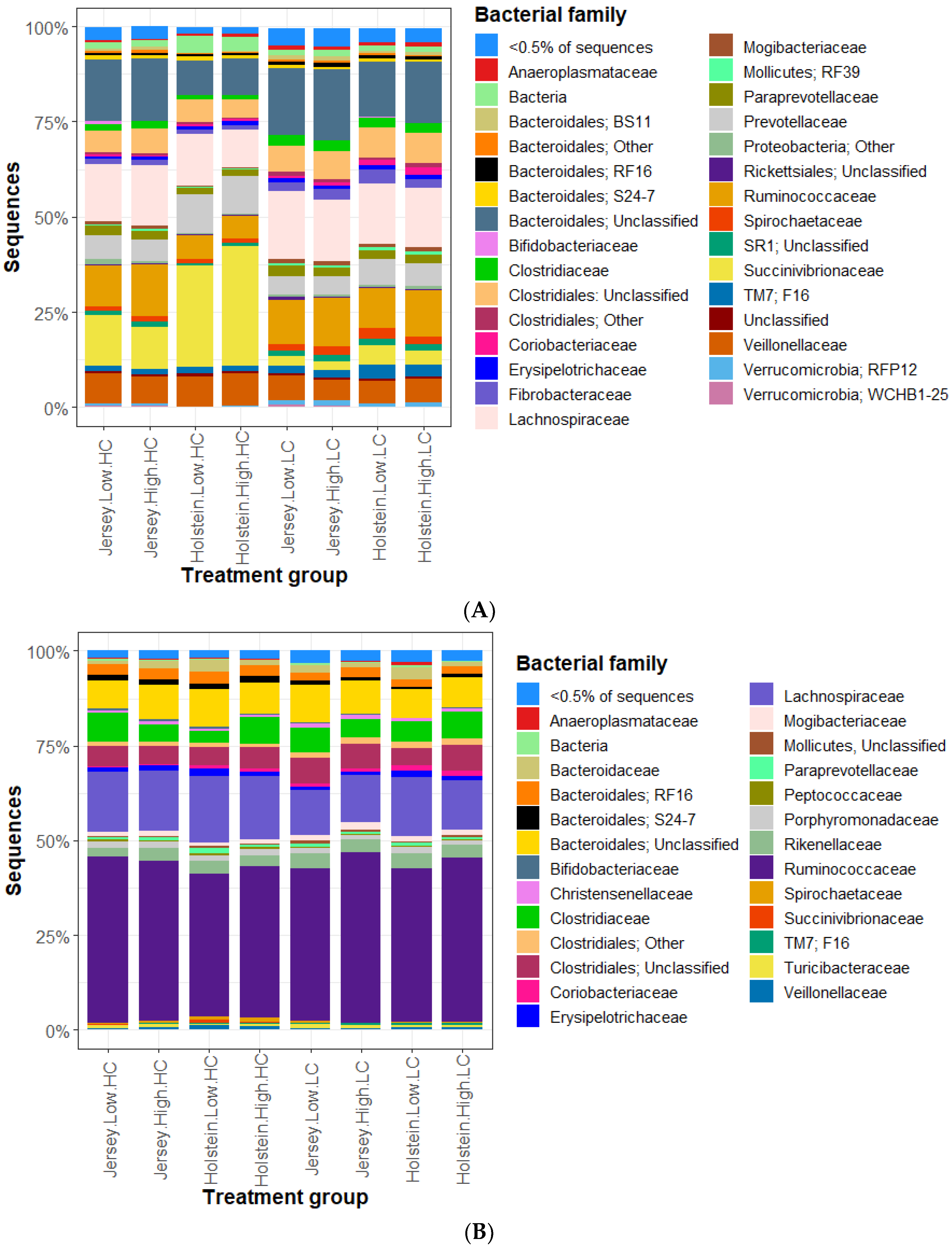
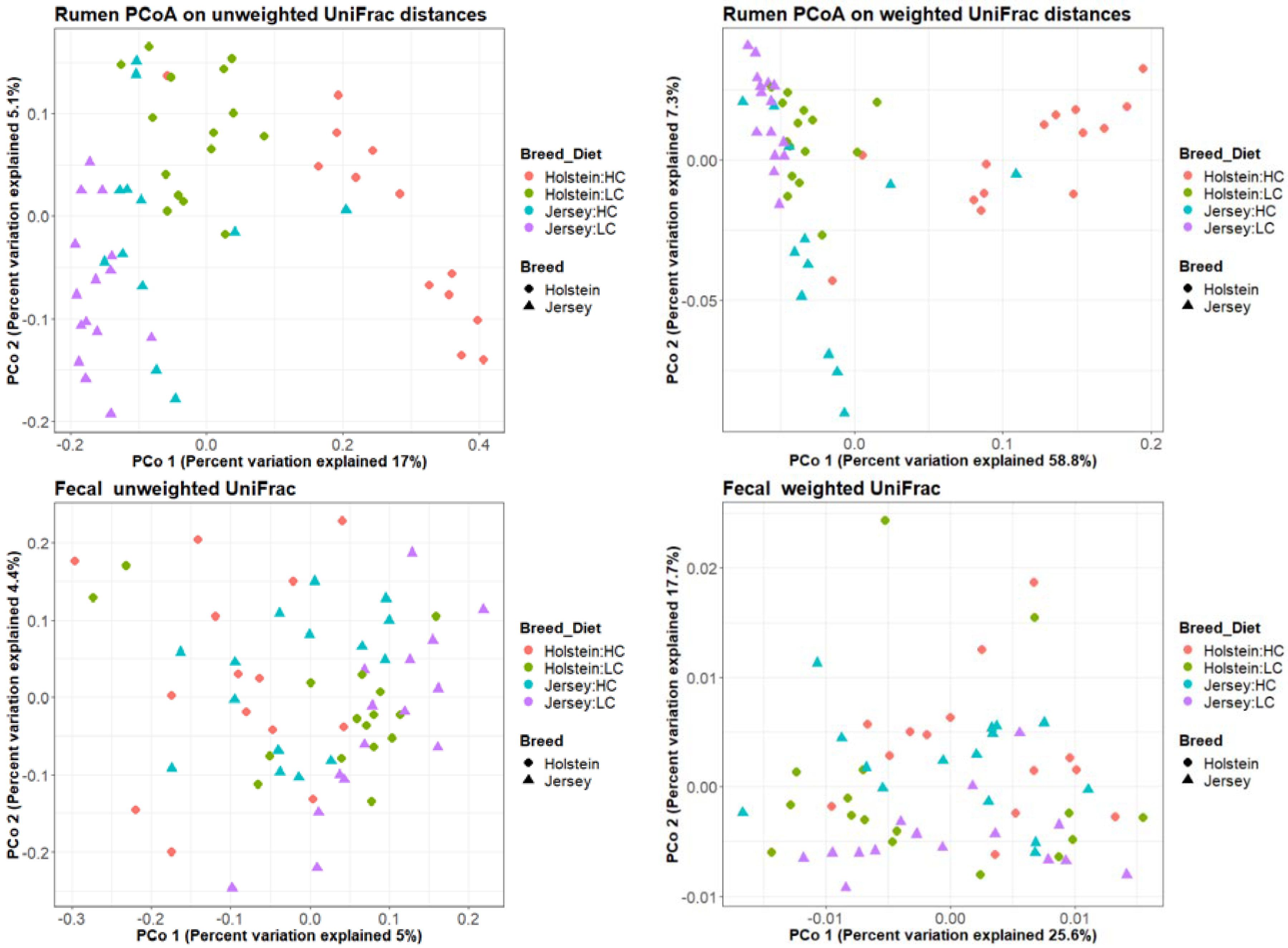
3.1. Rumen Methanogen Community
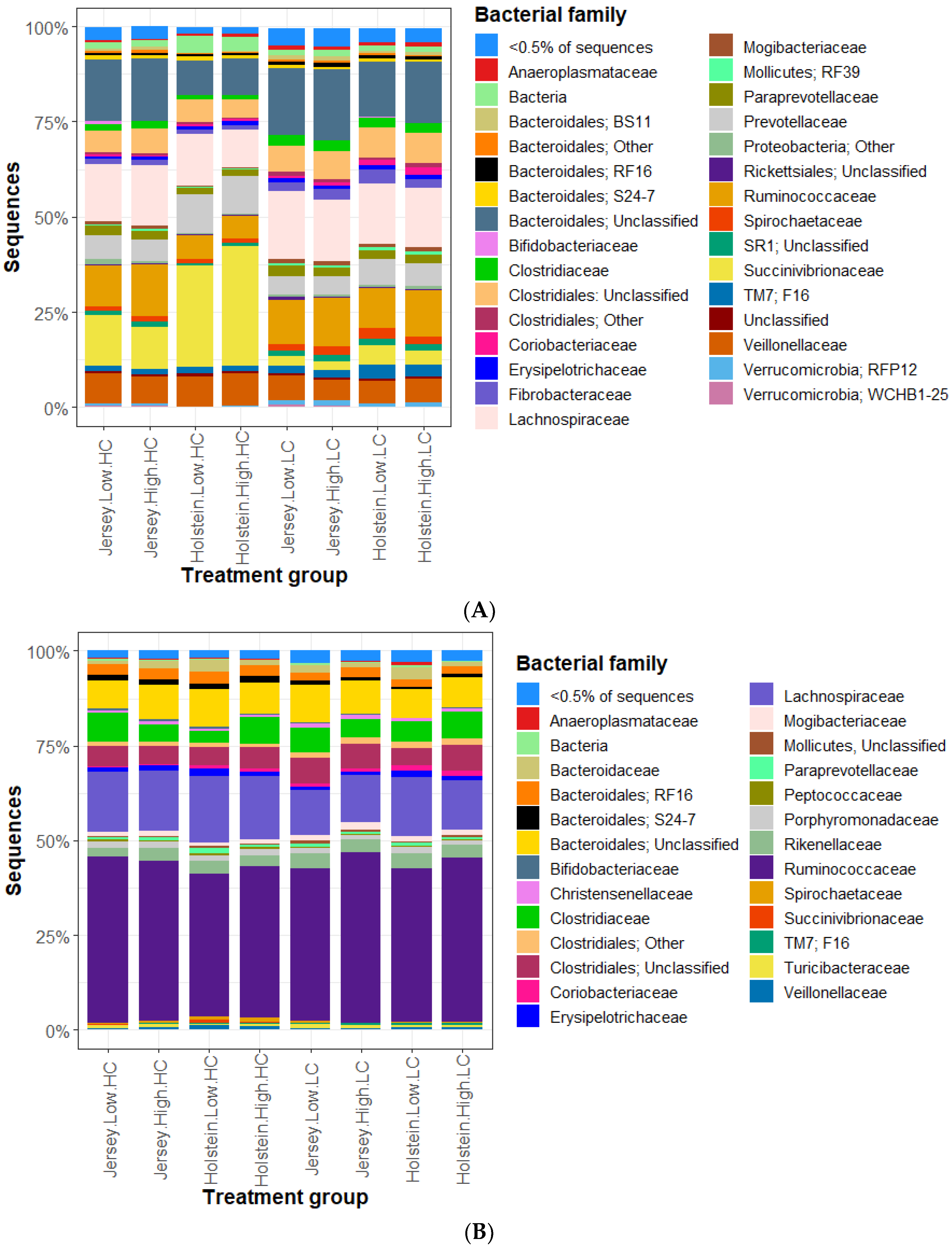
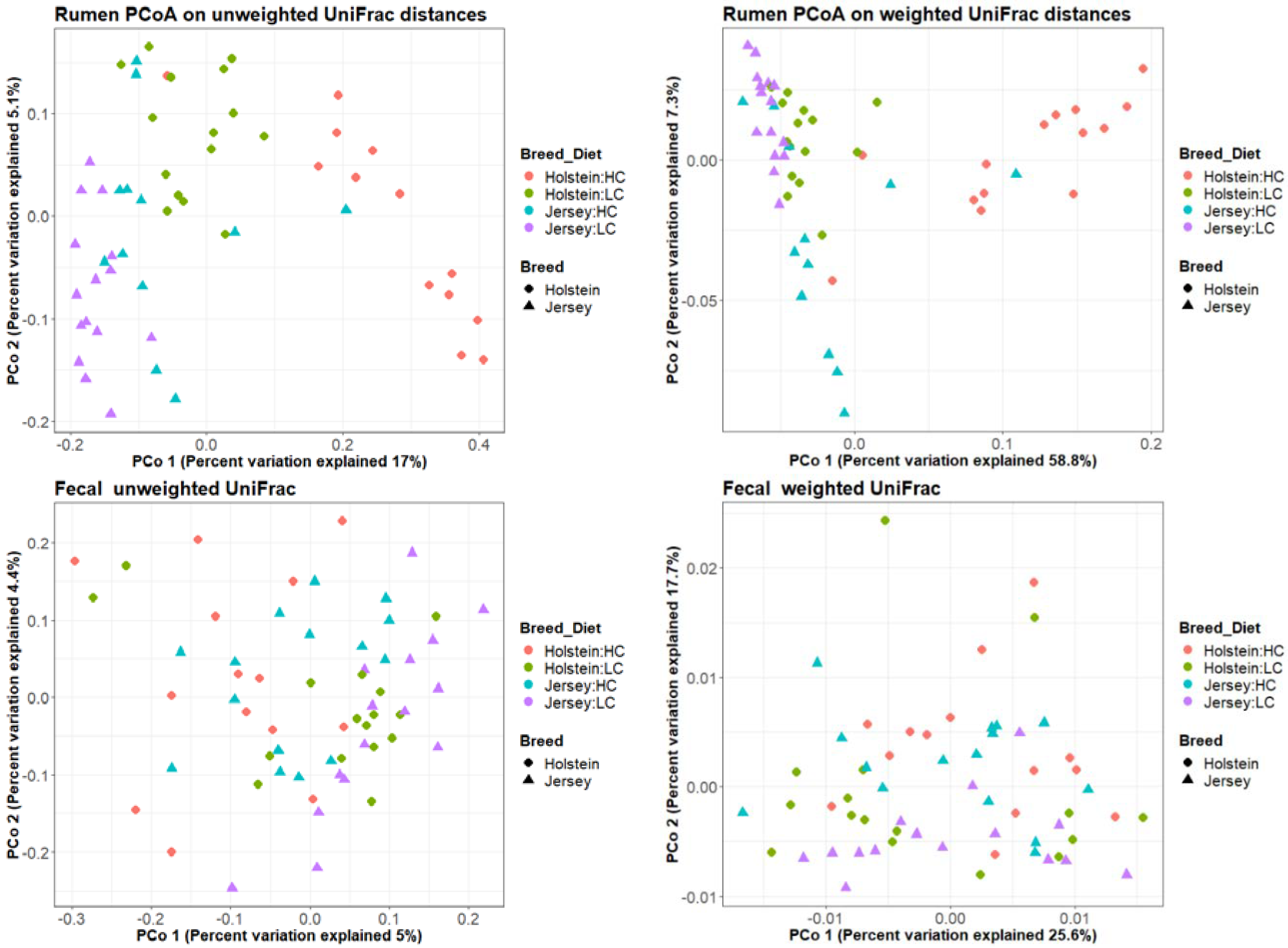
3.2. Bacterial Community in the Rumen and Feces
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Poulsen, M.; Schwab, C.; Jensen, B.B.; Engberg, R.M.; Spang, A.; Canibe, N.; Højberg, O.; Milinovich, G.; Fragner, L.; Schleper, C.; et al. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat. Commun. 2013, 4, 1428. [Google Scholar] [CrossRef] [PubMed]
- Roehe, R.; Dewhurst, R.J.; Duthie, C.-A.; Rooke, J.A.; McKain, N.; Ross, D.W.; Hyslop, J.J.; Waterhouse, A.; Freeman, T.C.; Watson, M.; et al. Bovine Host Genetic Variation Influences Rumen Microbial Methane Production with Best Selection Criterion for Low Methane Emitting and Efficiently Feed Converting Hosts Based on Metagenomic Gene Abundance. PLoS Genet. 2016, 12, e1005846. [Google Scholar] [CrossRef] [PubMed]
- Difford, G.F.; Plichta, D.R.; Løvendahl, P.; Lassen, J.; Noel, S.J.; Højberg, O.; Wright, A.G.; Zhu, Z.; Kristensen, L.; Nielsen, H.B.; et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. Plos Genet. 2018, 14, e1007580. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, I.; Jami, E. Review: The compositional variation of the rumen microbiome and its effect on host performance and methane emission. Animal 2018, 12, s220–s232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frey, J.C.; Pell, A.N.; Berthiaume, R.; Lapierre, H.; Lee, S.; Ha, J.K.; Mendell, J.E.; Angert, E.R. Comparative studies of microbial populations in the rumen, duodenum, ileum and faeces of lactating dairy cows. J. Appl. Microbiol. 2010, 108, 1982–1993. [Google Scholar] [CrossRef]
- Tapio, I.; Shingfield, K.J.; McKain, N.; Bonin, A.; Fischer, D.; Bayat, A.R.; Vilkki, J.; Taberlet, P.; Snelling, T.J.; Wallace, R.J. Oral Samples as Non-Invasive Proxies for Assessing the Composition of the Rumen Microbial Community. PLoS ONE 2016, 11, e0151220. [Google Scholar] [CrossRef]
- Callaway, T.R.; Dowd, S.E.; Edrington, T.S.; Anderson, R.C.; Krueger, N.; Bauer, N.; Kononoff, P.J.; Nisbet, D.J. Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J. Anim. Sci. 2010, 88, 3977–3983. [Google Scholar] [CrossRef]
- Bermingham, E.N.; Kittelmann, S.; Henderson, G.; Young, W.; Roy, N.C.; Thomas, D.G. Five-week dietary exposure to dry diets alters the faecal bacterial populations in the domestic cat (Felis catus). Br. J. Nutr. 2011, 106 (Suppl. 1), S49–S52. [Google Scholar] [CrossRef] [Green Version]
- Weimer, P.J.; Stevenson, D.M.; Mantovani, H.C.; Man, S.L. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents. J. Dairy Sci. 2010, 93, 5902–5912. [Google Scholar] [CrossRef]
- Olijhoek, D.W.; Løvendahl, P.; Lassen, J.; Hellwing, A.L.F.; Höglund, J.K.; Weisbjerg, M.R.; Noel, S.J.; McLean, F.; Højberg, O.; Lund, P. Methane production, rumen fermentation, and diet digestibility of Holstein and Jersey dairy cows being divergent in residual feed intake and fed at 2 forage-to-concentrate ratios. J. Dairy Sci. 2018, 101, 9926–9940. [Google Scholar] [CrossRef] [Green Version]
- Flay, H.E.; Kuhn-Sherlock, B.; Macdonald, K.A.; Camara, M.; Lopez-Villalobos, N.; Donaghy, D.J.; Roche, J.R. Hot topic: Selecting cattle for low residual feed intake did not affect daily methane production but increased methane yield. J. Dairy Sci. 2019, 102, 2708–2713. [Google Scholar] [CrossRef]
- Capper, J.L.; Cady, R.A.; Bauman, D.E. The environmental impact of dairy production: 1944 compared with 2007. J. Anim. Sci. 2009, 87, 2160–2167. [Google Scholar] [CrossRef]
- Hellwing, A.L.F.; Lund, P.; Weisbjerg, M.R.; Brask, M.; Hvelplund, T. Technical note: Test of a low-cost and animal-friendly system for measuring methane emissions from dairy cows. J. Dairy Sci. 2012, 95, 6077–6085. [Google Scholar] [CrossRef]
- Geishauser, T.; Linhart, N.; Neidl, A.; Reimann, A. Factors associated with ruminal pH at herd level. J. Dairy Sci. 2012, 95, 4556–4567. [Google Scholar] [CrossRef]
- Zhu, Z.G.; Kristensen, L.; Difford, G.F.; Poulsen, M.; Noel, S.J.; Abu Al-Soud, W.; Sørensen, S.J.; Lassen, J.; Løvendahl, P.; Højberg, O. Changes in rumen bacterial and archaeal communities over the transition period in primiparous Holstein dairy cows. J. Dairy Sci. 2018, 101, 9847–9862. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Illumina. 16S Metagenomic Sequencing Library Preparation. 27 November 2013. Available online: https://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 29 August 2018).
- Hildebrand, F.; Tadeo, R.; Voigt, A.Y.; Bork, P.; Raes, J. LotuS: An efficient and user-friendly OTU processing pipeline. Microbiome 2014, 2, 30. [Google Scholar] [CrossRef]
- Magoc, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- QIIME 2. Available online: https://qiime2.org/ (accessed on 9 October 2018).
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Zhou, M.; Hernandez-Sanabria, E.; Guan, L.L. Assessment of the microbial ecology of ruminal methanogens in cattle with different feed efficiencies. Appl. Environ. Microbiol. 2009, 75, 6524–6533. [Google Scholar] [CrossRef]
- Carberry, C.A.; Waters, S.M.; Kenny, D.A.; Creevey, C.J. Rumen methanogenic genotypes differ in abundance according to host residual feed intake phenotype and diet type. Appl. Environ. Microbiol. 2014, 80, 586–594. [Google Scholar] [CrossRef]
- Shi, W.B.; Moon, C.D.; Leahy, S.C.; Kang, D.W.; Froula, J.; Kittelmann, S.; Fan, C.; Deutsch, S.; Gagic, D.; Seedorf, H.; et al. Methane yield phenotypes linked to differential gene expression in the sheep rumen microbiome. Genome Res. 2014, 24, 1517–1525. [Google Scholar] [CrossRef] [Green Version]
- Jeyanathan, J.; Kirs, M.; Ronimus, R.S.; Hoskin, S.O.; Janssen, P.H. Methanogen community structure in the rumens of farmed sheep, cattle and red deer fed different diets. FEMS Microbiol. Ecol. 2011, 76, 311–326. [Google Scholar] [CrossRef]
- Cersosimo, L.M.; Bainbridge, M.L.; Kraft, J.; Wright, A.D.G. Influence of periparturient and postpartum diets on rumen methanogen communities in three breeds of primiparous dairy cows. BMC Microbiol. 2016, 16, 78. [Google Scholar] [CrossRef]
- Kumar, S.; Indugu, N.; Vecchiarelli, B.; Pitta, D.W. Associative patterns among anaerobic fungi, methanogenic archaea, and bacterial communities in response to changes in diet and age in the rumen of dairy cows. Front. Microbiol. 2015, 6, 781. [Google Scholar] [CrossRef]
- Henderson, G.; Cox, F.; Ganesh, S.; Jonker, A.; Young, W.; Janssen, P.H. Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range. Sci. Rep. 2015, 5, 14567. [Google Scholar] [CrossRef]
- McAllister, T.A.; Bae, H.D.; Jones, G.A.; Cheng, K.J. Microbial attachment and feed digestion in the rumen. J. Anim. Sci. 1994, 72, 3004–3018. [Google Scholar] [CrossRef] [Green Version]
- Song, J.; Choi, H.; Jeong, J.Y.; Lee, S.; Lee, H.J.; Baek, Y.; Ji, S.Y.; Kim, M. Effects of Sampling Techniques and Sites on Rumen Microbiome and Fermentation Parameters in Hanwoo Steers. J. Microbiol. Biotechnol. 2018, 28, 1700–1705. [Google Scholar] [CrossRef] [Green Version]
- Paz, H.A.; Anderson, C.L.; Muller, M.J.; Kononoff, P.J.; Fernando, S.C. Rumen Bacterial Community Composition in Holstein and Jersey Cows Is Different under Same Dietary Condition and Is Not Affected by Sampling Method. Front. Microbiol. 2016, 7, 1206. [Google Scholar] [CrossRef]
- Tajima, K.; Aminov, R.I.; Nagamine, T.; Matsui, H.; Nakamura, M.; Benno, Y. Diet-dependent shifts in the bacterial population of the rumen revealed with real-time PCR. Appl. Environ. Microbiol. 2001, 67, 2766–2774. [Google Scholar] [CrossRef]
- Fernando, S.C.; Purvis, H.T.; Najar, F.Z.; Sukharnikov, L.O.; Krehbiel, C.R.; Nagaraja, T.G.; Roe, B.A.; DeSilva, U. Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 2010, 76, 7482–7490. [Google Scholar] [CrossRef]
- Bainbridge, M.L.; Cersosimo, L.M.; Wright, A.D.G.; Kraft, J. Rumen bacterial communities shift across a lactation in Holstein, Jersey and Holstein x Jersey dairy cows and correlate to rumen function, bacterial fatty acid composition and production parameters. FEMS Microbiol. Ecol. 2016, 92, fiw059. [Google Scholar] [CrossRef]
- Mu, Y.; Lin, X.; Wang, Z.; Hou, Q.; Wang, Y.; Hu, Z. High-production dairy cattle exhibit different rumen and fecal bacterial community and rumen metabolite profile than low-production cattle. MicrobiologyOpen 2019, 8, e769. [Google Scholar] [CrossRef]
- Dill-McFarland, K.A.; Weimer, P.J.; Breaker, J.D.; Suen, G. Diet Influences Early Microbiota Development in Dairy Calves without Long-Term Impacts on Milk Production. Appl. Environ. Microbiol. 2019, 85, e02141-18. [Google Scholar] [CrossRef] [Green Version]
- Noel, S.J.; Attwood, G.T.; Rakonjac, J.; Moon, C.D.; Waghorn, G.C.; Janssen, P.H. Seasonal changes in the digesta-adherent rumen bacterial communities of dairy cattle grazing pasture. PLoS ONE 2017, 12, e0173819. [Google Scholar] [CrossRef]
- Kittelmann, S.; Pinares-Patino, C.S.; Seedorf, H.; Kirk, M.R.; Ganesh, S.; McEwan, J.C.; Janssen, P.H. Two Different Bacterial Community Types Are Linked with the Low-Methane Emission Trait in Sheep. PLoS ONE 2014, 9, e103171. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
| Factor | Rumen 1 | Fecal 1 | ||
|---|---|---|---|---|
| Unweighted UniFrac | Weighted UniFrac | Unweighted UniFrac | Weighted UniFrac | |
| Breed | 0.001 | 0.001 | 0.001 | 0.141 |
| Animal | 0.002 | 0.147 | 0.001 | 0.001 |
| Diet | 0.001 | 0.001 | 0.001 | 0.002 |
| RFI groups | 0.846 | 0.673 | 0.499 | 0.398 |
| Trait | Rumen 1 | Fecal 1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Unweighted UniFrac | Weighted UniFrac | Unweighted UniFrac | Weighted UniFrac | |||||
| r | p | r | p | r | p | r | p | |
| NDF digestibility | 0.384 | 0.001 | 0.475 | 0.001 | 0.33 | 0.001 | 0.188 | 0.002 |
| A:P ratio | 0.577 | 0.001 | 0.654 | 0.001 | 0.302 | 0.001 | 0.127 | 0.001 |
| Methane 2 | 0.492 | 0.001 | 0.556 | 0.001 | 0.364 | 0.001 | 0.163 | 0.006 |
| RFI values | 0.009 | 0.842 | 0.0001 | 0.99 | 0.054 | 0.347 | 0.008 | 0.891 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noel, S.J.; Olijhoek, D.W.; Mclean, F.; Løvendahl, P.; Lund, P.; Højberg, O. Rumen and Fecal Microbial Community Structure of Holstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake. Animals 2019, 9, 498. https://doi.org/10.3390/ani9080498
Noel SJ, Olijhoek DW, Mclean F, Løvendahl P, Lund P, Højberg O. Rumen and Fecal Microbial Community Structure of Holstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake. Animals. 2019; 9(8):498. https://doi.org/10.3390/ani9080498
Chicago/Turabian StyleNoel, Samantha J., Dana W. Olijhoek, Farran Mclean, Peter Løvendahl, Peter Lund, and Ole Højberg. 2019. "Rumen and Fecal Microbial Community Structure of Holstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake" Animals 9, no. 8: 498. https://doi.org/10.3390/ani9080498
APA StyleNoel, S. J., Olijhoek, D. W., Mclean, F., Løvendahl, P., Lund, P., & Højberg, O. (2019). Rumen and Fecal Microbial Community Structure of Holstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake. Animals, 9(8), 498. https://doi.org/10.3390/ani9080498