Production Phase Affects the Bioaerosol Microbial Composition and Functional Potential in Swine Confinement Buildings

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
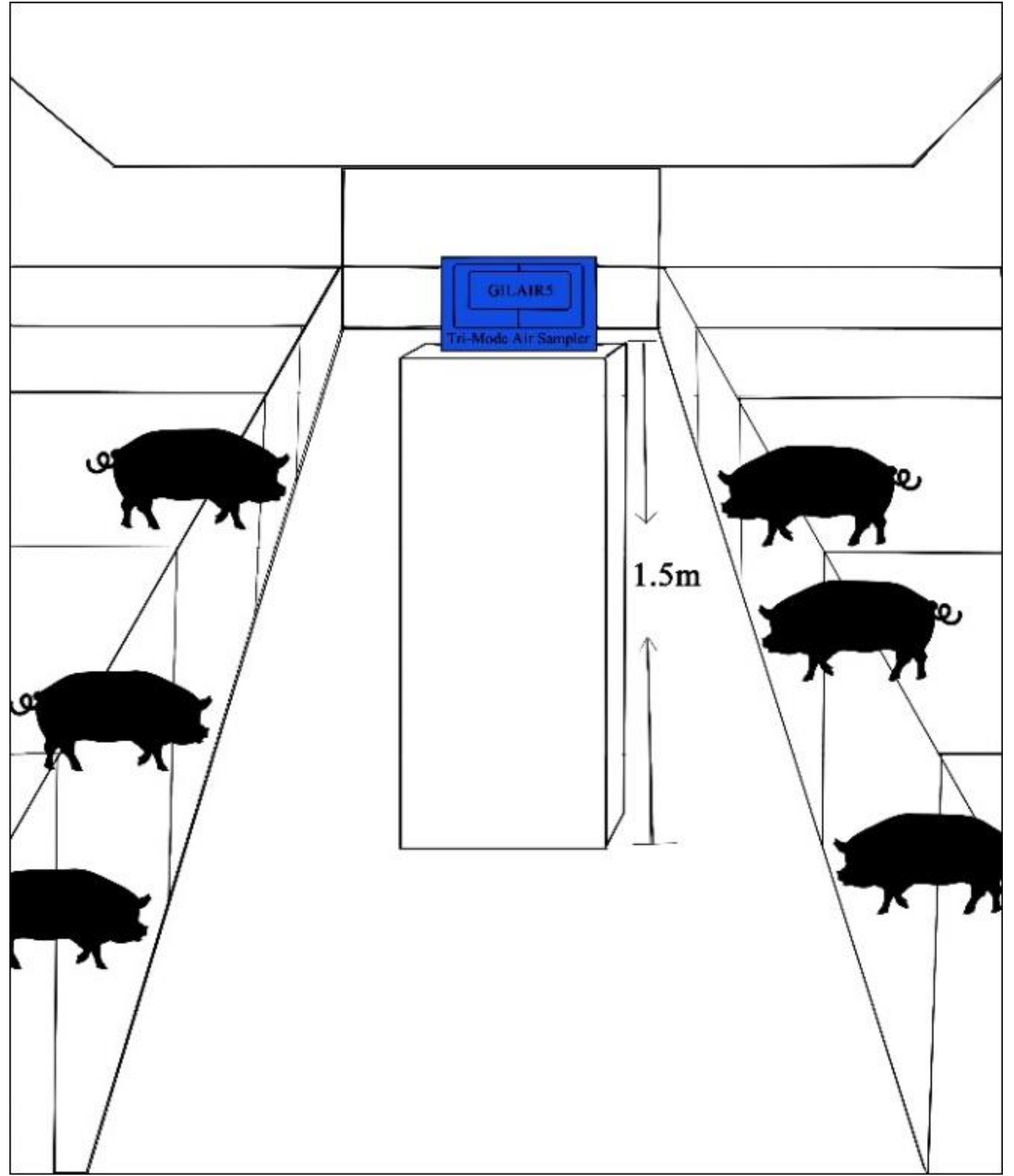
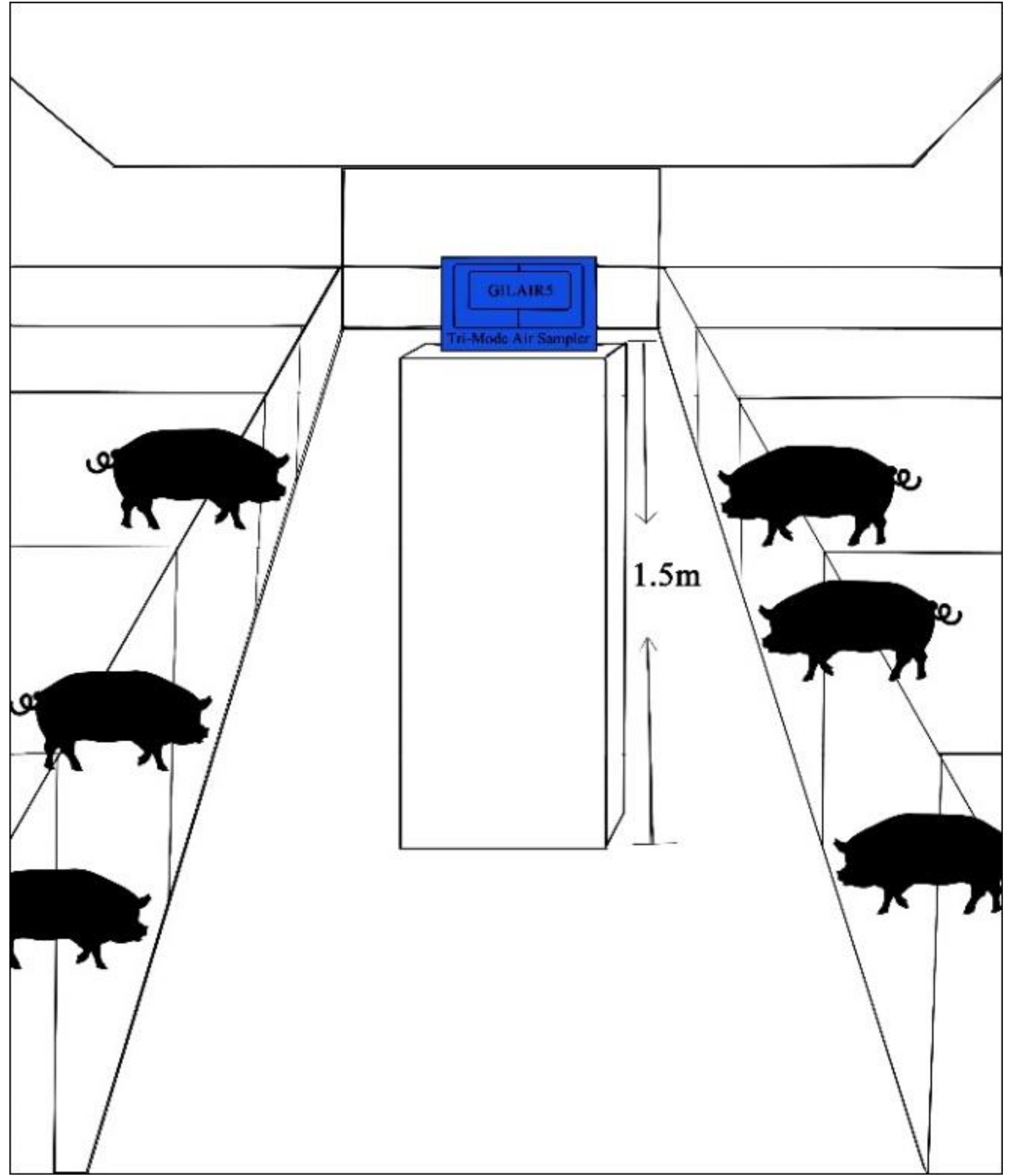
2.1. Sampling Sites
2.2. Sample Collection
2.3. Bioaerosol DNA Extraction and Metagenomic Library Preparation
2.4. Metagenomic Sequencing and Read Assembly
2.5. Taxonomical Prediction and Functional Annotation
2.6. Data Deposition
3. Results
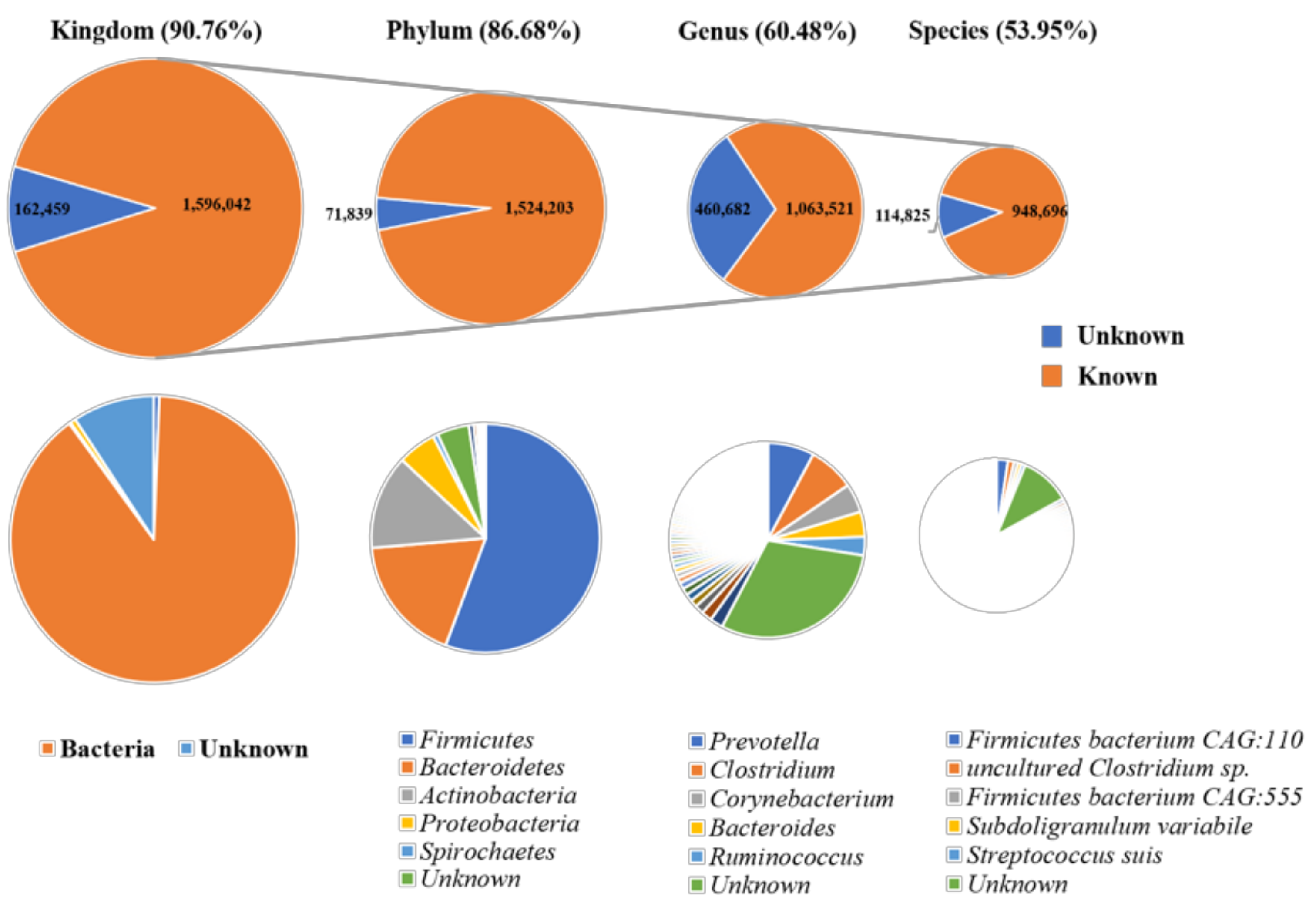
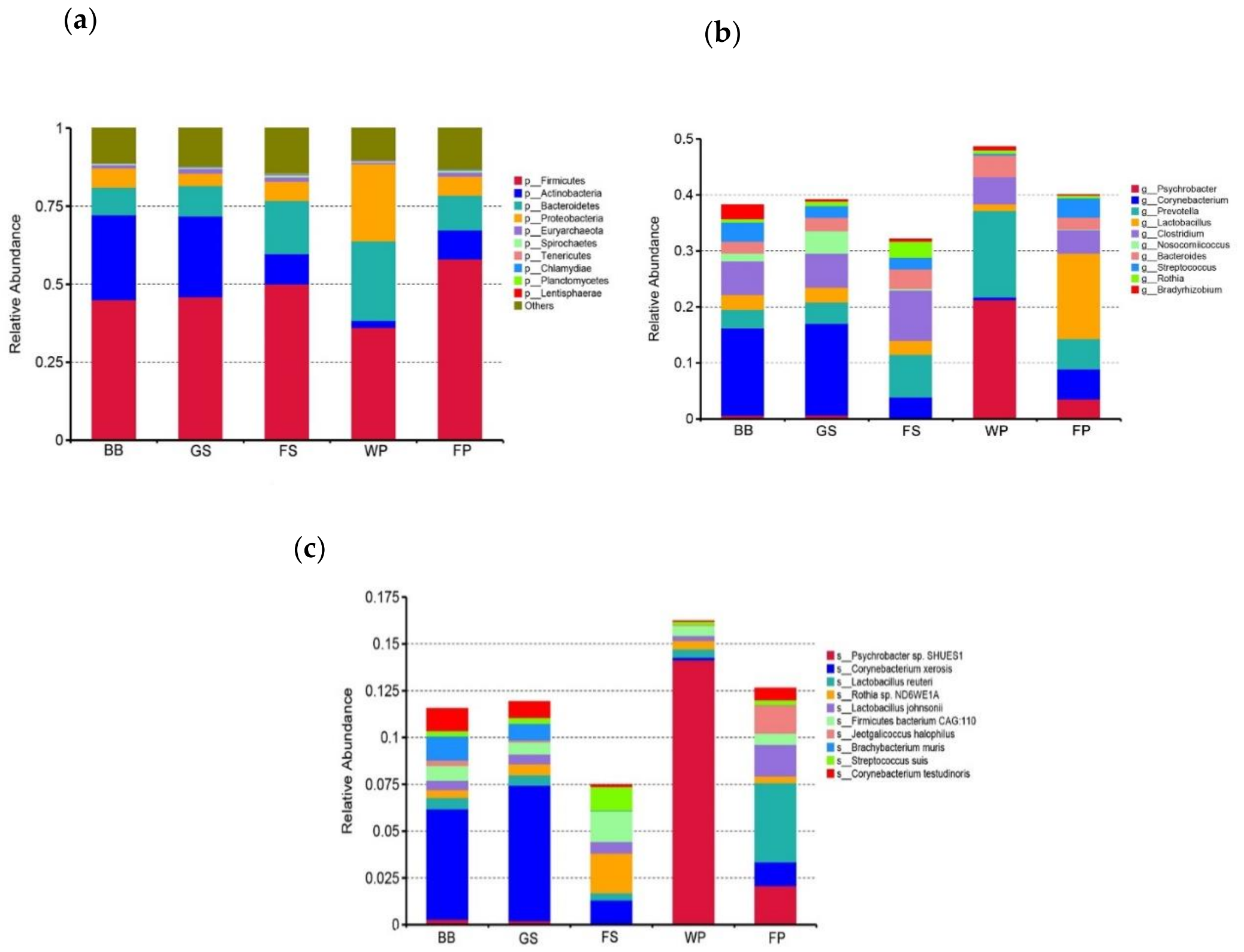
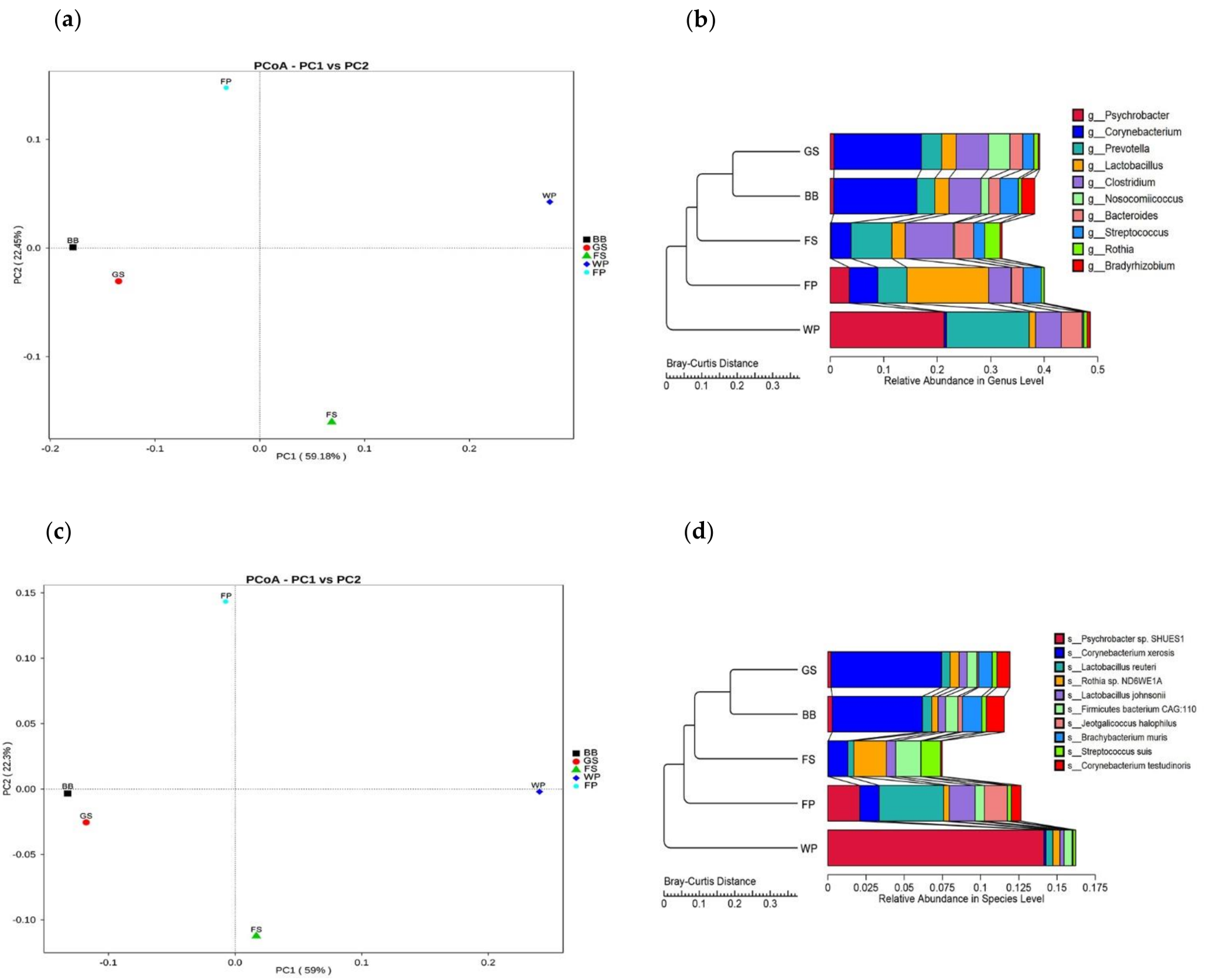
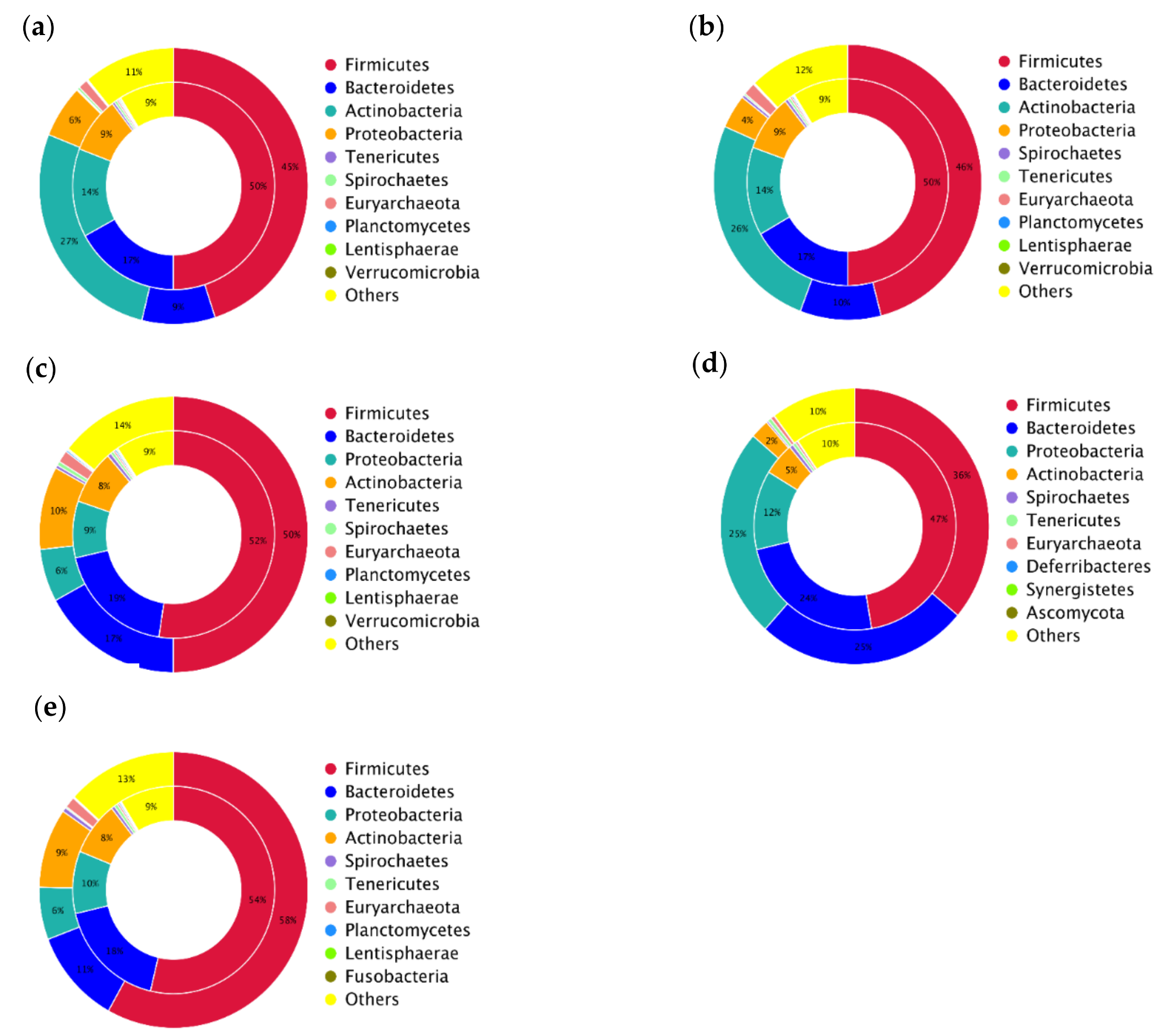
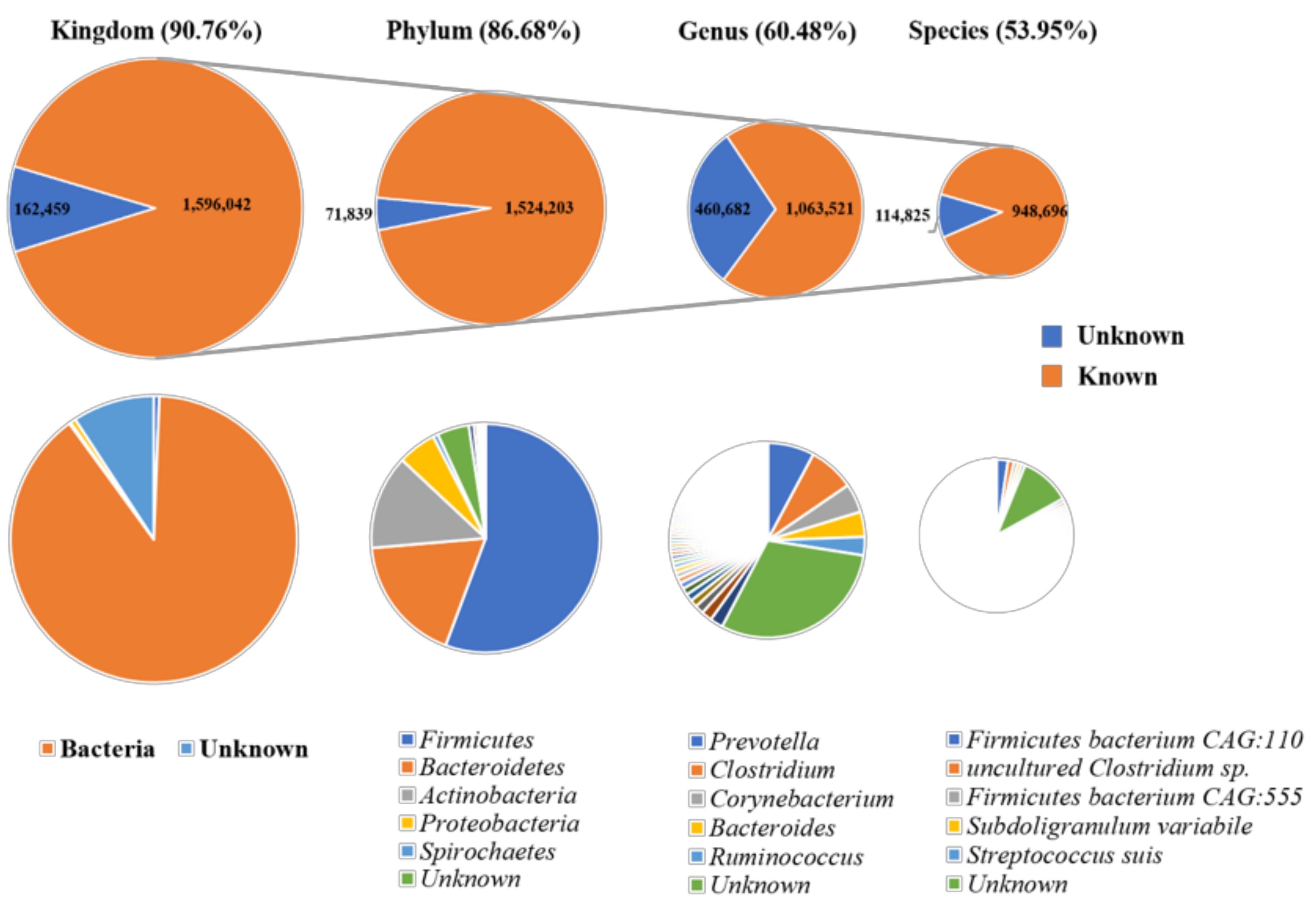
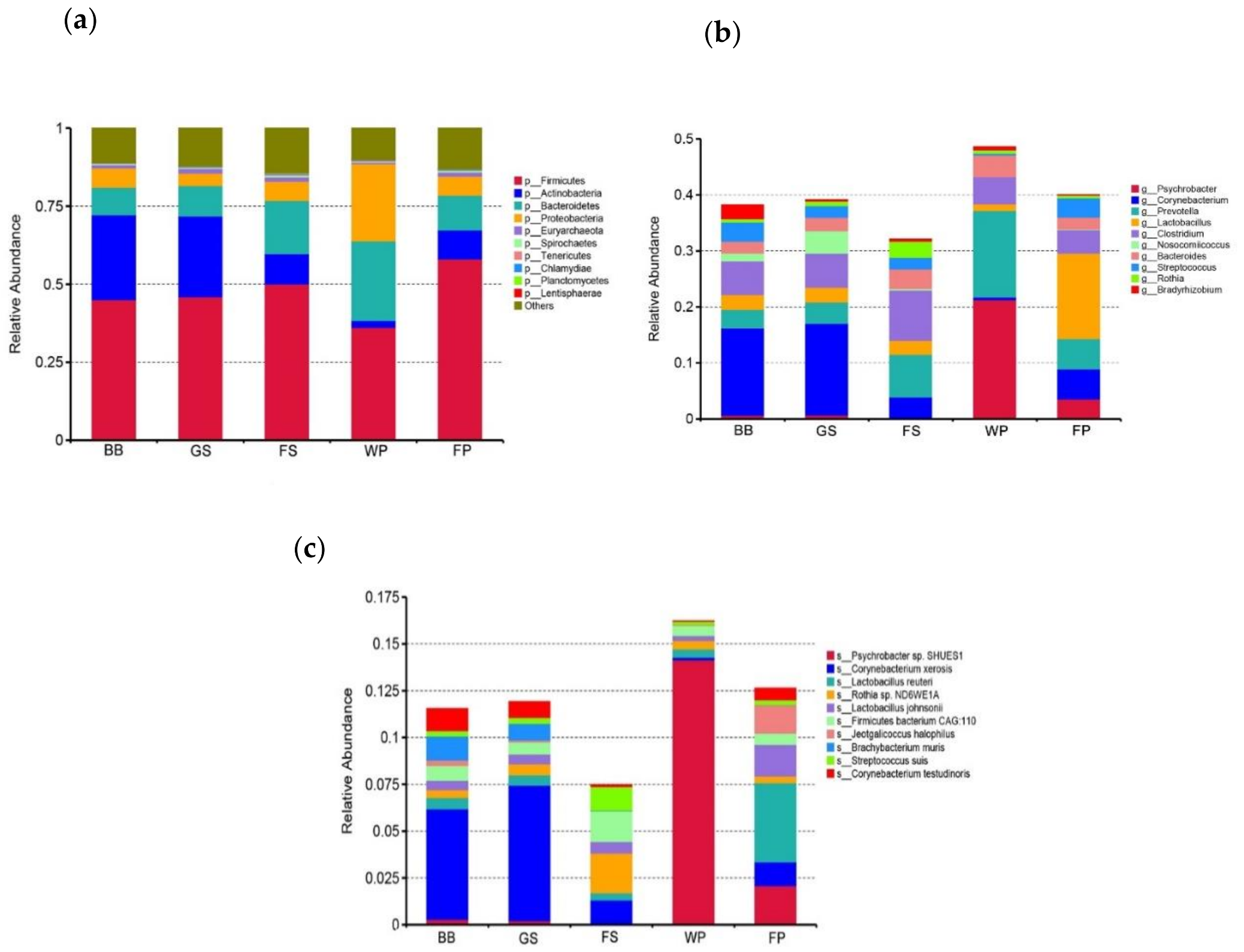
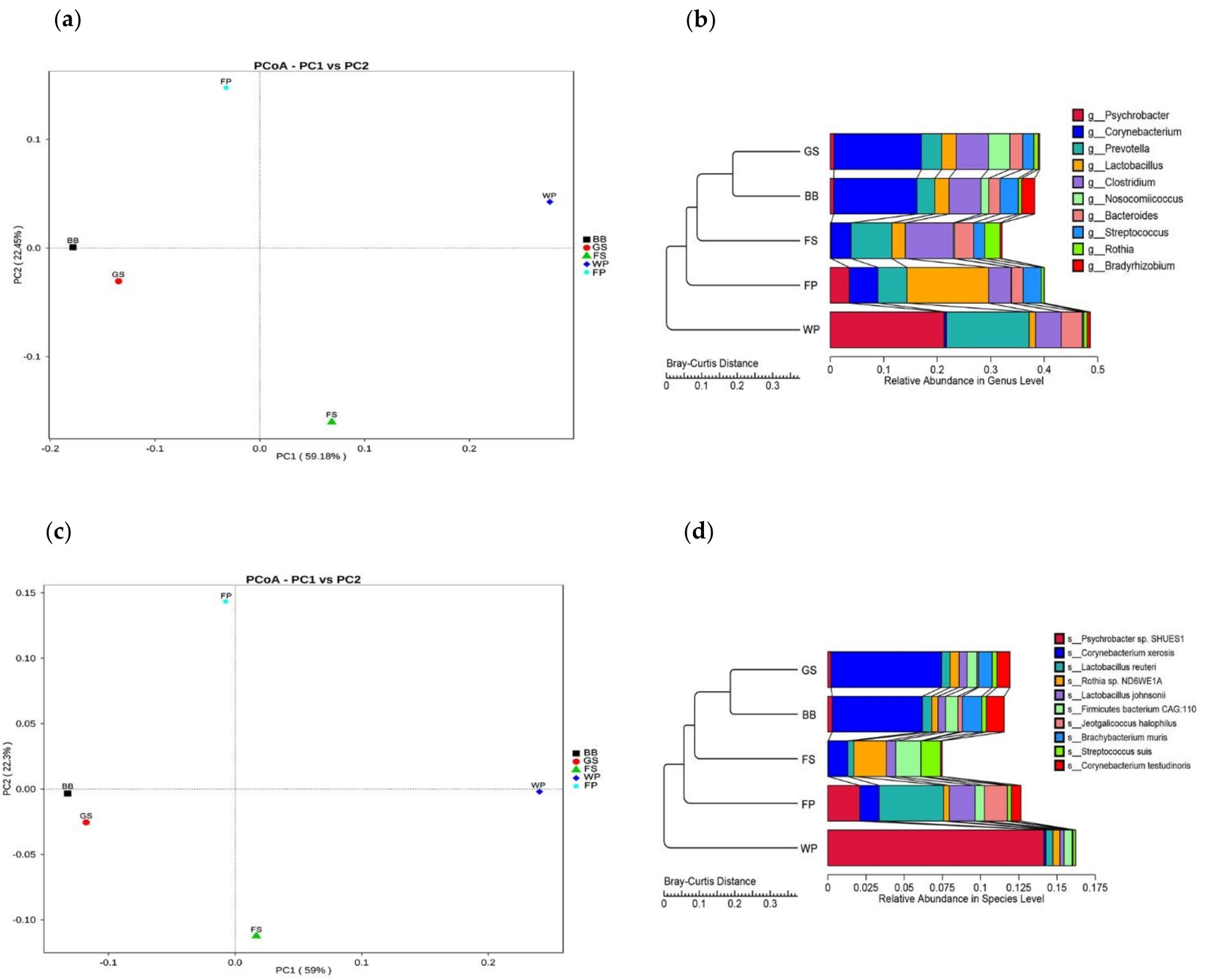
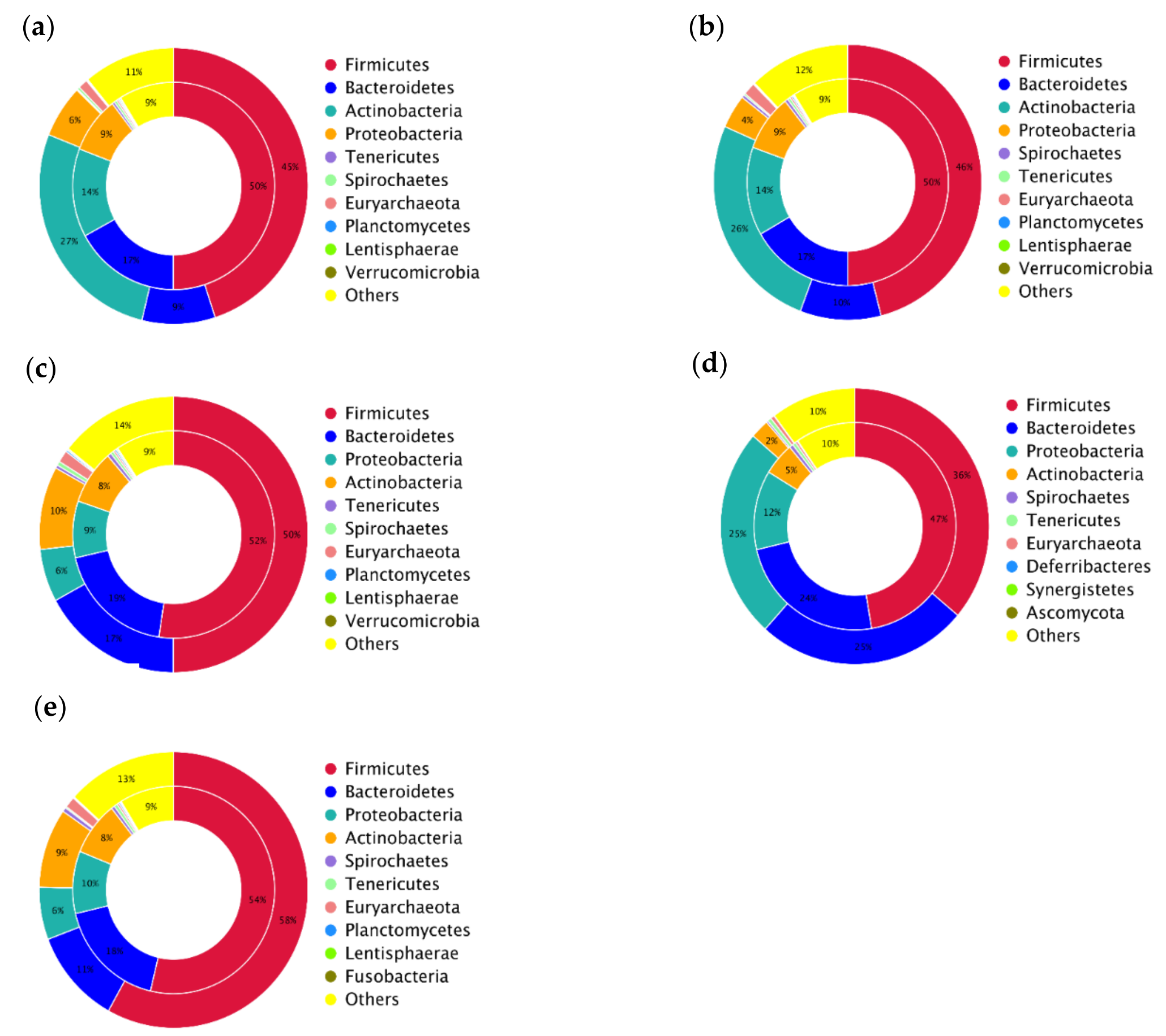
3.1. Taxonomical Characterization of the Metagenomic Profiles
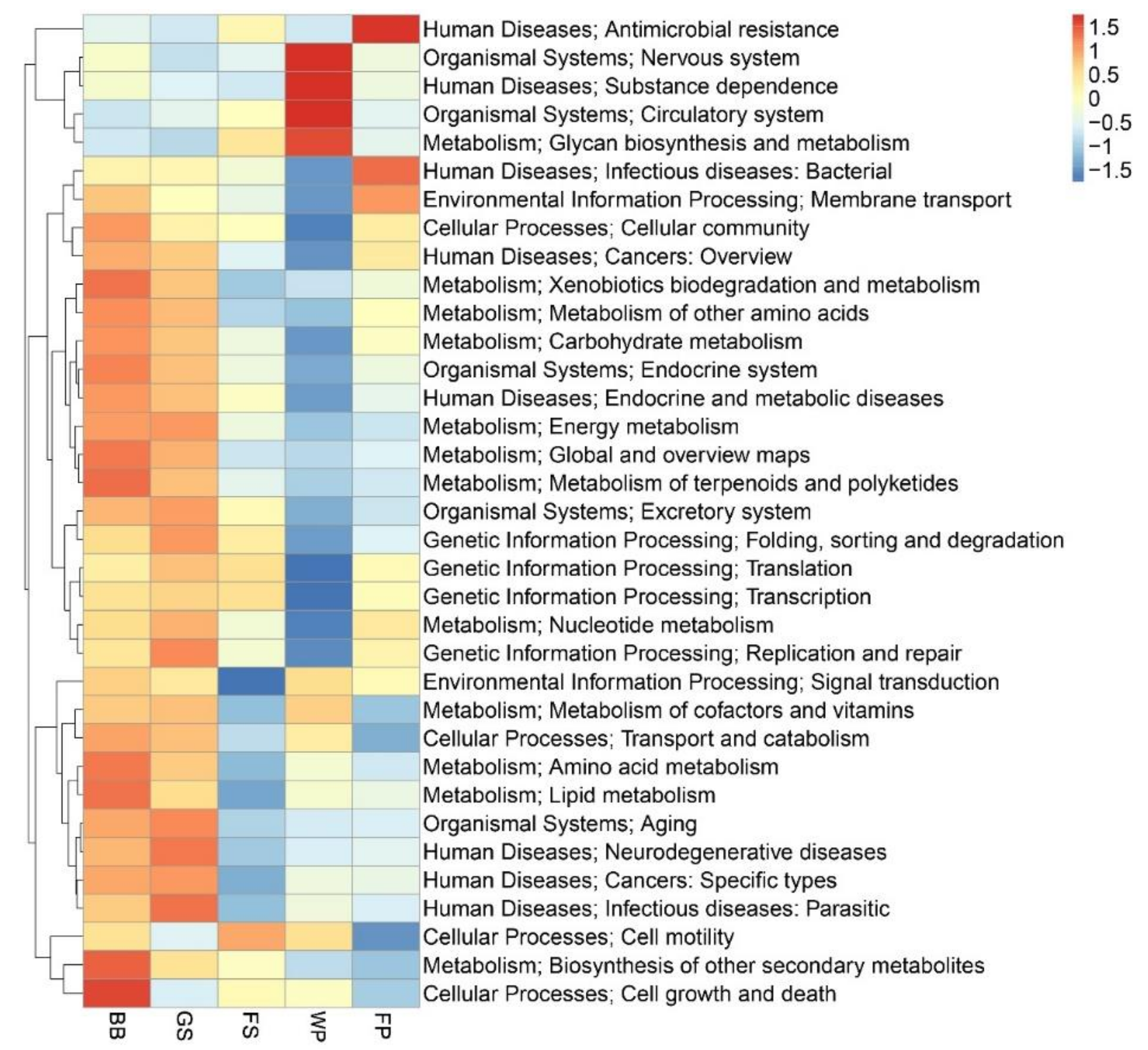
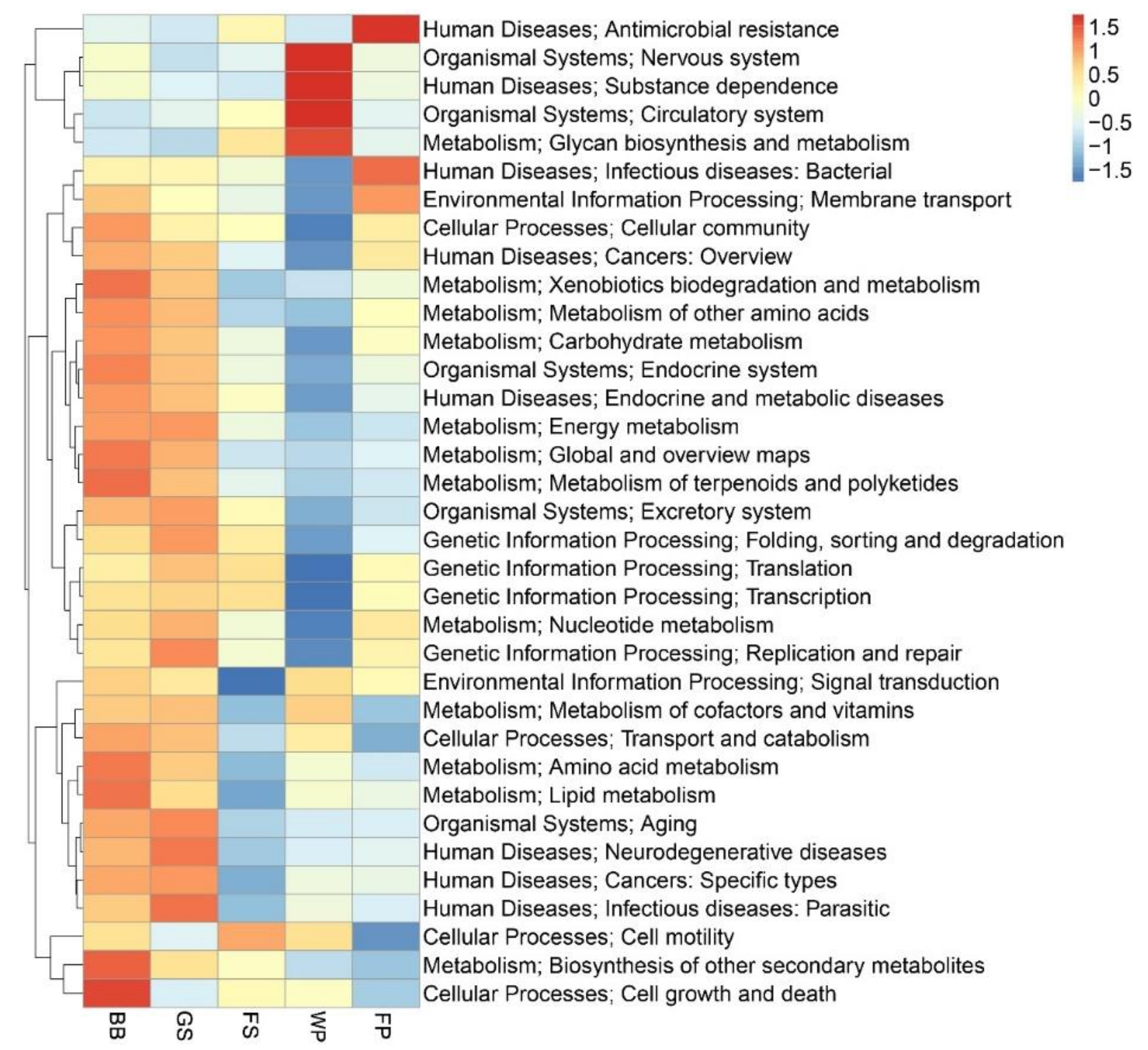
3.2. Functional Characterization of the Metagenomic Profile
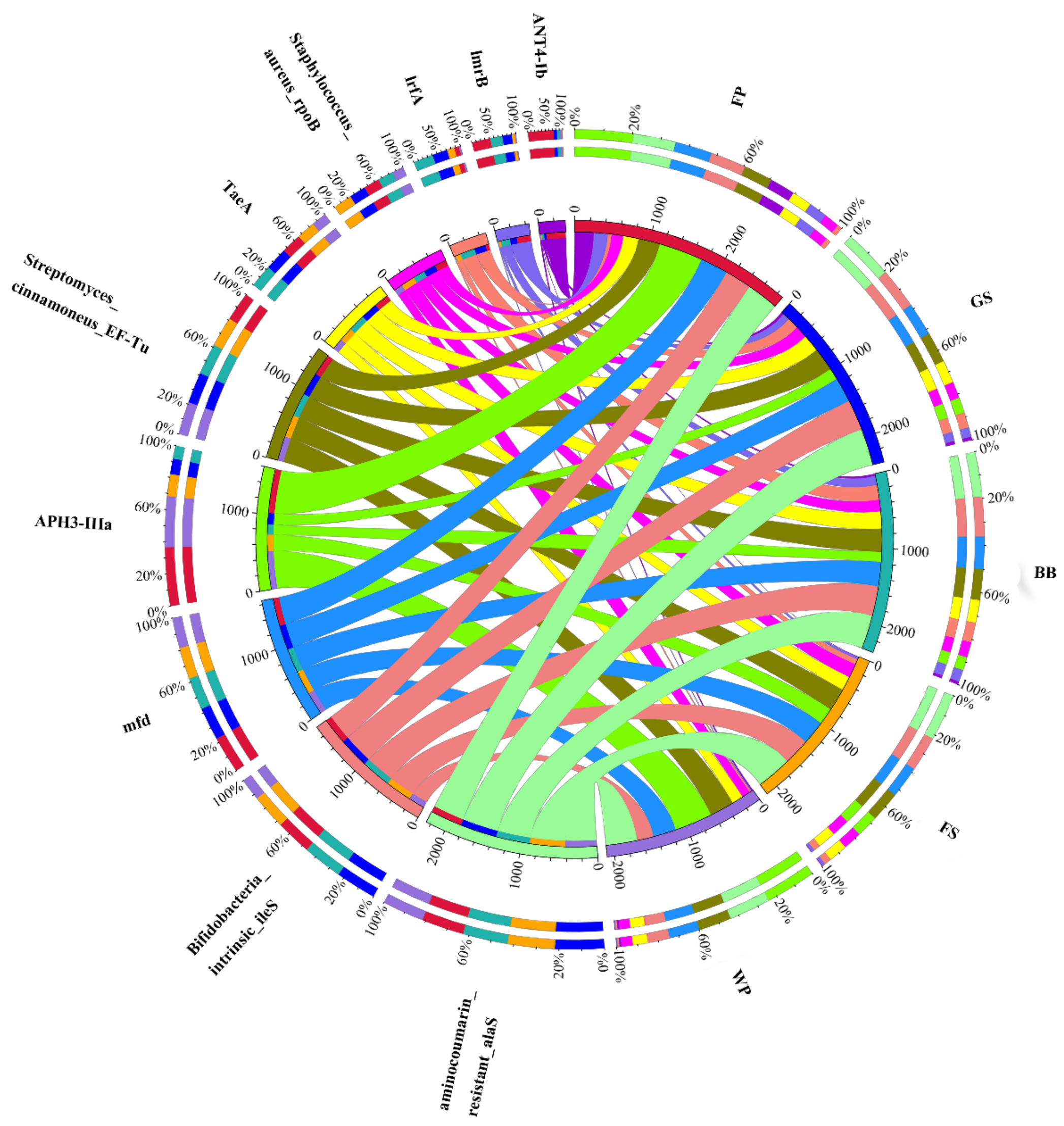
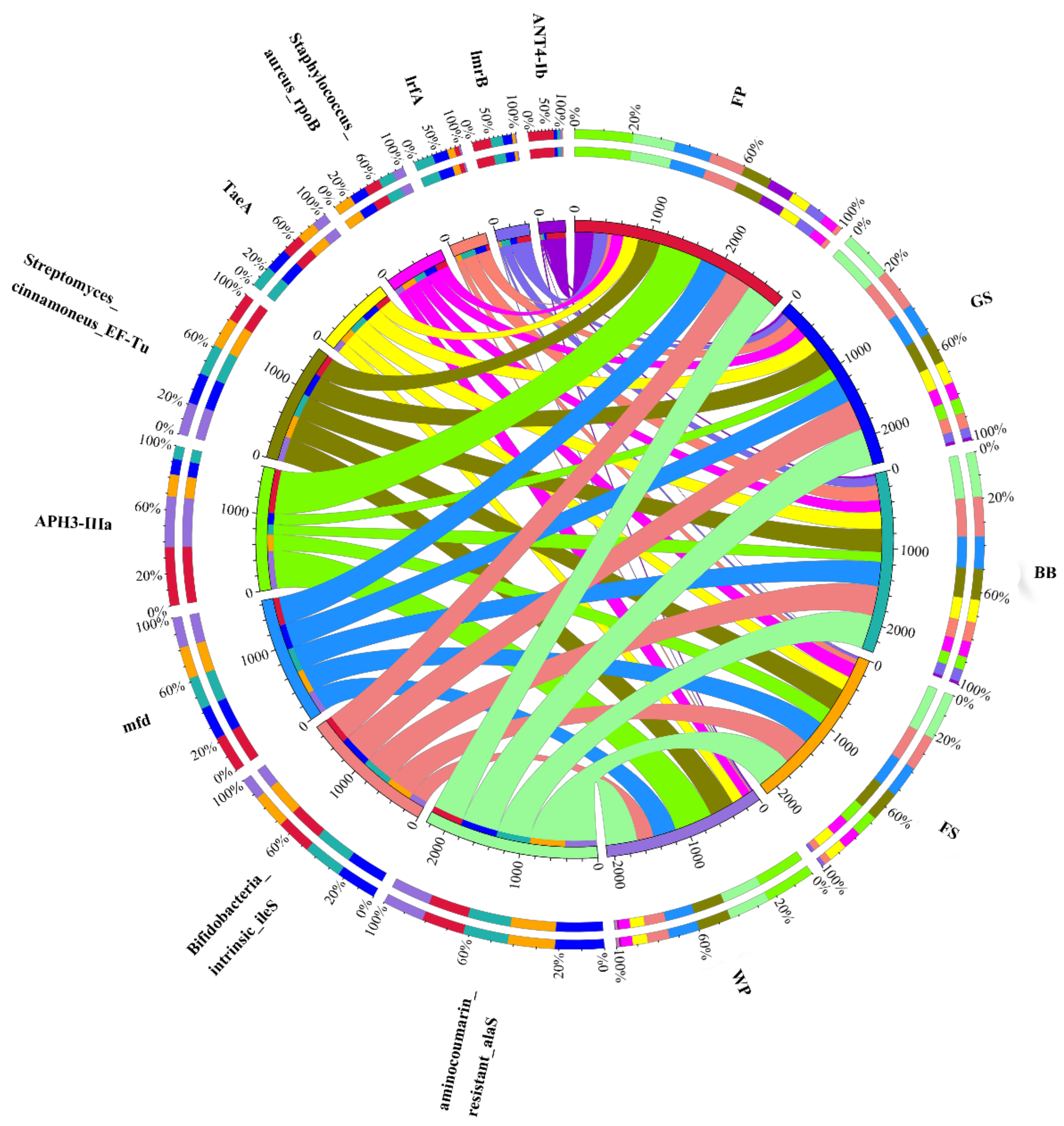
3.3. Occurrence, Abundance, and Diversity of ARGs
3.4. Co-Occurrence Pattern between ARGs and Microbial Community
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Heber, A.J.; Stroik, M. Size distribution and identification of aerial dust particles in swine finishing buildings. Trans. ASAE 1988, 31, 882–887. [Google Scholar] [CrossRef]
- Hong, P.Y.; Li, X. Monitoring airborne biotic contaminants in the indoor environment of pig and poultry confinement buildings. Environ. Microbiol. 2012, 14, 1420–1431. [Google Scholar] [CrossRef] [PubMed]
- Heederik, D.; Brouwer, R. Relationship of airborne endotoxin and bacteria levels in pig farms with the lung function and respiratory symptoms of farmers. Int. Arch. Occup. Environ. Health 1991, 62, 595–601. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.; Gant, T.W. A systematic review of the public health risks of bioaerosols from intensive farming. Int. J. Hyg. Environ. Health 2018, 221, 134–173. [Google Scholar] [CrossRef] [PubMed]
- Yoo, K.; Lee, T.K. Molecular approaches for the detection and monitoring of microbial communities in bioaerosols: A review. J. Environ. Sci. 2017, 51, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Nehme, B.; Létourneau, V. Culture-independent approach of the bacterial bioaerosol diversity in the standard swine confinement buildings, and assessment of the seasonal effect. Environ. Microbiol. 2008, 10, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, A.; Saunders, A.M. Community structure of bacteria and fungi in aerosols of a pig confinement building. FEMS Microbiol. Ecol. 2012, 80, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Kumari, P.; Choi, H.L. Seasonal variability in airborne biotic contaminants in swine confinement buildings. PLoS ONE 2014, 9, e112897. [Google Scholar] [CrossRef]
- Zhao, W.; Zhao, J. The dynamic distribution of porcine microbiota across different ages and gastrointestinal tract segments. PLoS ONE 2015, 10, e0117441. [Google Scholar] [CrossRef]
- Niu, Q.; Li, P. Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 2015, 5, 9938. [Google Scholar] [CrossRef]
- Kim, H.B.; Isaacson, R.E. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 2011, 153, 124–133. [Google Scholar] [CrossRef]
- Yang, Y.; Zhou, R. Characterization of airborne antibiotic resistance genes from typical bioaerosol emission sources in the urban environment using metagenomic approach. Chemosphere 2018, 213, 463–471. [Google Scholar] [CrossRef]
- Predicala, B.Z.; Goodband, R.D. Assessment of bioaerosols in swine barns by filtration and impaction. Curr. Microbiol. 2002, 44, 136–140. [Google Scholar] [CrossRef]
- Lee, S.A.; Reponen, T. Personal exposure to airborne dust and microorganisms in agricultural environments. J. Occup. Environ. Hyg. 2006, 3, 118–130. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.Q.; Zhu, K. Effect of microclimate on particulate matter, airborne bacteria, and odorous compounds in swine nursery houses. J. Anim. Sci. 2010, 88, 3707–3714. [Google Scholar] [CrossRef]
- Kumari, P.; Choi, H.L. Manure removal system influences the abundance and composition of airborne biotic contaminants in swine confinement buildings. Environ. Monit. Assess. 2015, 187, 537. [Google Scholar] [CrossRef]
- Poretsky, R.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, R.; Perkins, D.L. Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 2016, 469, 967–977. [Google Scholar] [CrossRef]
- Mbareche, H.; Brisebois, E. Bioaerosol sampling and detection methods based on molecular approaches: No pain no gain. Sci. Total Environ. 2017, 599–600, 2095–2104. [Google Scholar] [CrossRef] [PubMed]
- Taiganides, E.P. Pig Waste Management and Recycling: The Singapore Experience; IDRC: Ottawa, ON, Canada, 1992; Available online: https://www.unescap.org/cgi-bin/koha/opac-detail.pl?biblionumber=8704 (accessed on 20 December 2018).
- Liu, J.B.; Xue, P.C. Effects of dietary phosphorus concentration and body weight on postileal phosphorus digestion in pigs. Anim. Feed Sci. Technol. 2018, 242, 86–94. [Google Scholar] [CrossRef]
- Liu, J.B.; Yan, H.L. The response of performance in grower and finisher pigs to diets formulated to different tryptophan to lysine ratios. Livest. Sci. 2019, 222, 25–30. [Google Scholar] [CrossRef]
- Li, R.; Li, S. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 2010, 20, 265–272. [Google Scholar] [CrossRef]
- Noguchi, H.; Takagi, T. MetaGene: Prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 2006, 34, 5623–5630. [Google Scholar] [CrossRef]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef]
- Sunagawa, S.; Bork, P. Structure and function of the global ocean microbiome. Science 2015, 348, 1261359. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Wang, J. SOAP2: An improved ultrafast tool for short read alignment. Bioinformatics 2009, 25, 1966–1967. [Google Scholar] [CrossRef]
- Buchfink, B.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Schuster, S.C. MEGAN analysis of metagenomic data. Genome Res. 2007, 17, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Powell, S.; Szklarczyk, D.; Trachana, K.; Roth, A.; Kuhn, M.; Muller, J.; Arnold, R.; Rattei, T.; Letunic, I.; Doerks, T.; et al. eggNOG v3. 0: Orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2011, 40, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.W.; Su, H.J. Exposure of workers to airborne microorganisms in open-air swine houses. Appl. Environ. Microb. 2001, 67, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.L.; Shao, M.F. Airborne microbial communities in the atmospheric environment of urban hospitals in China. J. Hazard Mater. 2018, 349, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Yao, W. How host gender affects the bacterial community in pig feces and its correlation to skatole production. Ann. Microbiol. 2015, 65, 2379–2386. [Google Scholar] [CrossRef]
- Kim, J.; Unno, T. Analysis of swine fecal microbiota at various growth stages. Arch. Microbiol. 2015, 197, 753–759. [Google Scholar] [CrossRef] [PubMed]
- Leser, T.D.; Møller, K. Culture-independent analysis of gut bacteria: The pig gastrointestinal tract microbiota revisited. Appl. Environ. Microb. 2002, 68, 673–690. [Google Scholar] [CrossRef]
- Kim, H.B.; Isaacson, R.E. Microbial shifts in the swine distal gut in response to the treatment with antimicrobial growth promoter, tylosin. Proc. Natl. Acad. Sci. USA 2012, 6, 201205147. [Google Scholar] [CrossRef]
- Kim, S.J.; Park, H. Genome sequence of a novel member of the genus Psychrobacter isolated from Antarctic soil. J. Bacteriol. 2012, 194, 2403. [Google Scholar] [CrossRef] [PubMed]
- Dutkiewicz, J.; Wójtowicz, H. Airborne microorganisms and endotoxin in animal houses. Grana 1994, 33, 85–90. [Google Scholar] [CrossRef]
- Yooseph, S.; Fadrosh, D. A metagenomic framework for the study of airborne microbial communities. PLoS ONE 2013, 8, e81862. [Google Scholar] [CrossRef]
- Lamendella, R.; Oerther, D.B. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef]
- Fangman, T.J.; Coleman, J.L. Performance and disease status of pigs grown in a wean-to-finish facility compared to pigs grown in a conventional nursery and grower-finisher facility. J. Swine Health Prod. 2001, 9, 71–76. [Google Scholar]
- Zhu, Y.G.; Tiedje, J.M. Diverse and abundant antibiotic resistance genes in Chinese swine farms. Proc. Natl. Acad. Sci. India B 2013, 11, 201222743. [Google Scholar] [CrossRef] [PubMed]
- Farrington, L.A.; Inskip, P.D. Prevalence of antimicrobial resistance in Salmonellae isolated from market-age swine. J. Food Protect. 2001, 64, 1496–1502. [Google Scholar] [CrossRef]
- Jackson, C.R.; Ladely, S.R. High-level aminoglycoside resistant enterococci isolated from swine. Epidemiol. Infect. 2005, 133, 367–371. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Yuan, Z. Metagenomic analysis reveals wastewater treatment plants as hotspots of antibiotic resistance genes and mobile genetic elements. Water Res. 2017, 123, 468–478. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Lu, J. Metagenomic analysis of antibiotic resistance genes in coastal industrial mariculture systems. Bioresour. Technol. 2018, 253, 235–243. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, H.; Zhang, L.; Guo, Z.; Zhang, H.; Liu, J. Production Phase Affects the Bioaerosol Microbial Composition and Functional Potential in Swine Confinement Buildings. Animals 2019, 9, 90. https://doi.org/10.3390/ani9030090
Yan H, Zhang L, Guo Z, Zhang H, Liu J. Production Phase Affects the Bioaerosol Microbial Composition and Functional Potential in Swine Confinement Buildings. Animals. 2019; 9(3):90. https://doi.org/10.3390/ani9030090
Chicago/Turabian StyleYan, Honglin, Li Zhang, Zhendong Guo, Hongfu Zhang, and Jingbo Liu. 2019. "Production Phase Affects the Bioaerosol Microbial Composition and Functional Potential in Swine Confinement Buildings" Animals 9, no. 3: 90. https://doi.org/10.3390/ani9030090
APA StyleYan, H., Zhang, L., Guo, Z., Zhang, H., & Liu, J. (2019). Production Phase Affects the Bioaerosol Microbial Composition and Functional Potential in Swine Confinement Buildings. Animals, 9(3), 90. https://doi.org/10.3390/ani9030090