A Genome-Wide Detection of Copy Number Variations Using SNP Genotyping Arrays in Braque Français Type Pyrénées Dogs

 , , ,
, , ,  and
and
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Genotyping
2.2. CNV and CNVR Detection
2.3. Gene Contents and Functional Annotation
3. Results and Discussion
3.1. CNV and CNVR Detection
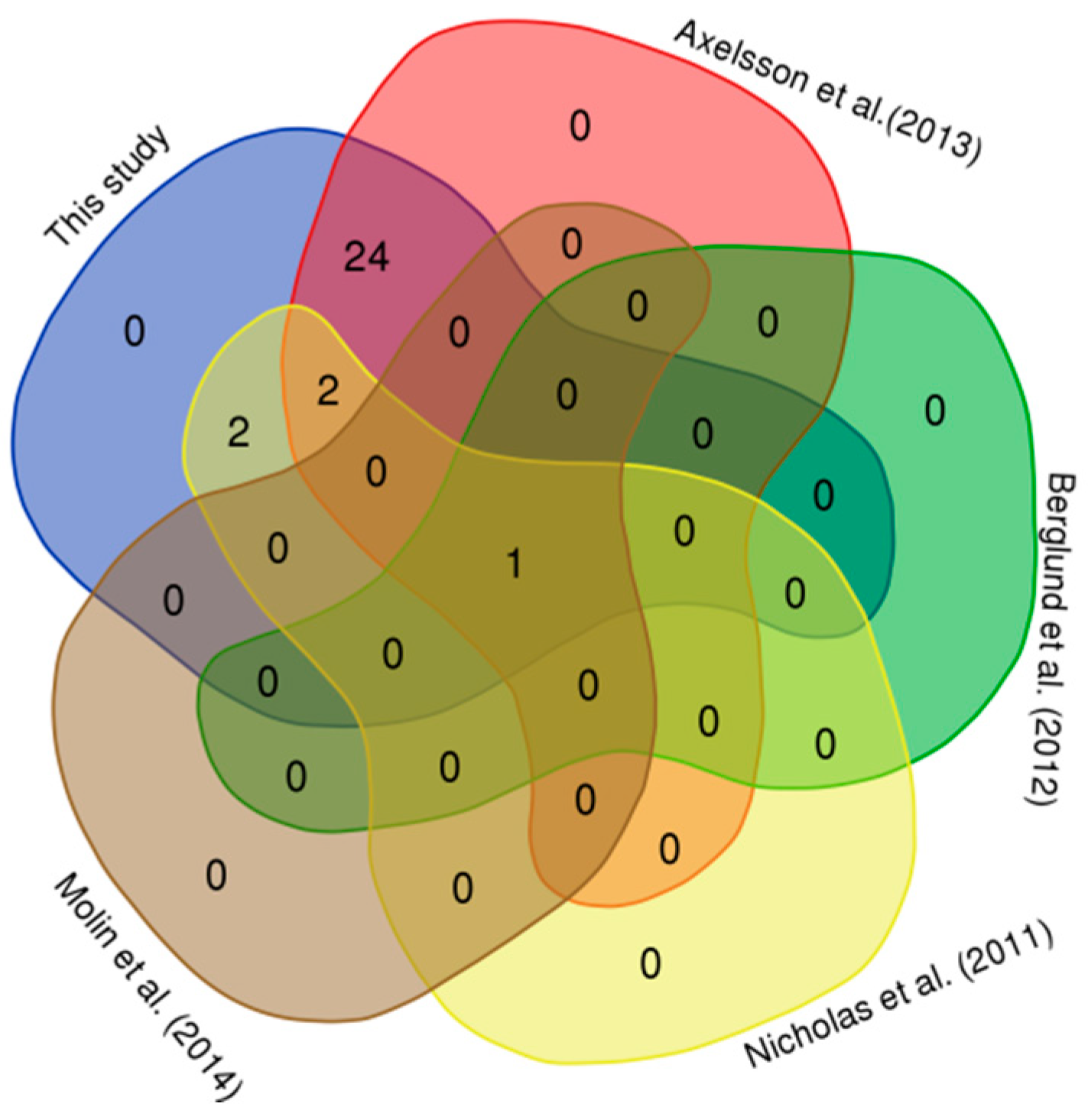
3.2. Comparison with Published CNVRs
3.3. CNVR Gene Content and Functional Annotations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Savolainen, P.; Zhang, Y.P.; Luo, J.; Lundeberg, J.; Leitner, T. Genetic evidence for an East Asian origin of domestic dogs. Science 2002, 298, 1610–1613. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, T.J.; Cheng, Z.; Ventura, M.; Mealey, K.; Eichler, E.E.; Akey, J.M. The genomic architecture of segmental duplications and associated copy number variants in dogs. Genome Res. 2009, 19, 491–499. [Google Scholar] [CrossRef] [PubMed]
- Vaysse, A.; Ratnakumar, A.; Derrien, T.; Axelsson, E.; Pielberg, G.R.; Sigurdsson, S.; Fall, T.; Seppälä, E.H.; Hansen, M.S.T.; Lawley, C.T.; et al. Identification of genomic regions associated with phenotypic variation between dog breeds using selection mapping. PLoS Genet. 2011, 7, e1002316. [Google Scholar] [CrossRef] [PubMed]
- Redon, R.; Ishikawa, S.; Fitch, K.R.; Feuk, L.; Perry, G.H.; Andrews, T.D.; Feigler, H.; Shapero, M.H.; Carson, A.R.; Chen, W.; et al. Global variation in copy number in the human genome. Nature 2006, 444, 444–454. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Jiang, J.; Yang, J.; Liu, X.; Wang, J.; Wang, H.; Fing, X.; Liu, J.; Zhang, Q. Genome-wide detection of copy number variations using high-density SNP genotyping platforms in Holsteins. BMC Genom. 2013, 14, 131. [Google Scholar] [CrossRef] [PubMed]
- Di Gerlando, R.; Sardina, M.T.; Tolone, M.; Sutera, A.M.; Mastrangelo, S.; Portolano, B. Genome-wide detection of copy-number variations in local cattle breeds. Anim. Prod. Sci. 2018. [Google Scholar] [CrossRef]
- Zhu, C.; Fan, H.; Yuan, Z.; Hu, S.; Ma, X.; Xuan, J.; Wang, H.; Zhang, L.; Wei, C.; Zhang, Q.; et al. Genome-wide detection of CNVs in Chinese indigenous sheep with different types of tails using ovine high-density 600 K SNP arrays. Sci. Rep. 2016, 6, 27822. [Google Scholar] [CrossRef] [PubMed]
- Paudel, Y.; Madsen, O.; Megens, H.J.; Frantz, L.A.; Bosse, M.; Bastiaansen, J.W.; Crooijmans, R.P.M.A.; Groenen, M.A.M. Evolutionary dynamics of copy number variation in pig genomes in the context of adaptation and domestication. BMC Genom. 2013, 14, 449. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.K.; Swartz, J.D.; Rush, L.J.; Alvarez, C.E. Mapping DNA structural variation in dogs. Genome Res. 2009, 19, 500–509. [Google Scholar] [CrossRef] [PubMed]
- Nicholas, T.J.; Baker, C.; Eichler, E.E.; Akey, J.M. A high-resolution integrated map of copy number polymorphisms within and between breeds of the modern domesticated dog. BMC Genom. 2011, 12, 414. [Google Scholar] [CrossRef] [PubMed]
- Berglund, J.; Nevalainen, E.M.; Molin, A.M.; Perloski, M.; André, C.; Zody, M.C.; Sharpe, T.; Hitte, C.; Lindblad-Toh, K.; The LUPA Consortium; et al. Novel origins of copy number variation in the dog genome. Genome Biol. 2012, 13, R73. [Google Scholar] [CrossRef] [PubMed]
- Molin, A.M.; Berglund, J.; Webster, M.T.; Lindblad-Toh, K. Genome-wide copy number variant discovery in dogs using the CanineHD genotyping array. BMC Genom. 2014, 15, 210. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Biscarini, F.; Auzino, B.; Ragatzu, M.; Spaterna, A.; Ciampolini, R. Genome-wide diversity and runs of homozygosity in the “Braque Français, type Pyrénées” dog breed. BMC Res. 2018, 11, 13. [Google Scholar] [CrossRef] [PubMed]
- Mastrangelo, S.; Biscarini, F.; Tolone, M.; Auzino, B.; Ragatzu, M.; Spaterna, A.; Ciampolini, R. Genomic characterisation of the Braque Français type Pyrénées dog and relationship with other breeds. PLoS ONE 2018, 13, e0208548. [Google Scholar] [CrossRef] [PubMed]
- Prinsen, R.T.M.M.; Strillacci, M.G.; Schiavini, F.; Santus, E.; Rossoni, A.; Maurer, V.; Bieber, A.; Gredler, B.; Dolezal, M.; Bagnato, A. A genome-wide scan of copy number variants using high-density SNPs in Brown Swiss dairy cattle. Livest. Sci. 2016, 191, 153–160. [Google Scholar] [CrossRef]
- Strillacci, M.G.; Gorla, E.; Cozzi, M.C.; Vevey, M.; Genova, F.; Scienski, K.; Longeri, M.; Bagnato, A. A copy number variant scan in the autochthonous Valdostana Red Pied cattle breed and comparison with specialized dairy populations. PLoS ONE 2018, 13, e0204669. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Wain, L.V.; Armour, J.A.; Tobin, M.D. Genomic copy number variation, human health, and disease. Lancet 2009, 374, 340–350. [Google Scholar] [CrossRef]
- Bagnato, A.; Strillacci, M.G.; Pellegrino, L.; Schiavini, F.; Frigo, E.; Rossoni, A.; Fontanesi, L.; Maltecca, C.; Prinsen, R.T.M.M.; Dolezal, M.A. Identification and validation of copy number variants in Italian Brown Swiss Dairy cattle using Illumina Bovine SNP50 BeadChip. Italian J. Anim. Sci. 2015, 14, 552–558. [Google Scholar] [CrossRef]
- Winchester, L.; Yau, C.; Ragoussis, J. Comparing CNV detection methods for SNP arrays. Brief. Funct. Genom. Proteomics 2009, 8, 353–366. [Google Scholar] [CrossRef] [PubMed]
- Axelsson, E.; Ratnakumar, A.; Arendt, M.L.; Maqbool, K.; Webster, M.T.; Perloski, M.; Liberg, O.; Arnemo, J.M.; Hedhammar, A.; Lindblad-Toh, K. The genomic signature of dog domestication reveals adaptation to a starch-rich diet. Nature 2013, 495, 360. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, C.E.; Akey, J.M. Copy number variation in the domestic dog. Mamm. Genome 2012, 23, 144–163. [Google Scholar] [CrossRef] [PubMed]
- Chaboissier, M.C.; Kobayashi, A.; Vidal, V.I.; Lützkendorf, S.; van de Kant, H.J.; Wegner, M.; de Rooij, D.G.; Richard, R.; Behringer, R.R.; Schedl, A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development 2004, 131, 1891–1901. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wu, D.D.; Boyko, A.R.; Wang, G.D.; Wu, S.F.; Irwin, D.M.; Zhang, Y.P. Population variation revealed high-altitude adaptation of Tibetan mastiffs. Mol. Biol. Evol. 2014, 31, 1200–1205. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Yang, X.; Li, Q.; Ma, Y.; Cui, S.; He, D.; Chang, J. Protein tyrosine phosphatase-like A regulates myoblast proliferation and differentiation through MyoG and the cell cycling signaling pathway. Mol. Cell. Biol. 2012, 32, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Tong, H.L.; Jiang, R.Y.; Zhang, W.W.; Yan, Y.Q. MiR-2425–5p targets RAD9A and MYOG to regulate the proliferation and differentiation of bovine skeletal muscle-derived satellite cells. Sci. Rep. 2017, 7, 418. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Fang, M.; Xu, H.; Xing, H.; Nie, Q. Transcriptome comparison in the pituitary–adrenal axis between Beagle and Chinese Field dogs after chronic stress exposure. Anim. Genet. 2015, 46, 522–534. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CNVRs | Chromosomes | Start Position (bp) | End Position (bp) | Nicholas et al. (2011) | Berglund et al. (2012) | Axelsson et al. (2013) | Molin et al. (2014) |
|---|---|---|---|---|---|---|---|
| 1 | chr1 | 11,200,412 | 11,215,077 | * | |||
| 4 | chr4 | 33,991,898 | 34,067,769 | * | |||
| 5 | chr6 | 39,482,383 | 39,941,098 | * | |||
| 6 | chr6 | 40,137,436 | 40,261,501 | * | |||
| 9 | chr7 | 790,325 | 836,217 | * | |||
| 10 | chr7 | 846,445 | 871,741 | * | |||
| 12 | chr9 | 615,993 | 795,784 | * | |||
| 13 | chr9 | 660,066 | 909,141 | * | |||
| 14 | chr9 | 916,372 | 997,748 | * | |||
| 17 | chr10 | 16,943,758 | 1,705,5639 | * | |||
| 18 | chr10 | 17,172,641 | 1,728,4700 | * | |||
| 20 | chr10 | 18,100,399 | 18,220,792 | * | * | ||
| 25 | chr14 | 853,721 | 1,000,647 | * | |||
| 26 | chr15 | 8,373,335 | 8,384,052 | * | |||
| 27 | chr16 | 383,954 | 442,252 | * | |||
| 28 | chr17 | 749,155 | 818,590 | * | |||
| 29 | chr17 | 39,880,141 | 3,990,5486 | * | |||
| 30 | chr18 | 25,644,344 | 25,740,090 | * | |||
| 32 | chr23 | 52,156,177 | 5,215,9756 | * | * | ||
| 33 | chr25 | 48,611,803 | 48,662,322 | * | |||
| 34 | chr25 | 50,314,369 | 50,482,865 | * | |||
| 35 | chr27 | 2,869,179 | 2,886,074 | * | |||
| 36 | chr27 | 25,695,551 | 2,581,0881 | * | * | * | * |
| 37 | chr28 | 40,299,547 | 40,325,378 | * | |||
| 38 | chr28 | 40,715,077 | 40,730,141 | * | |||
| 40 | chr30 | 39,280,736 | 39,318,243 | * | |||
| 42 | chr36 | 13,722,385 | 13,742,102 | * | |||
| 43 | chr37 | 30,065,232 | 30,112,232 | * | |||
| 44 | chr38 | 4,831,886 | 4,885,582 | * |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Gerlando, R.; Mastrangelo, S.; Sardina, M.T.; Ragatzu, M.; Spaterna, A.; Portolano, B.; Biscarini, F.; Ciampolini, R. A Genome-Wide Detection of Copy Number Variations Using SNP Genotyping Arrays in Braque Français Type Pyrénées Dogs. Animals 2019, 9, 77. https://doi.org/10.3390/ani9030077
Di Gerlando R, Mastrangelo S, Sardina MT, Ragatzu M, Spaterna A, Portolano B, Biscarini F, Ciampolini R. A Genome-Wide Detection of Copy Number Variations Using SNP Genotyping Arrays in Braque Français Type Pyrénées Dogs. Animals. 2019; 9(3):77. https://doi.org/10.3390/ani9030077
Chicago/Turabian StyleDi Gerlando, Rosalia, Salvatore Mastrangelo, Maria Teresa Sardina, Marco Ragatzu, Andrea Spaterna, Baldassare Portolano, Filippo Biscarini, and Roberta Ciampolini. 2019. "A Genome-Wide Detection of Copy Number Variations Using SNP Genotyping Arrays in Braque Français Type Pyrénées Dogs" Animals 9, no. 3: 77. https://doi.org/10.3390/ani9030077
APA StyleDi Gerlando, R., Mastrangelo, S., Sardina, M. T., Ragatzu, M., Spaterna, A., Portolano, B., Biscarini, F., & Ciampolini, R. (2019). A Genome-Wide Detection of Copy Number Variations Using SNP Genotyping Arrays in Braque Français Type Pyrénées Dogs. Animals, 9(3), 77. https://doi.org/10.3390/ani9030077