Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle

 ,
,  , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Korean Hanwoo Cattle Data
2.2. Statistical Analysis
2.3. Models
2.4. Variance Component and Heritability Estimation
2.5. Correlation
3. Results
3.1. Trait Summary and Heritability
3.1.1. Single-Trait Model
3.1.2. Multi-Trait Model
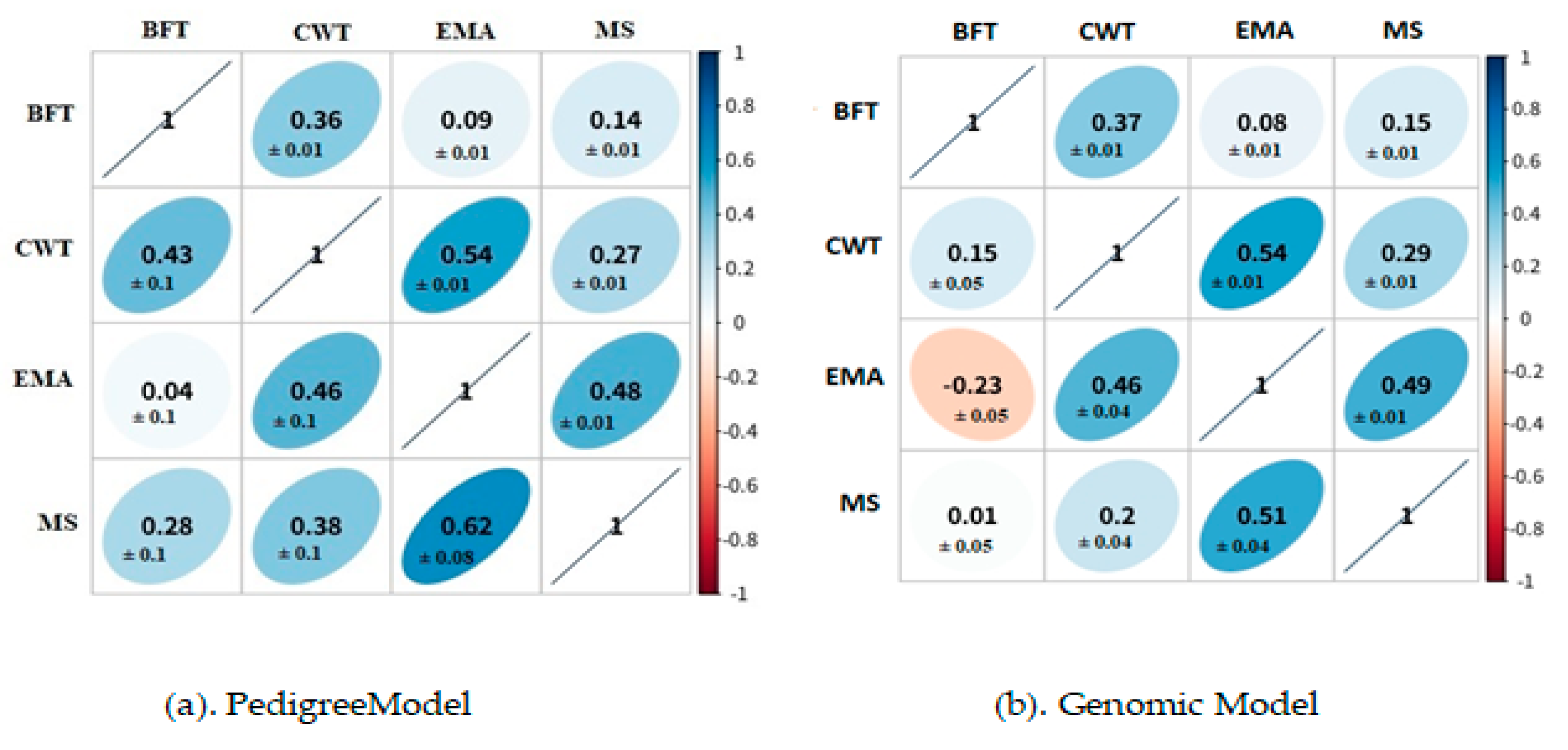
3.2. Genetic and Phenotypic Correlation
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lee, S.-H.; Park, B.-H.; Sharma, A.; Dang, C.-G.; Lee, S.-S.; Choi, T.-J.; Choy, Y.-H.; Kim, H.-C.; Jeon, K.-J.; Kim, S.-D. Hanwoo cattle: Origin, domestication, breeding strategies and genomic selection. J. Anim. Sci. Technol. 2014, 56, 2. [Google Scholar] [CrossRef] [PubMed]
- Soller, M.; Beckmann, J. Genetic polymorphism in varietal identification and genetic improvement. Theor. Appl. Genet. 1983, 67, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Calus, M.P.; Veerkamp, R.F. Accuracy of multi-trait genomic selection using different methods. Genet. Sel. Evol. 2011, 43, 26. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Jannink, J.-L. Multiple-Trait Genomic Selection Methods Increase Genetic Value Prediction Accuracy. Genetics 2012, 192, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Zhao, F.; Wang, Y.; Zhang, Y.; Du, L.; Su, G. Comparison of single-trait and multiple-trait genomic prediction models. BMC Genet. 2014, 15, 30. [Google Scholar] [CrossRef]
- Christensen, O.F.; Madsen, P.; Nielsen, B.; Ostersen, T.; Su, G. Single-step methods for genomic evaluation in pigs. Animal 2012, 6, 1565–1571. [Google Scholar] [CrossRef]
- Åkesson, M.; Bensch, S.; Hasselquist, D.; Tarka, M.; Hansson, B. Estimating heritabilities and genetic correlations: Comparing the ‘animal model’with parent-offspring regression using data from a natural population. PLoS ONE 2008, 3, e1739. [Google Scholar] [CrossRef]
- Hayes, B.; Goddard, M. Prediction of total genetic value using genome-wide dense marker maps. Genetics 2001, 157, 1819–1829. [Google Scholar]
- Ayalew, W.; Aliy, M.; Negussie, E. Estimation of genetic parameters of the productive and reproductive traits in Ethiopian Holstein using multi-trait models. Asian Australas. J. Anim. Sci. 2017, 30, 1550. [Google Scholar] [CrossRef]
- Sawyer, S.F. Analysis of variance: The fundamental concepts. J. Man. Manip. Ther. 2009, 17, 27E–38E. [Google Scholar] [CrossRef]
- Agresti, A. Categorical Data Analysis; John Wiley and Sons: New York, NY, USA, 1990. [Google Scholar]
- Misztal, I.; Tsuruta, S.; Lourenco, D.; Aguilar, I.; Legarra, A.; Vitezica, Z. Manual for BLUPF90 Family of Programs; University of Georgia: Athens, GA, USA, 2014. [Google Scholar]
- Misztal, I.; Tsuruta, S.; Strabel, T.; Auvray, B.; Druet, T.; Lee, D. BLUPF90 and related programs (BGF90). In Proceedings of the 7th World Congress on Genetics Applied to Livestock Production, Montpellier, France, 19–23 August 2002. [Google Scholar]
- VanRaden, P.M. Efficient methods to compute genomic predictions. J. Dairy Sci. 2008, 91, 4414–4423. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Christensen, O.F.; Ostersen, T.; Wang, Y.; Lund, M.S.; Su, G. Improving genetic evaluation of litter size and piglet mortality for both genotyped and nongenotyped individuals using a single-step method. J. Anim. Sci. 2015, 93, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Choi, T.; Alam, M.; Cho, C.; Lee, J.; Park, B.; Kim, S.; Koo, Y.; Roh, S. Genetic parameters for yearling weight, carcass traits, and primal-cut yields of Hanwoo cattle. J. Anim. Sci. 2015, 93, 1511–1521. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.H.; Lim, D.; Gondro, C.; Cho, Y.M.; Dang, C.G.; Sharma, A.; Jang, G.W.; Lee, K.T.; Yoon, D. Genome-wide association study identifies major loci for carcass weight on BTA14 in Hanwoo (Korean cattle). PLoS ONE 2013, 8, e74677. [Google Scholar] [CrossRef]
- Park, B.; Choi, T.; Kim, S.; Oh, S.-H. National genetic evaluation (system) of Hanwoo (Korean native cattle). Asian-Australas. J. Anim. Sci. 2013, 26, 151. [Google Scholar] [CrossRef]
- Bhuiyan, M.; Kim, H.; Lee, D.; Lee, S.; Cho, S.; Yang, B.; Kim, S.; Lee, S. Genetic parameters of carcass and meat quality traits in different muscles (longissimus dorsi and semimembranosus) of Hanwoo (Korean cattle). J. Anim. Sci. 2017, 95, 3359–3369. [Google Scholar]
- Oikawa, T.; Sanehira, T.; Sato, K.; Mizoguchi, Y.; Yamamoto, H.; Baba, M. Genetic parameters for growth and carcass traits of Japanese Black (Wagyu) cattle. Anim. Sci. 2000, 71, 59–64. [Google Scholar] [CrossRef]
- Júnior, G.A.F.; Rosa, G.J.; Valente, B.D.; Carvalheiro, R.; Baldi, F.; Garcia, D.A.; Gordo, D.G.; Espigolan, R.; Takada, L.; Tonussi, R.L. Genomic prediction of breeding values for carcass traits in Nellore cattle. Genet. Sel. Evol. 2016, 48, 7. [Google Scholar] [CrossRef]
- Do, C.; Park, B.; Kim, S.; Choi, T.; Yang, B.; Park, S.; Song, H. Genetic parameter estimates of carcass traits under national scale breeding scheme for beef cattle. Asian-Australas. J. Anim. Sci. 2016, 29, 1083. [Google Scholar] [CrossRef]
- Conner, J.K. quantitative genetic approaches to evolutionary constraint: how useful? Evolution 2012, 66, 3313–3320. [Google Scholar] [CrossRef]
- Hayashi, T.; Iwata, H. A Bayesian method and its variational approximation for prediction of genomic breeding values in multiple traits. BMC Bioinform. 2013, 14, 34. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
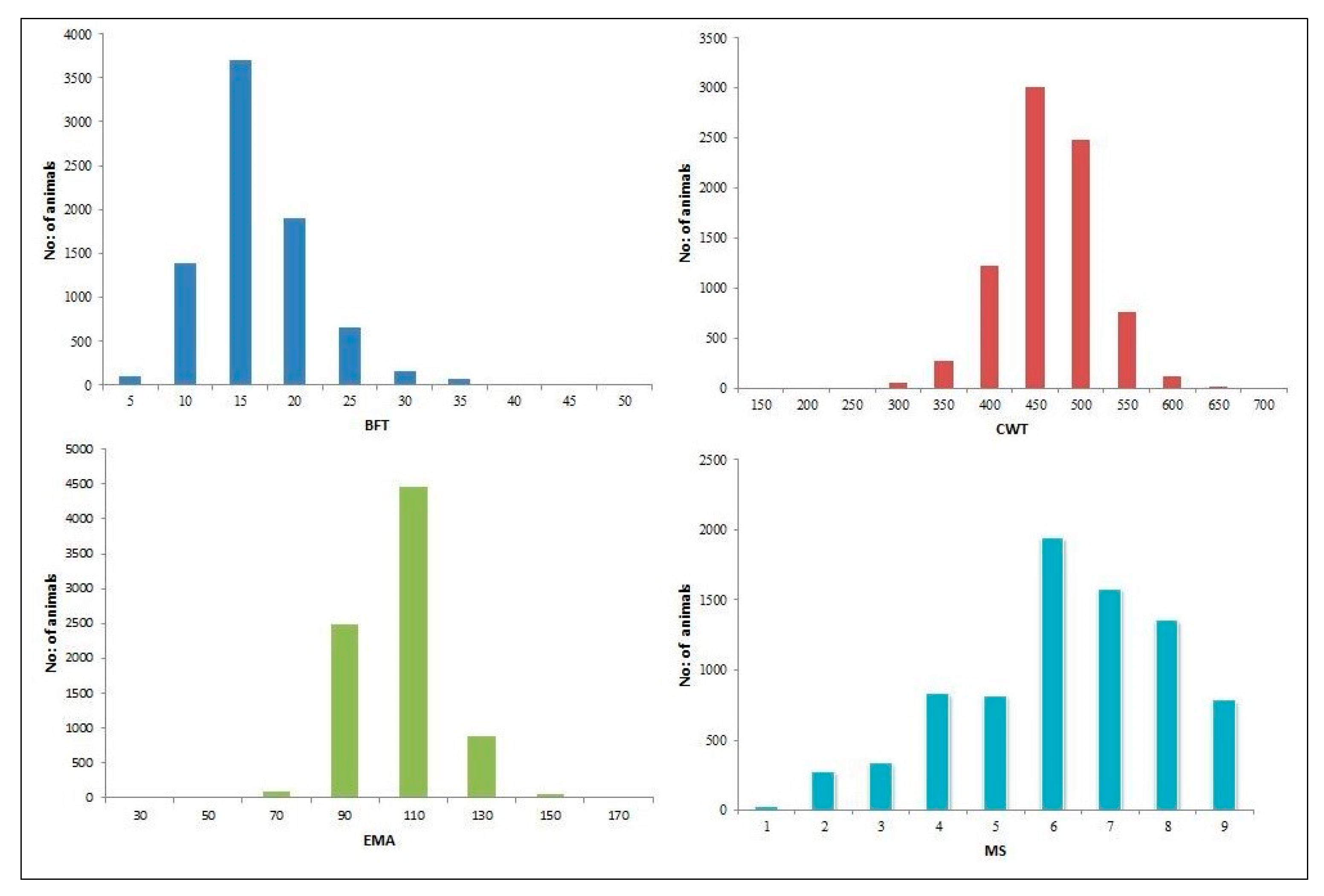
| Carcass Traits | Mean | SD | Min | Max | Number of Animals |
|---|---|---|---|---|---|
| BFT (mm) | 14.48 | 4.98 | 2 | 47 | 7991 |
| CWT (kg) | 440.94 | 52.36 | 159 | 692 | 7991 |
| EMA (cm2) | 96.29 | 12.13 | 34 | 156 | 7991 |
| MS (1–9) | 6.23 | 1.81 | 1 | 9 | 7991 |
| Model | Pedigree | Phenotype | Genotype | Single Trait | All Trait |
|---|---|---|---|---|---|
| STPM | Yes | Yes | No | Yes | No |
| MTPM | Yes | Yes | No | No | Yes |
| STGM | Yes | Yes | Yes | Yes | No |
| MTGM | Yes | Yes | Yes | No | Yes |
| Heading | Pedigree Model (PM) | Genomic Model (GM) | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BFT | CWT | EMA | MS | BFT | CWT | EMA | MS | |||||||||
| ST | MT | ST | MT | ST | MT | ST | MT | ST | MT | ST | MT | ST | MT | ST | MT | |
| 0.40 | 0.41 | 0.33 | 0.34 | 0.36 | 0.37 | 0.35 | 0.38 | 0.39 | 0.39 | 0.39 | 0.39 | 0.39 | 0.39 | 0.46 | 0.46 | |
| s.e | 0.06 | 0.06 | 0.05 | 0.04 | 0.05 | 0.06 | 0.05 | 0.05 | 0.03 | 0.02 | 0.02 | 0.02 | 0.02 | 0.02 | 0.03 | 0.03 |
| 9.5 | 9.7 | 684.6 | 704.2 | 47.9 | 49.1 | 0.98 | 1.08 | 8.85 | 8.99 | 789.78 | 785.59 | 49.81 | 49.87 | 1.31 | 1.31 | |
| 14.2 | 14.1 | 1407.5 | 1391.6 | 83.7 | 82.7 | 1.84 | 1.76 | 14.18 | 14.08 | 1220.2 | 1222.4 | 78.7 | 78.64 | 1.52 | 1.52 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Srivastava, S.; Lopez, B.I.; Heras-Saldana, S.d.l.; Park, J.-E.; Shin, D.-H.; Chai, H.-H.; Park, W.; Lee, S.-H.; Lim, D. Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle. Animals 2019, 9, 1061. https://doi.org/10.3390/ani9121061
Srivastava S, Lopez BI, Heras-Saldana Sdl, Park J-E, Shin D-H, Chai H-H, Park W, Lee S-H, Lim D. Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle. Animals. 2019; 9(12):1061. https://doi.org/10.3390/ani9121061
Chicago/Turabian StyleSrivastava, Swati, Bryan Irvine Lopez, Sara de las Heras-Saldana, Jong-Eun Park, Dong-Hyun Shin, Han-Ha Chai, Woncheol Park, Seung-Hwan Lee, and Dajeong Lim. 2019. "Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle" Animals 9, no. 12: 1061. https://doi.org/10.3390/ani9121061
APA StyleSrivastava, S., Lopez, B. I., Heras-Saldana, S. d. l., Park, J.-E., Shin, D.-H., Chai, H.-H., Park, W., Lee, S.-H., & Lim, D. (2019). Estimation of Genetic Parameters by Single-Trait and Multi-Trait Models for Carcass Traits in Hanwoo Cattle. Animals, 9(12), 1061. https://doi.org/10.3390/ani9121061