

Development and Application of a 40 K Liquid Capture Chip for Beef Cattle
 , , and
, , and 
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Sampling
2.2. SNP Discovery
2.3. Development of Liquid Capture Chip Panel
2.4. Validation of the 40K Chip Panel
2.5. Population Genetic Analysis
2.6. Identification of Runs of Homozygosity
2.7. Detection and Analyses of Common Runs of Homozygosity
3. Results
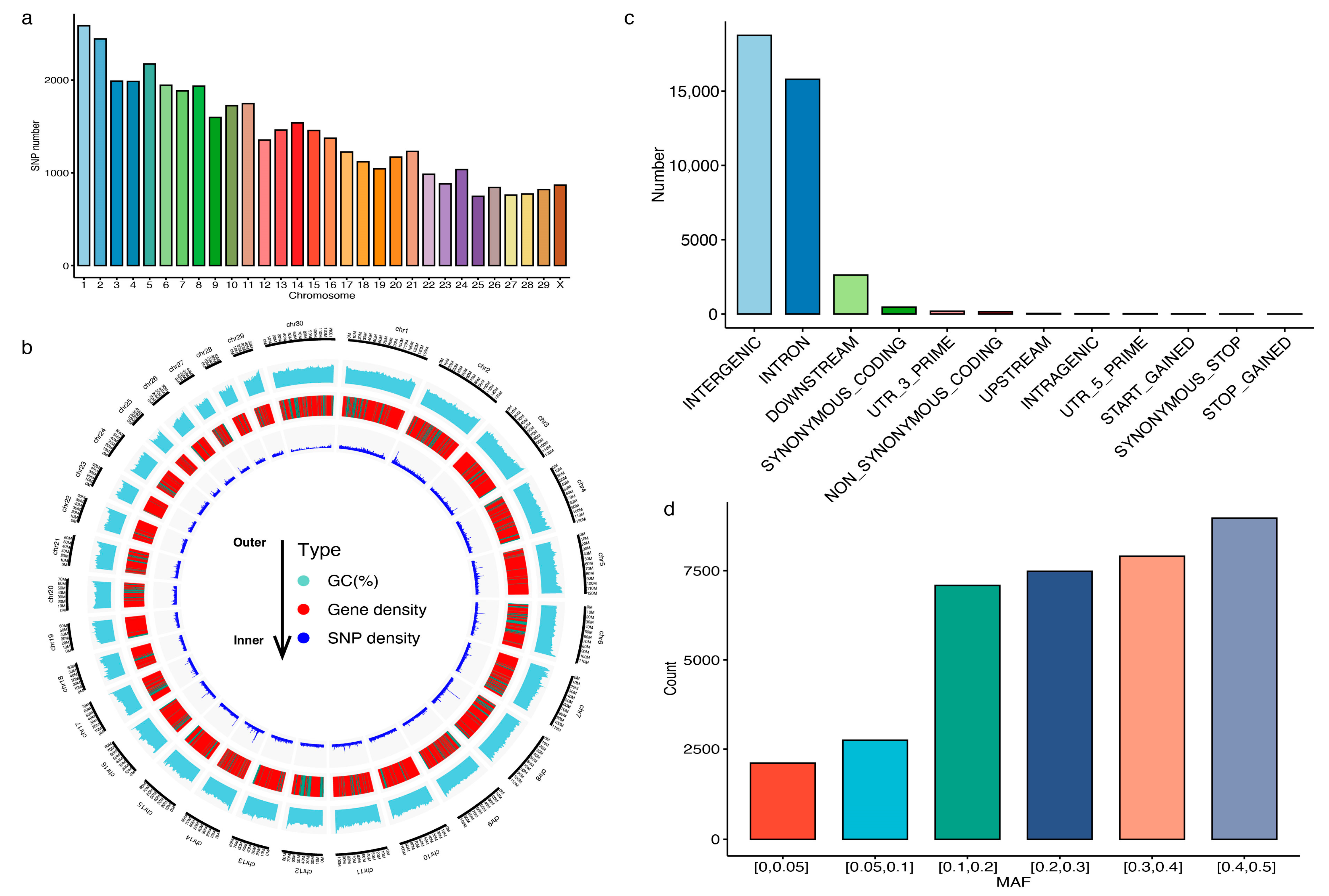
3.1. Characterization of the Customed 40K Chip Panel
3.2. Genotyping Performance of the 40K Chip Panel
3.3. Population Structure Analysis Based on the Chip Data
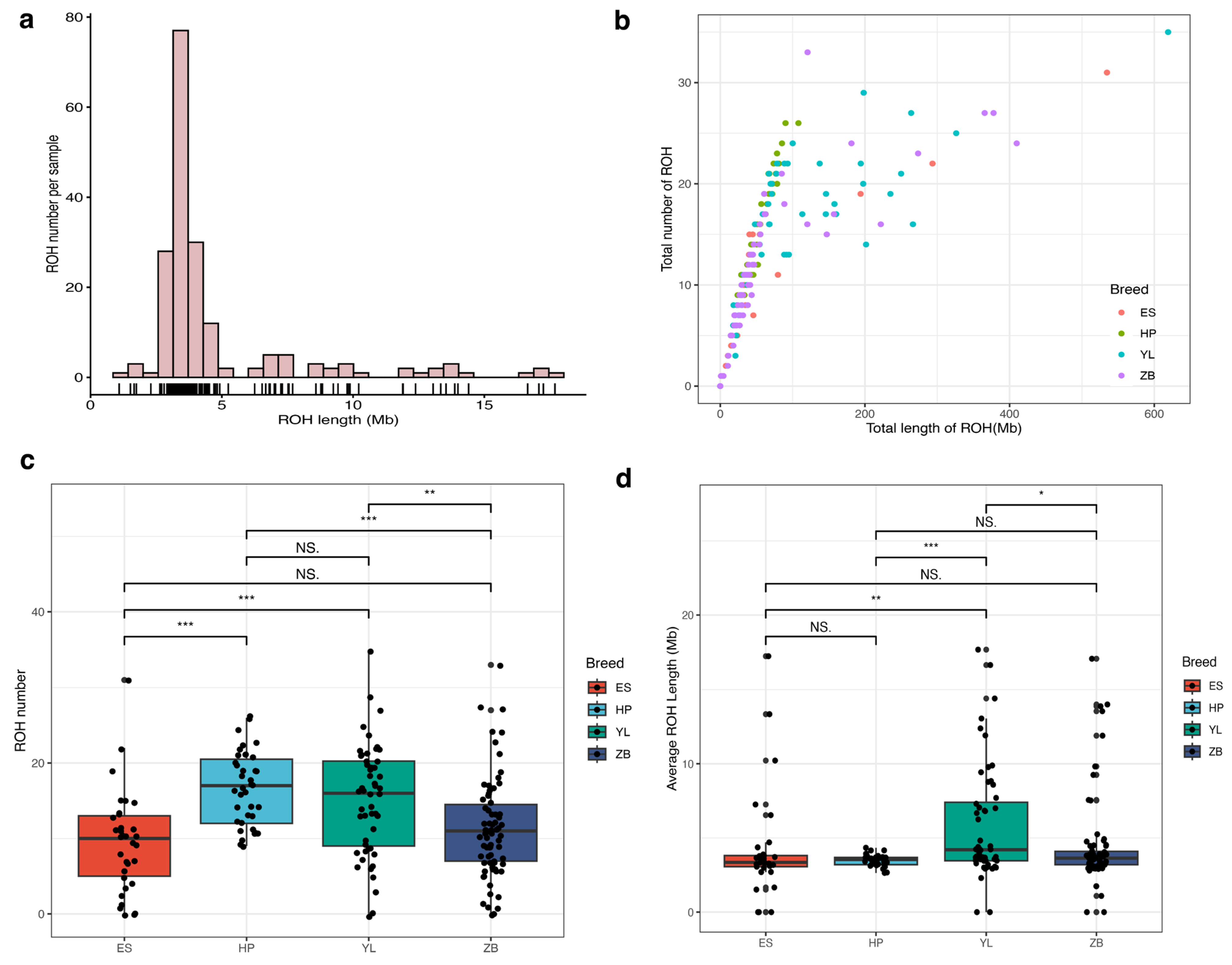
3.4. Runs of Homozygosity Analysis Based on Chip Data
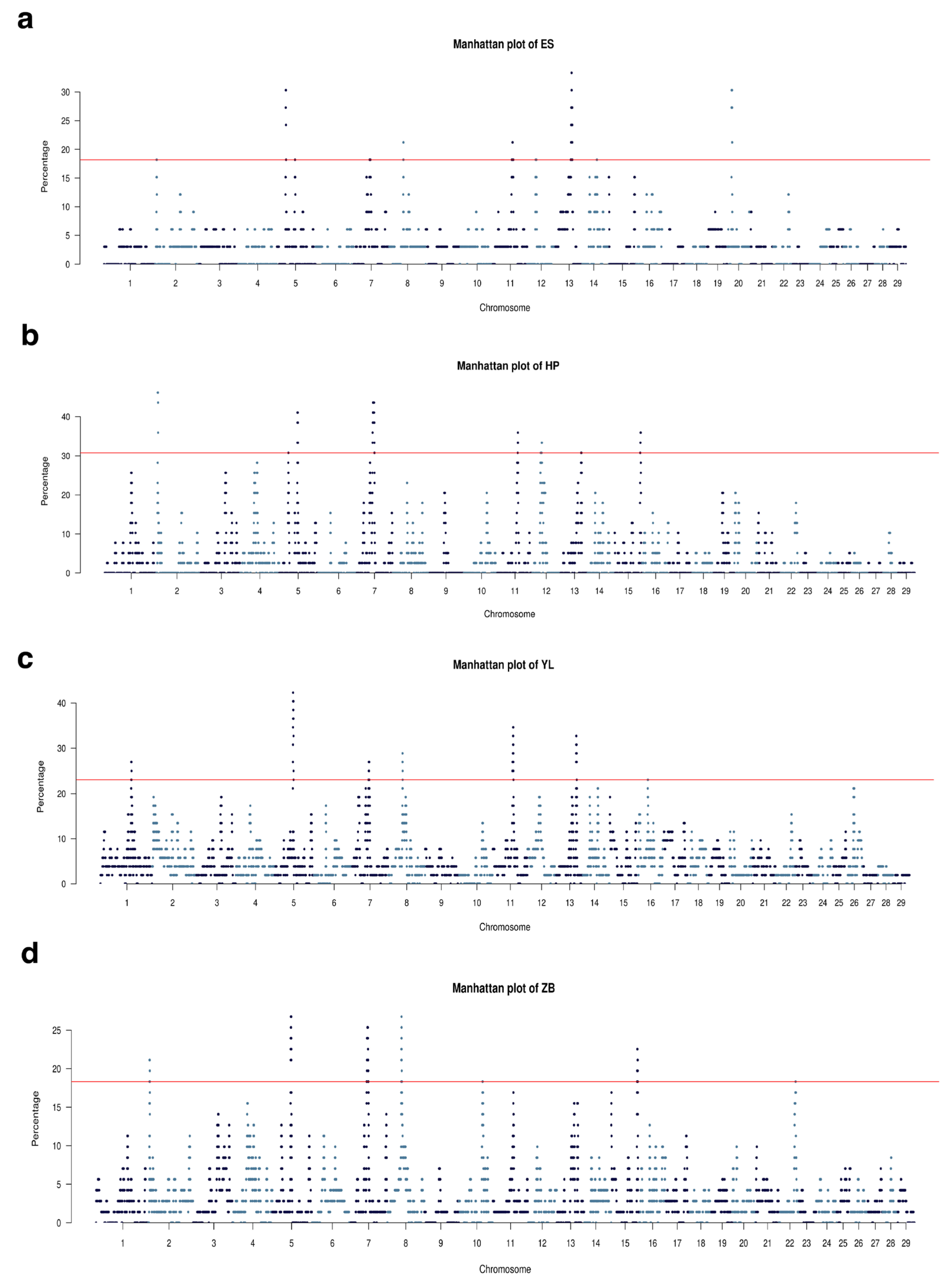
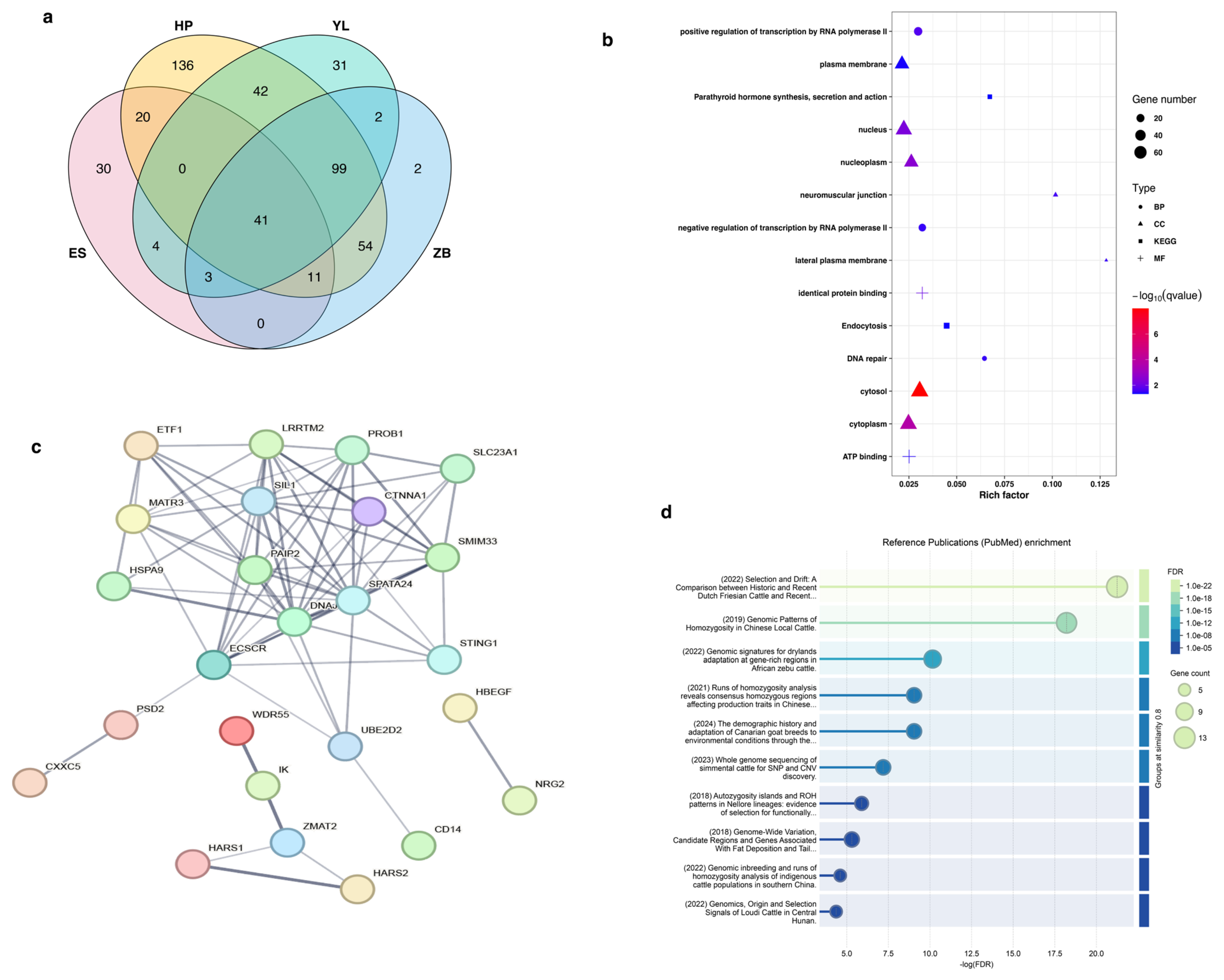
3.5. Runs of Homozygosity Involved in Economically Important Traits
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, H.; Wu, H.; Zhang, W.; Jiang, J.; Qian, H.; Man, C.; Gao, H.; Chen, Q.; Du, L.; Chen, S.; et al. Development and Validation of a 5K Low-Density SNP Chip for Hainan Cattle. BMC Genom. 2024, 25, 873. [Google Scholar] [CrossRef] [PubMed]
- Burrow, H.; Goddard, M. Application of Genetics and Genomics in Livestock Production. Agriculture 2023, 13, 386. [Google Scholar] [CrossRef]
- Nayak, S.S.; Rajawat, D.; Jain, K.; Sharma, A.; Gondro, C.; Tarafdar, A.; Dutt, T.; Panigrahi, M. A Comprehensive Review of Livestock Development: Insights into Domestication, Phylogenetics, Diversity, and Genomic Advances. Mamm. Genome 2024, 35, 577–599. [Google Scholar] [CrossRef] [PubMed]
- Jones, H.E.; Wilson, P.B. Progress and Opportunities through Use of Genomics in Animal Production. Trends Genet. 2022, 38, 1228–1252. [Google Scholar] [CrossRef]
- Deng, T.X.; Ma, X.Y.; Lu, X.R.; Duan, A.Q.; Shokrollahi, B.; Shang, J.H. Signatures of Selection Reveal Candidate Genes Involved in Production Traits in Chinese Crossbred Buffaloes. J. Dairy. Sci. 2022, 105, 1327–1337. [Google Scholar] [CrossRef]
- Tolone, M.; Sardina, M.T.; Criscione, A.; Lasagna, E.; Senczuk, G.; Rizzuto, I.; Riggio, S.; Moscarelli, A.; Macaluso, V.; Di Gerlando, R.; et al. High-Density Single Nucleotide Polymorphism Markers Reveal the Population Structure of 2 Local Chicken Genetic Resources. Poult. Sci. 2023, 102, 102692. [Google Scholar] [CrossRef]
- More, M.; Veli, E.; Cruz, A.; Gutiérrez, J.P.; Gutiérrez, G.; Ponce de León, F.A. Genome-Wide Association Study of Fiber Diameter in Alpacas. Animal 2023, 13, 3316. [Google Scholar] [CrossRef]
- Neumann, G.B.; Korkuć, P.; Arends, D.; Wolf, M.J.; May, K.; Reißmann, M.; Elzaki, S.; König, S.; Brockmann, G.A. Design and Performance of a Bovine 200 k SNP Chip Developed for Endangered German Black Pied Cattle (DSN). BMC Genom. 2021, 22, 905. [Google Scholar] [CrossRef]
- Anderson, R.M. Development of a High Density (600K) Illumina Ovine SNP Chip and Its Use to Fine Map the Yellow Fat Locus. In Proceedings of the Plant and Animal Genome XXII Conference, San Diego, CA, USA, 12–17 January 2014. [Google Scholar]
- Qiao, X.; Su, R.; Wang, Y.; Wang, R.; Yang, T.; Li, X.; Chen, W.; He, S.; Jiang, Y.; Xu, Q.; et al. Genome-Wide Target Enrichment-Aided Chip Design: A 66 K SNP Chip for Cashmere Goat. Sci. Rep. 2017, 7, 8621. [Google Scholar] [CrossRef]
- Ramos, A.M.; Crooijmans, R.P.M.A.; Affara, N.A.; Amaral, A.J.; Archibald, A.L.; Beever, J.E.; Bendixen, C.; Churcher, C.; Clark, R.; Dehais, P.; et al. Design of a High Density SNP Genotyping Assay in the Pig Using SNPs Identified and Characterized by next Generation Sequencing Technology. PLoS ONE 2009, 4, e6524. [Google Scholar] [CrossRef]
- Iamartino, D.; Nicolazzi, E.L.; Van Tassell, C.P.; Reecy, J.M.; Fritz-Waters, E.R.; Koltes, J.E.; Biffani, S.; Sonstegard, T.S.; Schroeder, S.G.; Ajmone-Marsan, P.; et al. Design and Validation of a 90K SNP Genotyping Assay for the Water Buffalo (Bubalus Bubalis). PLoS ONE 2017, 12, e0185220. [Google Scholar] [CrossRef]
- Cañas-Álvarez, J.J.; González-Rodríguez, A.; Munilla, S.; Varona, L.; Díaz, C.; Baro, J.A.; Altarriba, J.; Molina, A.; Piedrafita, J. Genetic Diversity and Divergence among Spanish Beef Cattle Breeds Assessed by a Bovine High-Density SNP Chip. J. Anim. Sci. 2015, 93, 5164–5174. [Google Scholar] [CrossRef]
- Liu, B.; Tao, W.; Feng, D.; Wang, Y.; Heizatuola, N.; Ahemetbai, T.; Wu, W. Revealing Genetic Diversity and Population Structure of Endangered Altay White-Headed Cattle Population Using 100 k SNP Markers. Animals 2022, 12, 3214. [Google Scholar] [CrossRef]
- Ramey, H.R.; Decker, J.E.; McKay, S.D.; Rolf, M.M.; Schnabel, R.D.; Taylor, J.F. Detection of Selective Sweeps in Cattle Using Genome-Wide SNP Data. BMC Genom. 2013, 14, 382. [Google Scholar] [CrossRef]
- Schwarz, L.; Križanac, A.-M.; Schneider, H.; Falker-Gieske, C.; Heise, J.; Liu, Z.; Bennewitz, J.; Thaller, G.; Tetens, J. Genetic and Genomic Analysis of Reproduction Traits in Holstein Cattle Using SNP Chip Data and Imputed Sequence Level Genotypes. BMC Genom. 2024, 25, 880. [Google Scholar] [CrossRef]
- Teng, J.; Wang, D.; Zhao, C.; Zhang, X.; Chen, Z.; Liu, J.; Sun, D.; Tang, H.; Wang, W.; Li, J.; et al. Longitudinal Genome-Wide Association Studies of Milk Production Traits in Holstein Cattle Using Whole-Genome Sequence Data Imputed from Medium-Density Chip Data. J. Dairy. Sci. 2023, 106, 2535–2550. [Google Scholar] [CrossRef]
- Ghoreishifar, S.M.; Moradi-Shahrbabak, H.; Fallahi, M.H.; Jalil Sarghale, A.; Moradi-Shahrbabak, M.; Abdollahi-Arpanahi, R.; Khansefid, M. Genomic Measures of Inbreeding Coefficients and Genome-Wide Scan for Runs of Homozygosity Islands in Iranian River Buffalo, Bubalus Bubalis. BMC Genet. 2020, 21, 16. [Google Scholar] [CrossRef]
- Oliveira, C.S.; Camargo, L.S.A.; da Silva, M.V.G.B.; Saraiva, N.Z.; Quintão, C.C.; Machado, M.A. Embryo Biopsies for Genomic Selection in Tropical Dairy Cattle. Anim. Reprod. 2023, 20, e20230064. [Google Scholar] [CrossRef]
- Balog, K.; Mizeranschi, A.E.; Wanjala, G.; Sipos, B.; Kusza, S.; Bagi, Z. Application Potential of Chicken DNA Chip in Domestic Pigeon Species - Preliminary Results. Saudi J. Biol. Sci. 2023, 30, 103594. [Google Scholar] [CrossRef]
- Chen, Y.; Guo, Y.; Ge, F.; Gao, H.; Zhou, J.; Wu, X.; Qian, C.; Wang, Z.; Wang, Z.; Zhu, B.; et al. Developing a Liquid Capture Chip to Accelerate the Genetic Progress of Cattle. Anim. Res. One Health 2024, 2, 204–216. [Google Scholar] [CrossRef]
- Wang, F.; Guo, Y.; Liu, Z.; Wang, Q.; Jiang, Y.; Zhao, G. New Insights into the Novel Sequences of the Chicken Pan-Genome by Liquid Chip. J. Anim. Sci. 2022, 100, skac336. [Google Scholar] [CrossRef]
- Zhang, Z.; Xing, S.; Qiu, A.; Zhang, N.; Wang, W.; Qian, C.; Zhang, J.; Wang, C.; Zhang, Q.; Ding, X. The Development of a Porcine 50K SNP Panel Using Genotyping by Target Sequencing and Its Application1. J. Integr. Agric. 2023, 24, 1930–1943. [Google Scholar] [CrossRef]
- Guo, Y.; Bai, F.; Wang, J.; Fu, S.; Zhang, Y.; Liu, X.; Zhang, Z.; Shao, J.; Li, R.; Wang, F.; et al. Design and Characterization of a High-Resolution Multiple-SNP Capture Array by Target Sequencing for Sheep. J. Anim. Sci. 2023, 101, skac383. [Google Scholar] [CrossRef]
- Guan, S.; Li, W.; Jin, H.; Zhang, L.; Liu, G. Development and Validation of a 54K Genome-Wide Liquid SNP Chip Panel by Target Sequencing for Dairy Goat. Genes 2023, 14, 1122. [Google Scholar] [CrossRef]
- Meng, Y.; Zhang, W.; Cheng, Y.; Wu, Y.; Wu, H.; He, M.; Chen, S.; Man, C.; Gao, H.; Du, L.; et al. Development and Verification of a 10K Liquid Chip for Hainan Black Goat Based on Genotyping by Pinpoint Sequencing of Liquid Captured Targets. BMC Genom. Data 2024, 25, 44. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Vasimuddin, M.; Misra, S.; Li, H.; Aluru, S. Efficient Architecture-Aware Acceleration of BWA-MEM for Multicore Systems. In Proceedings of the 2019 IEEE international parallel and distributed processing symposium (IPDPS), Rio de Janeiro, Brazil, 20–24 May 2019; pp. 314–324. [Google Scholar]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce Framework for Analyzing next-Generation DNA Sequencing Data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Kavakiotis, I.; Triantafyllidis, A.; Ntelidou, D.; Alexandri, P.; Megens, H.-J.; Crooijmans, R.P.M.A.; Groenen, M.A.M.; Tsoumakas, G.; Vlahavas, I. TRES: Identification of Discriminatory and Informative SNPs from Population Genomic Data. J. Hered. 2015, 106, 672–676. [Google Scholar] [CrossRef]
- Cai, W.; Zhang, Y.; Chang, T.; Wang, Z.; Zhu, B.; Chen, Y.; Gao, X.; Xu, L.; Zhang, L.; Gao, H.; et al. The eQTL Colocalization and Transcriptome-Wide Association Study Identify Potentially Causal Genes Responsible for Economic Traits in Simmental Beef Cattle. J. Anim. Sci. Biotechnol. 2023, 14, 78. [Google Scholar] [CrossRef]
- Liu, R.; Xing, S.; Wang, J.; Zheng, M.; Cui, H.; Crooijmans, R.P.M.A.; Li, Q.; Zhao, G.; Wen, J. A New Chicken 55K SNP Genotyping Array. BMC Genom. 2019, 20, 410. [Google Scholar] [CrossRef]
- Weimin, H. VCF2Dis. Available online: https://github.com/BGI-shenzhen/VCFDis (accessed on 12 December 2024).
- Alexander, D.H.; Novembre, J.; Lange, K. Fast Model-Based Estimation of Ancestry in Unrelated Individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A Fast and Effective Tool for Linkage Disequilibrium Decay Analysis Based on Variant Call Format Files. Bioinformatics 2018, 35, 1786–1788. [Google Scholar] [CrossRef]
- Xu, L.; Zhao, G.; Yang, L.; Zhu, B.; Chen, Y.; Zhang, L.; Gao, X.; Gao, H.; Liu, G.E.; Li, J. Genomic Patterns of Homozygosity in Chinese Local Cattle. Sci. Rep. 2019, 9, 16977. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A Flexible Suite of Utilities for Comparing Genomic Features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Bu, D.; Luo, H.; Huo, P.; Wang, Z.; Zhang, S.; He, Z.; Wu, Y.; Zhao, L.; Liu, J.; Guo, J.; et al. KOBAS-i: Intelligent Prioritization and Exploratory Visualization of Biological Functions for Gene Enrichment Analysis. Nucleic Acids Res. 2021, 49, W317–W325. [Google Scholar] [CrossRef]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of Homozygosity and Population History in Cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Cole, J.B.; Wiggans, G.R.; Ma, L.; Sonstegard, T.S.; Lawlor, T.J.; Crooker, B.A.; Van Tassell, C.P.; Yang, J.; Wang, S.; Matukumalli, L.K.; et al. Genome-Wide Association Analysis of Thirty One Production, Health, Reproduction and Body Conformation Traits in Contemporary U.S. Holstein Cows. BMC Genom. 2011, 12, 408. [Google Scholar] [CrossRef]
- Li, B.; VanRaden, P.M.; Null, D.J.; O’Connell, J.R.; Cole, J.B. Major Quantitative Trait Loci Influencing Milk Production and Conformation Traits in Guernsey Dairy Cattle Detected on Bos Taurus Autosome 19. J. Dairy. Sci. 2021, 104, 550–560. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Han, B.; Liu, L.; Zhao, F.; Liang, W.; Jiang, J.; Yang, Y.; Ma, Z.; Sun, D. Genetic Association of DDIT3, RPL23A, SESN2 and NR4A1 Genes with Milk Yield and Composition in Dairy Cattle. Anim. Genet. 2019, 50, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Liu, Y.; Zheng, W.; Han, B.; Wang, K.; Sun, D. Identification of Genetic Effects of ACADVL and IRF6 Genes with Milk Production Traits of Holstein Cattle in China. Genes 2022, 13, 2393. [Google Scholar] [CrossRef] [PubMed]
- Baeza, M.C.; Corva, P.M.; Soria, L.A.; Rincon, G.; Medrano, J.F.; Pavan, E.; Villarreal, E.L.; Schor, A.; Melucci, L.; Mezzadra, C.; et al. Genetic Markers of Body Composition and Carcass Quality in Grazing Brangus Steers. Genet. Mol. Res. 2011, 10, 3146–3156. [Google Scholar] [CrossRef]
- Rincon, G.; Farber, E.A.; Farber, C.R.; Nkrumah, J.D.; Medrano, J.F. Polymorphisms in the STAT6 Gene and Their Association with Carcass Traits in Feedlot Cattle. Anim. Genet. 2009, 40, 878–882. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Itoh, T.; Fujimori, Y.; Satoh, M.; Miyazaki, Y.; Takahashi, H.; Shimizu, K.; Malau-Aduli, A.E.O.; Morita, M. Genetic Association between GHSR1a 5’UTR-Microsatellite and Nt-7(C>A) Loci and Growth and Carcass Traits in Japanese Black Cattle. Anim. Sci. J. 2011, 82, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fan, T.; Du, Z.; Xu, L.; Chen, Y.; Zhang, L.; Gao, H.; Li, J.; Ma, Y.; Gao, X. Genome-Wide Association Analysis Identifies the PMEL Gene Affecting Coat Color and Birth Weight in Simmental × Holstein. Animal 2023, 13, 3821. [Google Scholar] [CrossRef]
- Porto-Neto, L.R.; Edwards, S.; Fortes, M.R.S.; Lehnert, S.A.; Reverter, A.; McGowan, M. Genome-Wide Association for the Outcome of Fixed-Time Artificial Insemination of Brahman Heifers in Northern Australia. J. Anim. Sci. 2015, 93, 5119–5127. [Google Scholar] [CrossRef]
- Vanvanhossou, S.F.U.; Scheper, C.; Dossa, L.H.; Yin, T.; Brügemann, K.; König, S. A Multi-Breed GWAS for Morphometric Traits in Four Beninese Indigenous Cattle Breeds Reveals Loci Associated with Conformation, Carcass and Adaptive Traits. BMC Genom. 2020, 21, 783. [Google Scholar] [CrossRef]
- Hulsegge, I.; Oldenbroek, K.; Bouwman, A.; Veerkamp, R.; Windig, J. Selection and Drift: A Comparison between Historic and Recent Dutch Friesian Cattle and Recent Holstein Friesian Using WGS Data. Animal 2022, 12, 329. [Google Scholar] [CrossRef]
- Sölkner, J.; Ferencakovic, M.; Karimi, Z.; OBrien, A.M.P.; Mészáros, G.; Eaglen, S.A.E.; Boison, S.A.; Curik, I. Extremely Non-Uniform: Patterns of Runs of Homozygosity in Bovine Populations. In Proceedings of the Proceedings of the 10th World Congress on Genetics Applied to Livestock Production, Vancouver, BC, Canada, 17–22 August 2014; pp. 17–22. [Google Scholar]
- Signer-Hasler, H.; Casanova, L.; Barenco, A.; Maitre, B.; Bagnato, A.; Vevey, M.; Berger, B.; Simčič, M.; Boichon, D.; Capitan, A.; et al. Genomic Regions Underlying Positive Selection in Local, Alpine Cattle Breeds. Anim. Genet. 2023, 54, 239–253. [Google Scholar] [CrossRef]
- Jang, J.; Terefe, E.; Kim, K.; Lee, Y.H.; Belay, G.; Tijjani, A.; Han, J.-L.; Hanotte, O.; Kim, H. Correction to: Population Differentiated Copy Number Variation of Bos Taurus, Bos Indicus and Their African Hybrids. BMC Genom. 2022, 23, 207. [Google Scholar] [CrossRef]
- Bongiorni, S.; Gruber, C.E.M.; Bueno, S.; Chillemi, G.; Ferrè, F.; Failla, S.; Moioli, B.; Valentini, A. Transcriptomic Investigation of Meat Tenderness in Two Italian Cattle Breeds. Anim. Genet. 2016, 47, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, G.; Lin, X.; Zhang, J.; Hou, G.; Zhang, L.; Liu, D.; Li, Y.; Li, J.; Xu, L. Genomic Inbreeding and Runs of Homozygosity Analysis of Indigenous Cattle Populations in Southern China. PLoS ONE 2022, 17, e0271718. [Google Scholar] [CrossRef]
- Delbes, G.; Yanagiya, A.; Sonenberg, N.; Robaire, B. PABP Interacting Protein 2 (Paip2) Is a Major Translational Regulator Involved in the Maturation of Male Germ Cells and Male Fertility. Biol. Reprod. 2009, 81, 167. [Google Scholar] [CrossRef]
- Guan, D.; Luo, N.; Tan, X.; Zhao, Z.; Huang, Y.; Na, R.; Zhang, J.; Zhao, Y. Scanning of Selection Signature Provides a Glimpse into Important Economic Traits in Goats (Capra Hircus). Sci. Rep. 2016, 6, 36372. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, S.A.; Reverter, A.; Byrne, K.A.; Wang, Y.; Nattrass, G.S.; Hudson, N.J.; Greenwood, P.L. Gene Expression Studies of Developing Bovine Longissimus Muscle from Two Different Beef Cattle Breeds. BMC Dev. Biol. 2007, 7, 95. [Google Scholar] [CrossRef]
- Akakabe, Y.; Koide, M.; Kitamura, Y.; Matsuo, K.; Ueyama, T.; Matoba, S.; Yamada, H.; Miyata, K.; Oike, Y.; Ikeda, K. Ecscr Regulates Insulin Sensitivity and Predisposition to Obesity by Modulating Endothelial Cell Functions. Nat. Commun. 2013, 4, 2389. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed | Sample Number | ROH Number | SNP Number | Gene Number | Longest ROH (Mb) | Mean ROH (Mb) |
|---|---|---|---|---|---|---|
| ES | 33 | 8 | 338 | 175 | 533.612 | 57.100 |
| HP | 39 | 9 | 487 | 617 | 106.031 | 57.807 |
| YL | 52 | 7 | 424 | 352 | 619.931 | 105.322 |
| ZB | 71 | 6 | 308 | 327 | 410.002 | 61.872 |
| Breed | CHR | Start (bp) | End (bp) | Region (Kb) | Number of SNPs | Genes |
|---|---|---|---|---|---|---|
| HP | 7 | 48,542,265 | 54,706,580 | 6164.316 | 103 | DNAJC18, PAIP2, SLC23A1, LRRTM2, SPATA24, SMIM33, SIL1, UBE2D2, MATR3, PROB1, STING1, CTNNA1, ECSCR |
| ES | 7 | 48,542,265 | 53,412,209 | 4869.945 | 78 | DNAJC18, PAIP2, SLC23A1, LRRTM2, SPATA24, SMIM33, SIL1, UBE2D2, MATR3, PROB1, STING1, CTNNA1, ECSCR |
| YL | 7 | 48,542,265 | 53,053,673 | 4511.409 | 74 | DNAJC18, PAIP2, SLC23A1, LRRTM2, SPATA24, SMIM33, SIL1, UBE2D2, MATR3, PROB1, STING1, CTNNA1, ECSCR |
| ZB | 7 | 48,542,265 | 54,768,354 | 6226.09 | 108 | DNAJC18, PAIP2, SLC23A1, LRRTM2, SPATA24, SMIM33, SIL1, UBE2D2, MATR3, PROB1, STING1, CTNNA1, ECSCR |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Q.; Shi, L.; Zhang, P.; Yu, B.; Liu, C.; Xiang, M.; Li, S.; Liu, L.; Cheng, L.; Chen, H. Development and Application of a 40 K Liquid Capture Chip for Beef Cattle. Animals 2025, 15, 1346. https://doi.org/10.3390/ani15091346
Liu Q, Shi L, Zhang P, Yu B, Liu C, Xiang M, Li S, Liu L, Cheng L, Chen H. Development and Application of a 40 K Liquid Capture Chip for Beef Cattle. Animals. 2025; 15(9):1346. https://doi.org/10.3390/ani15091346
Chicago/Turabian StyleLiu, Qing, Liangyu Shi, Pu Zhang, Bo Yu, Chenhui Liu, Min Xiang, Shuilian Li, Lei Liu, Lei Cheng, and Hongbo Chen. 2025. "Development and Application of a 40 K Liquid Capture Chip for Beef Cattle" Animals 15, no. 9: 1346. https://doi.org/10.3390/ani15091346
APA StyleLiu, Q., Shi, L., Zhang, P., Yu, B., Liu, C., Xiang, M., Li, S., Liu, L., Cheng, L., & Chen, H. (2025). Development and Application of a 40 K Liquid Capture Chip for Beef Cattle. Animals, 15(9), 1346. https://doi.org/10.3390/ani15091346