
Whole Genome Insights into Genetic Diversity, Introgression, and Adaptation of Hunan Cattle

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Sequence Processing and SNP Calling
2.3. Population Genomic Analysis
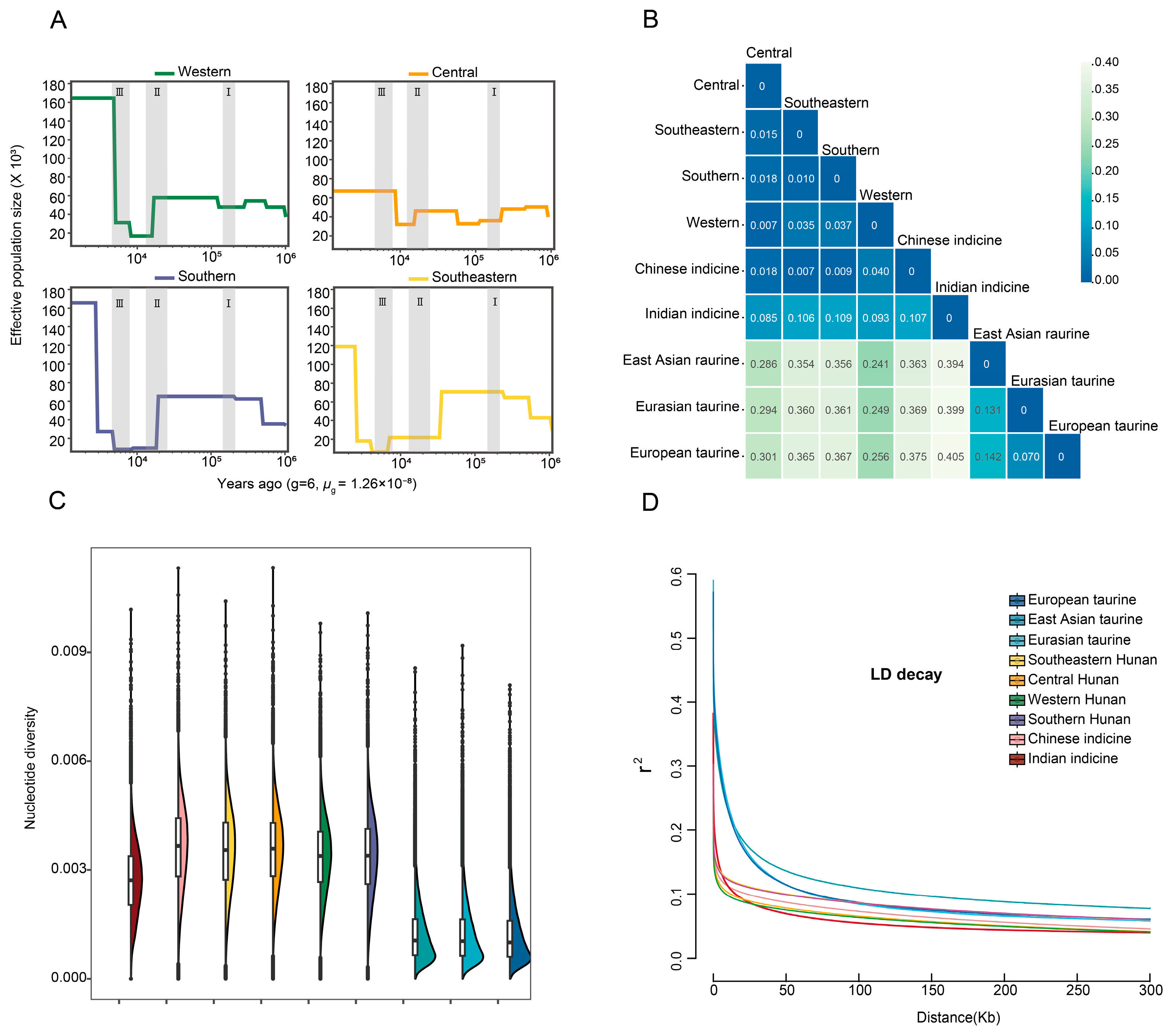
2.4. Detection of Genetic Diversity
2.5. Extract, Auto-Classify and Analyze ROH Region
2.6. Selective Sweep Identification
2.7. Introgression Analysis
2.8. Paternal Analysis
3. Results
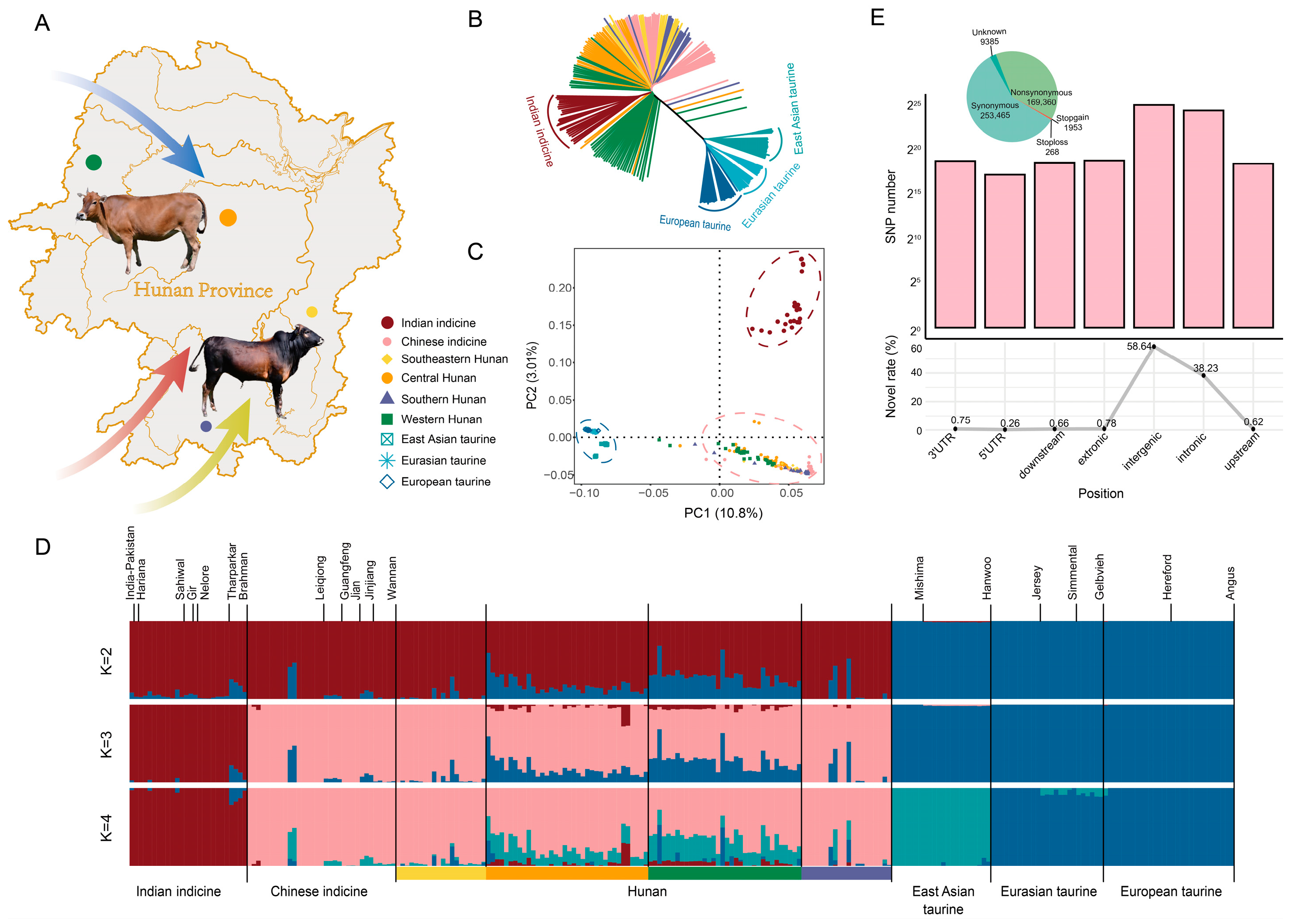
3.1. Detecting and Classifying the Single Nucleotide Polymorphism (SNP)
3.2. Detecting, Classifying and Genomic Variation Analysis of the SNPs
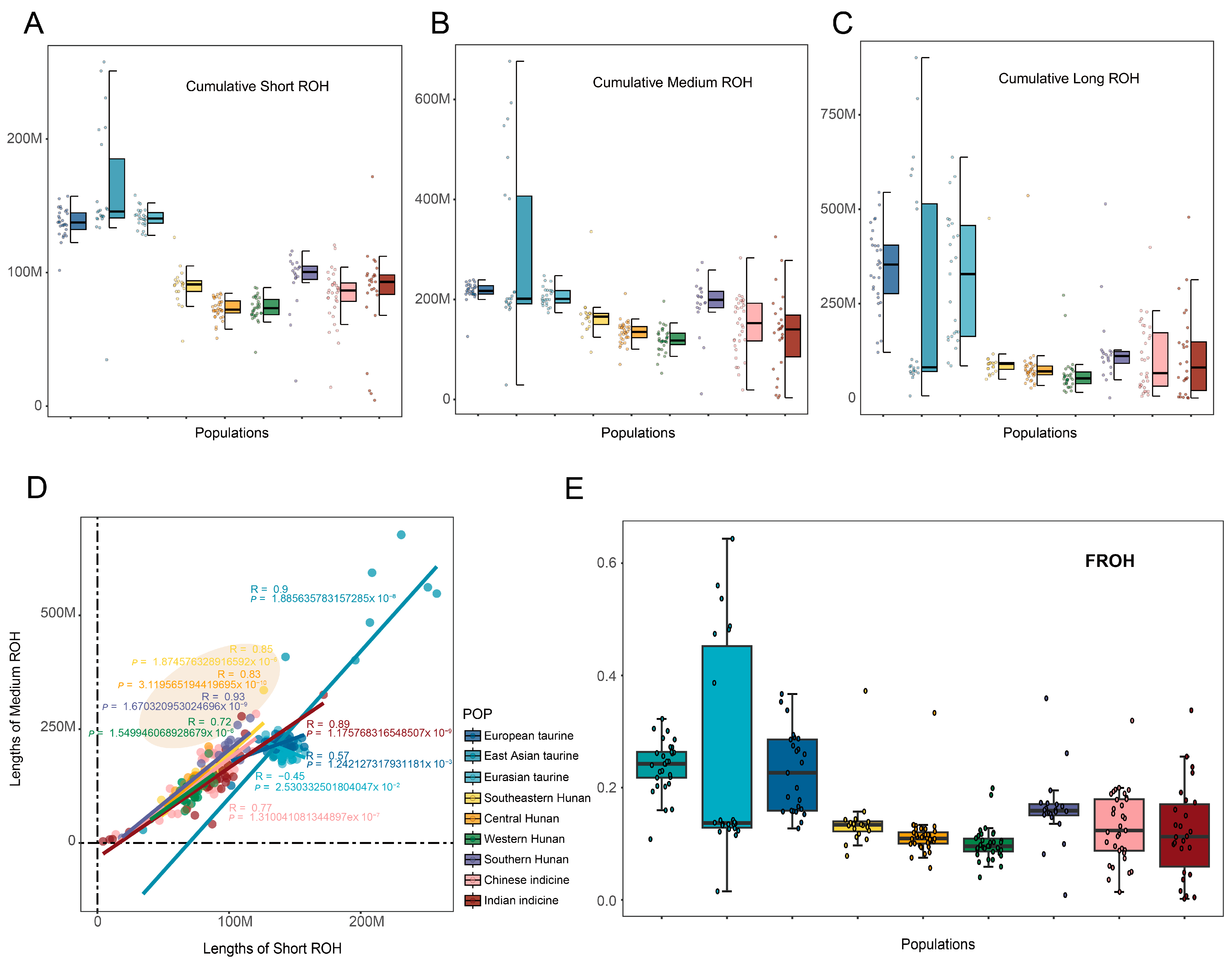
3.3. Run of Homozygosity (ROH) Classification, Calculation, and Inbreeding
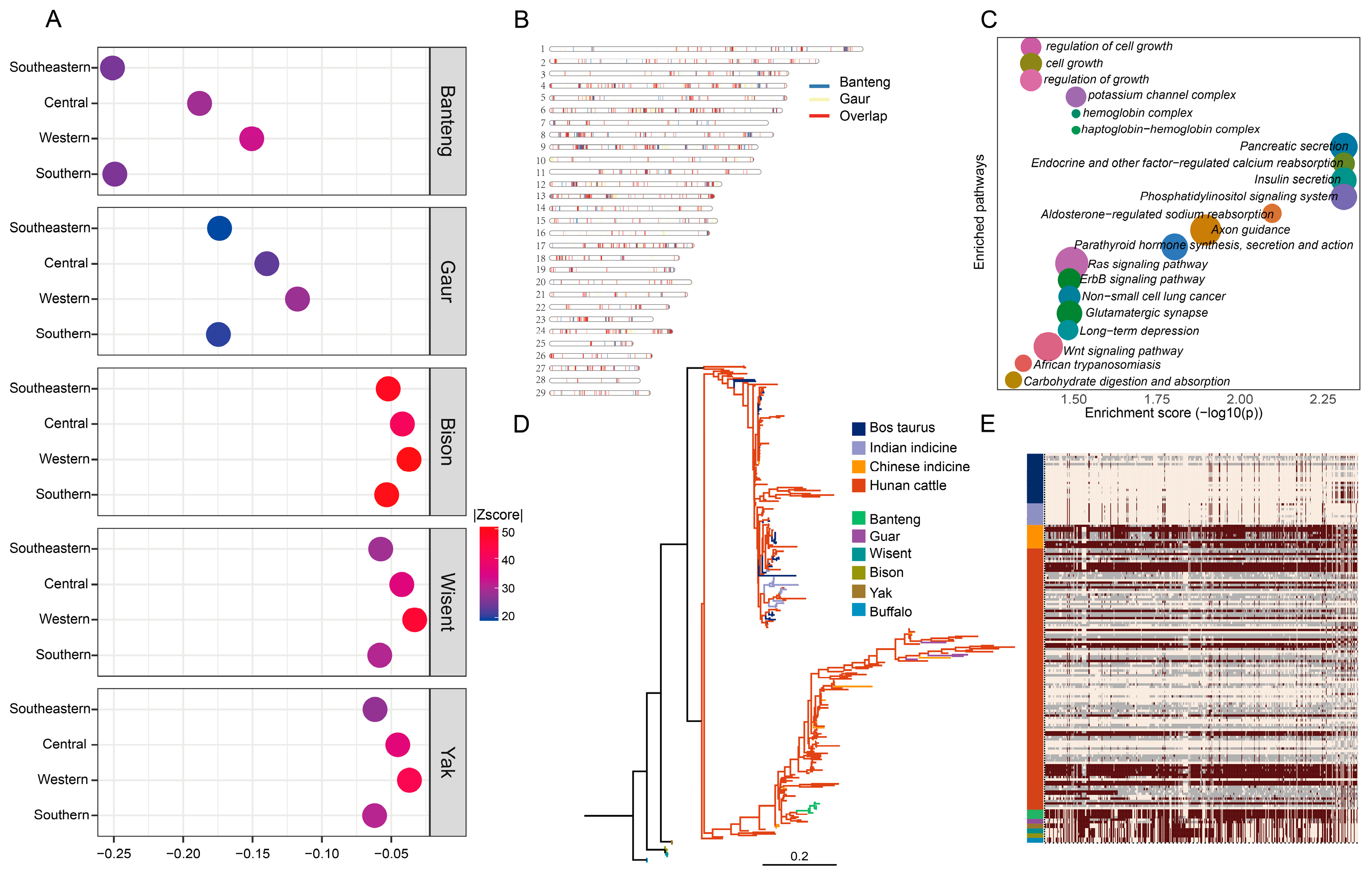
3.4. Selection Signatures in Hunan Cattle
3.5. The Introgressed Events Form Other Wild Species
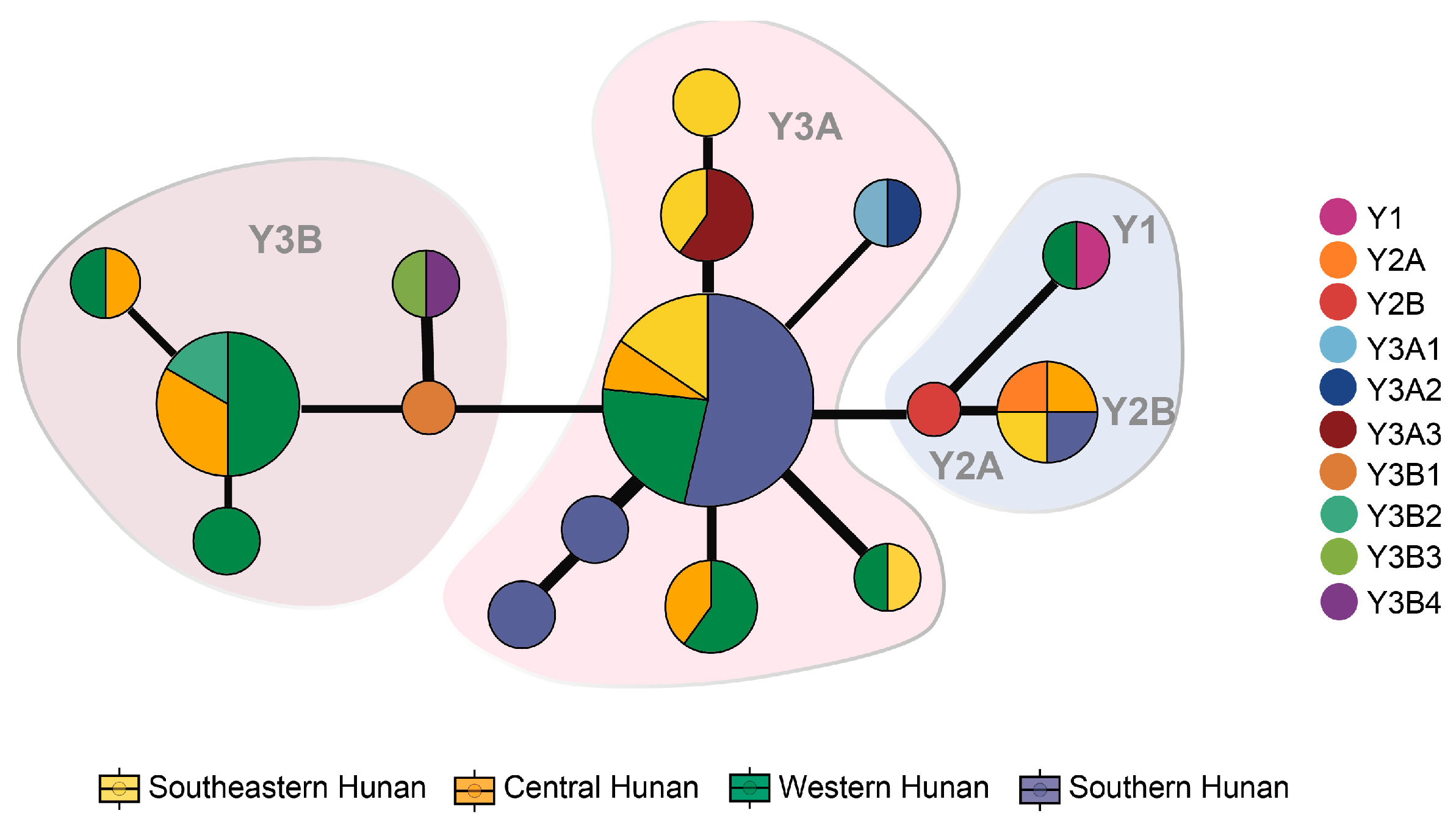
3.6. Parental Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| SNP | Single nucleotide polymorphism |
| WGS | Whole-genome sequencing |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| GO | Gene Ontology |
| NJ | Neighbor-joining |
| ML | Maximum Likelihood |
| ROH | Runs of homozygosity |
| FROH | Genomic inbreeding coefficient based on Runs of Homozygosity |
| LD | Linkage disequilibrium |
| PCA | Principle component analysis |
References
- Achilli, A.; Olivieri, A.; Pellecchia, M.; Uboldi, C.; Colli, L.; Al-Zahery, N.; Accetturo, M.; Pala, M.; Kashani, B.H.; Perego, U.A.; et al. Mitochondrial genomes of extinct aurochs survive in domestic cattle. Curr. Biol. 2008, 18, R157–R158. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.-D.; Ding, X.-D.; Wang, S.; Wójcik, J.M.; Zhang, Y.; Tokarska, M.; Li, Y.; Wang, M.-S.; Faruque, O.; Nielsen, R.; et al. Pervasive introgression facilitated domestication and adaptation in the Bos species complex. Nat. Ecol. Evol. 2018, 2, 1139–1145. [Google Scholar] [CrossRef]
- Chen, N.; Xia, X.; Hanif, Q.; Zhang, F.; Dang, R.; Huang, B.; Lyu, Y.; Luo, X.; Zhang, H.; Yan, H.; et al. Global genetic diversity, introgression, and evolutionary adaptation of indicine cattle revealed by whole genome sequencing. Nat. Commun. 2023, 14, 7803. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhang, B.; Luo, J.; Li, J.; Liang, J.; Wu, W.; Xie, Y.; Li, F.; Lei, C.; Yi, K. Genomics, Origin and Selection Signals of Loudi Cattle in Central Hunan. Biology 2022, 11, 1775. [Google Scholar] [CrossRef]
- Luo, X.; Li, J.; Xiao, C.; Sun, L.; Xiang, W.; Chen, N.; Lei, C.; Lei, H.; Long, Y.; Long, T.; et al. Whole-Genome Resequencing of Xiangxi Cattle Identifies Genomic Diversity and Selection Signatures. Front. Genet. 2022, 13, 816379. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lei, H.; Li, J.; Sun, A.; Ahmed, Z.; Duan, H.; Chen, L.; Zhang, B.; Lei, C.; Yi, K. Analysis of genetic diversity and selection signals in Chaling cattle of southern China using whole-genome scan. Anim. Genet. 2023, 54, 284–294. [Google Scholar] [CrossRef]
- Yan, H.; Li, J.; Zhang, K.; Duan, H.; Sun, A.; Zhang, B.; Li, F.; Chen, N.; Lei, C.; Yi, K. Local Ancestry and Adaptive Introgression in Xiangnan Cattle. Biology 2024, 13, 1000. [Google Scholar] [CrossRef]
- Chen, N.; Cai, Y.; Chen, Q.; Li, R.; Wang, K.; Huang, Y.; Hu, S.; Huang, S.; Zhang, H.; Zheng, Z.; et al. Whole-genome resequencing reveals world-wide ancestry and adaptive introgression events of domesticated cattle in East Asia. Nat. Commun. 2018, 9, 2337. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Houtgast, E.J.; Sima, V.M.; Bertels, K.; Al-Ars, Z. Hardware acceleration of BWA-MEM genomic short read mapping for longer read lengths. Compu. Biol. Chem. 2018, 75, 54–64. [Google Scholar] [CrossRef]
- Anton, N.; James, T. Next-generation sequencing data interpretation: Enhancing reproducibility and accessibility. Nat. Rev. Genet. 2012, 13, 667–672. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Patterson, N.; Price, A.L.; Reich, D. Population structure and eigenanalysis. PLoS Genet. 2006, 2, e190. [Google Scholar] [CrossRef]
- Alexander, D.H.; Lange, K. Enhancements to the ADMIXTURE algorithm for individual ancestry estimation. BMC Bioinf. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Zhang, C.; Dong, S.-S.; Xu, J.-Y.; He, W.-M.; Yang, T.-L. PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files. Bioinformatics 2019, 35, 1786–1788. [Google Scholar] [CrossRef]
- Browning, S.R.; Browning, B.L. Rapid and Accurate Haplotype Phasing and Missing-Data Inference for Whole-Genome Association Studies by Use of Localized Haplotype Clustering. Am. J. Hum. Genet. 2007, 81, 1084–1097. [Google Scholar] [CrossRef]
- McQuillan, R.; Leutenegger, A.-L.; Abdel-Rahman, R.; Franklin, C.S.; Pericic, M.; Barac-Lauc, L.; Smolej-Narancic, N.; Janicijevic, B.; Polasek, O.; Tenesa, A.; et al. Runs of homozygosity in European populations. Am. J. Hum. Genet. 2008, 83, 359–372. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Hernandez, R.D. selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 2014, 31, 2824–2827. [Google Scholar] [CrossRef] [PubMed]
- Maples, B.K.; Gravel, S.; Kenny, E.E.; Bustamante, C.D. RFMix: A Discriminative Modeling Approach for Rapid and Robust Local-Ancestry Inference. Am. J. Hum. Genet. 2013, 93, 278–288. [Google Scholar] [CrossRef]
- Milan, M.; Michael, M.; Hannes, S. Dsuite-fast D-statistics and related admixture evidence from VCF files. Mole. Ecol. Resour. 2020, 21, 584–595. [Google Scholar]
- Huerta-Sanchez, E.; Jin, X.; Asan; Bianba, Z.; Peter, B.M.; Vinckenbosch, N.; Liang, Y.; Yi, X.; He, M.; Somel, M.; et al. Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 2014, 512, 194–197. [Google Scholar] [CrossRef]
- Lyu, Y.; Wang, F.; Cheng, H.; Han, J.; Dang, R.; Xia, X.; Wang, H.; Zhong, J.; Lenstra, J.A.; Zhang, H.; et al. Recent selection and introgression facilitated high-altitude adaptation in cattle. Sci. Bull. 2024, 69, 3415–3424. [Google Scholar] [CrossRef]
- Racimo, F.; Marnetto, D.; Huerta-Sanchez, E. Signatures of Archaic Adaptive Introgression in Present-Day Human Populations. Mol. Biol. Evol. 2017, 34, 296–317. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Lenstra, J.A.; Zhang, S.; Liu, W.; Liu, J. Evolution and domestication of the Bovini species. Anim. Genet. 2020, 51, 637–657. [Google Scholar] [CrossRef]
- Terhorst, J.; Kamm, J.A.; Song, Y.S. Robust and scalable inference of population history from hundreds of unphased whole genomes. Nat. Genet. 2017, 49, 303–309. [Google Scholar] [CrossRef]
- Ceballos, F.C.; Joshi, P.K.; Clark, D.W.; Ramsay, M.; Wilson, J.F. Runs of homozygosity: Windows into population history and trait architecture. Nat. Rev. Genet. 2018, 19, 220–234. [Google Scholar] [CrossRef]
- Santer, R.; Kinner, M.; Lassen, C.L.; Schneppenheim, R.; Eggert, P.; Bald, M.; Brodehl, J.; Daschner, M.; Ehrich, J.H.; Kemper, M.; et al. Molecular analysis of the SGLT2 gene in patients with renal glucosuria. J. Am. Soc. Nephrol. 2003, 14, 2873–2882. [Google Scholar] [CrossRef]
- Zhao, F.Q.; McFadden, T.; Wall, E.; Dong, B.; Zheng, Y.-C. Cloning and expression of bovine sodium/glucose cotransporter SGLT2. J. Dairy. Sci. 2005, 88, 2738–2748. [Google Scholar] [CrossRef] [PubMed]
- Melville, M.W.; Tan, S.L.; Wambach, M.; Song, J.; Morimoto, R.I.; Katze, M.G. The cellular inhibitor of the PKR protein kinase, P58(IPK), is an influenza virus-activated co-chaperone that modulates heat shock protein 70 activity. J. Biol. Chem. 1999, 274, 3797–3803. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Truong, Q.A.; Park, S.; Breslow, J.L. Two Hsp70 family members expressed in atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 2003, 100, 1256–1261. [Google Scholar] [CrossRef]
- Khan, I.; Maldonado, E.; Vasconcelos, V.; O’brien, S.J.; E Johnson, W.; Antunes, A. Mammalian keratin associated proteins (KRTAPs) subgenomes: Disentangling hair diversity and adaptation to terrestrial and aquatic environments. BMC Genom. 2014, 15, 779. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Li, J.; Duan, H.; Sun, A.; Hu, R.; Gao, S.; Zhang, B.; Huang, B.; Lei, C.; Yi, K. Whole Genome Insights into Genetic Diversity, Introgression, and Adaptation of Hunan Cattle. Animals 2025, 15, 1287. https://doi.org/10.3390/ani15091287
Li Y, Li J, Duan H, Sun A, Hu R, Gao S, Zhang B, Huang B, Lei C, Yi K. Whole Genome Insights into Genetic Diversity, Introgression, and Adaptation of Hunan Cattle. Animals. 2025; 15(9):1287. https://doi.org/10.3390/ani15091287
Chicago/Turabian StyleLi, Yushan, Jianbo Li, Hongfeng Duan, Ao Sun, Renke Hu, Shuai Gao, Baizhong Zhang, Bizhi Huang, Chuzhao Lei, and Kangle Yi. 2025. "Whole Genome Insights into Genetic Diversity, Introgression, and Adaptation of Hunan Cattle" Animals 15, no. 9: 1287. https://doi.org/10.3390/ani15091287
APA StyleLi, Y., Li, J., Duan, H., Sun, A., Hu, R., Gao, S., Zhang, B., Huang, B., Lei, C., & Yi, K. (2025). Whole Genome Insights into Genetic Diversity, Introgression, and Adaptation of Hunan Cattle. Animals, 15(9), 1287. https://doi.org/10.3390/ani15091287