
Molecular Mechanism of the Grid Gene Family Regulating Growth Size Heteromorphism in Cynoglossus semilaevis


Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Fish and Tissue Collection
2.2. Experimental Program
2.3. Total RNA Extraction and cDNA Synthesis
2.4. Sequence Analysis and Comparison of Grid Gene Families
2.5. Genomic DNA Extraction and Genetic Characterization
2.6. QPCR Detection of Gene Expression
2.7. Grid1 and Grid2 siRNA Interference
2.8. Transcriptome Analysis
3. Results
3.1. GWAS Analysis Confirmed the Growth-Related Genes
3.2. Cloning and Sequence Analysis of the Grid Gene Family
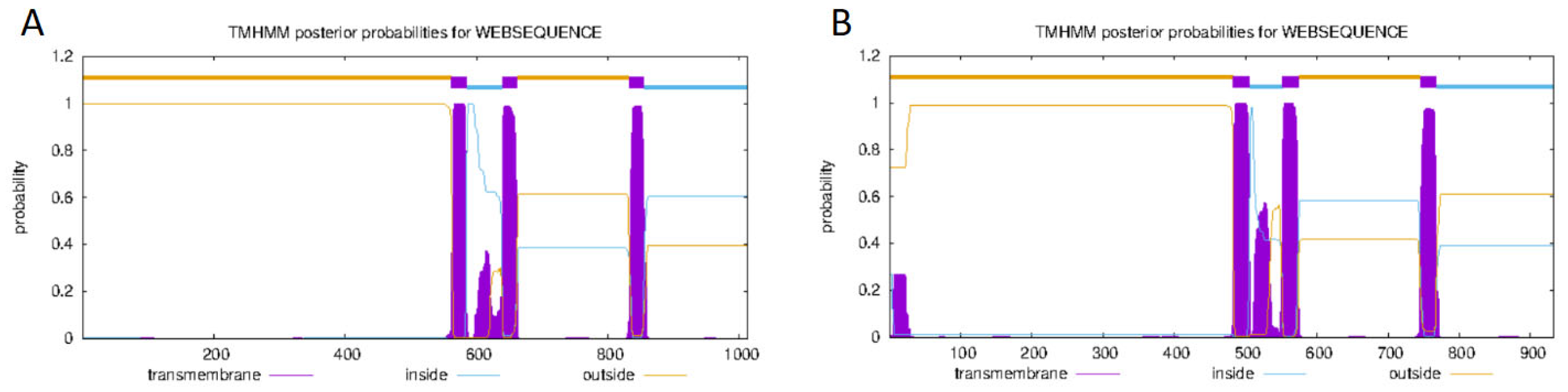
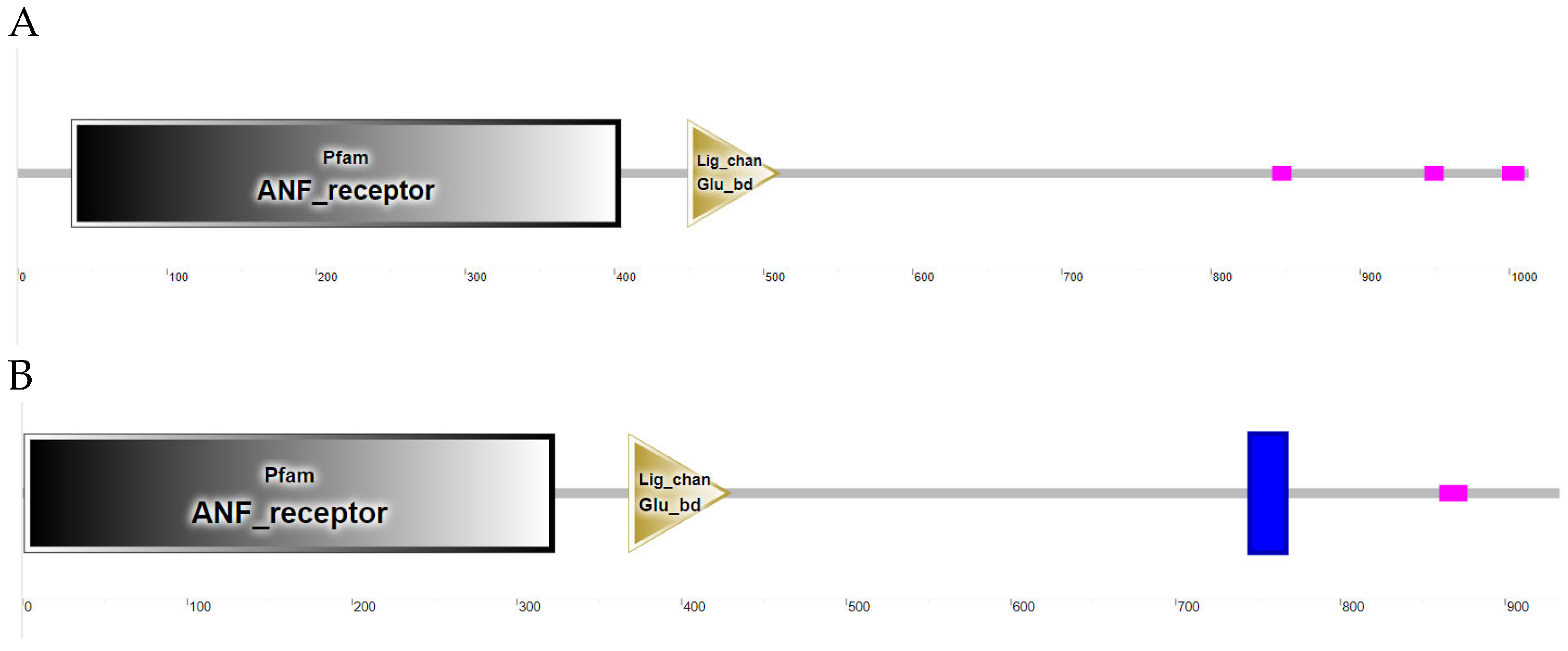
3.2.1. Grid Gene Family Sequence Analysis and Prediction
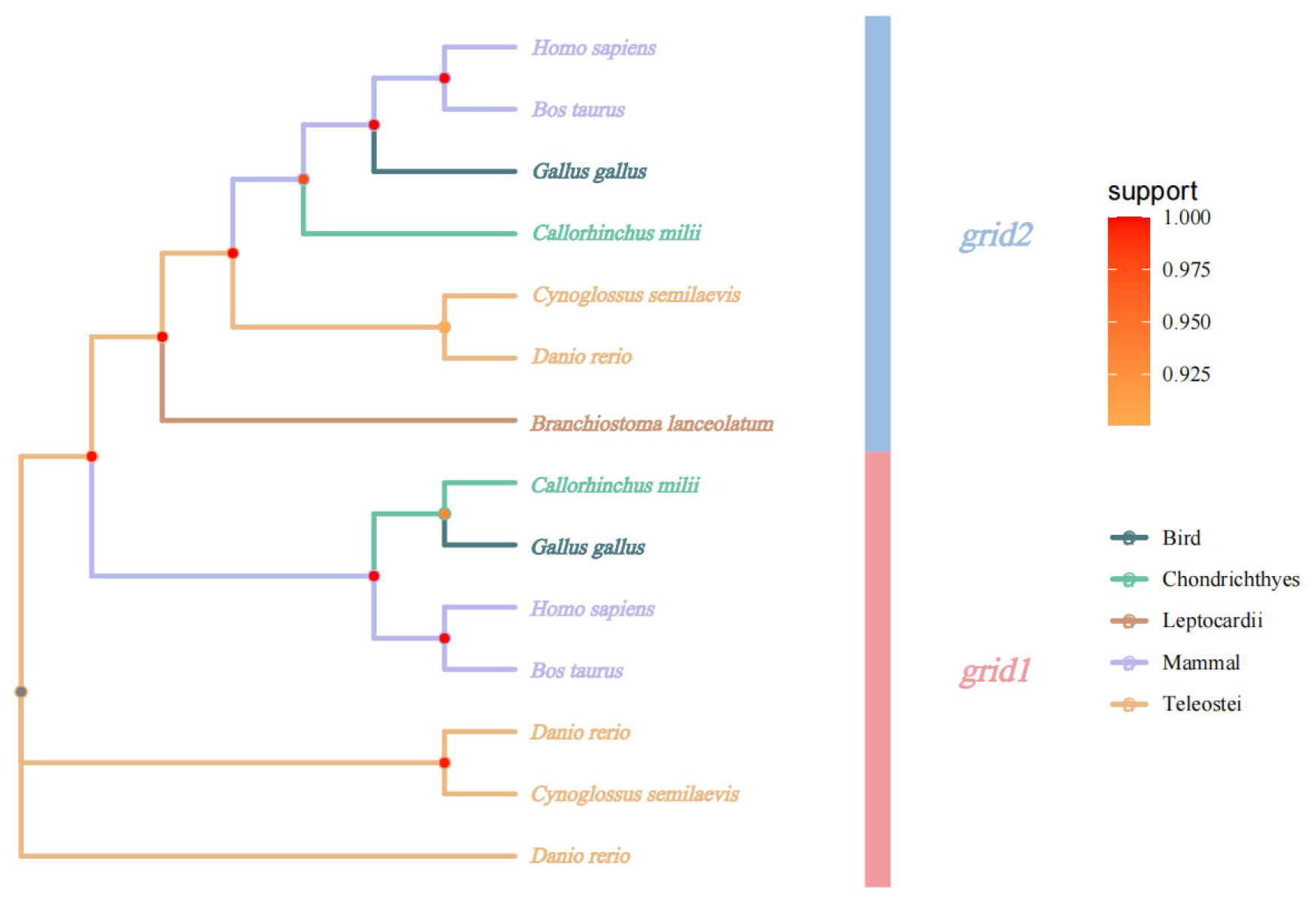
3.2.2. Evolutionary Analysis of Grid Gene Families
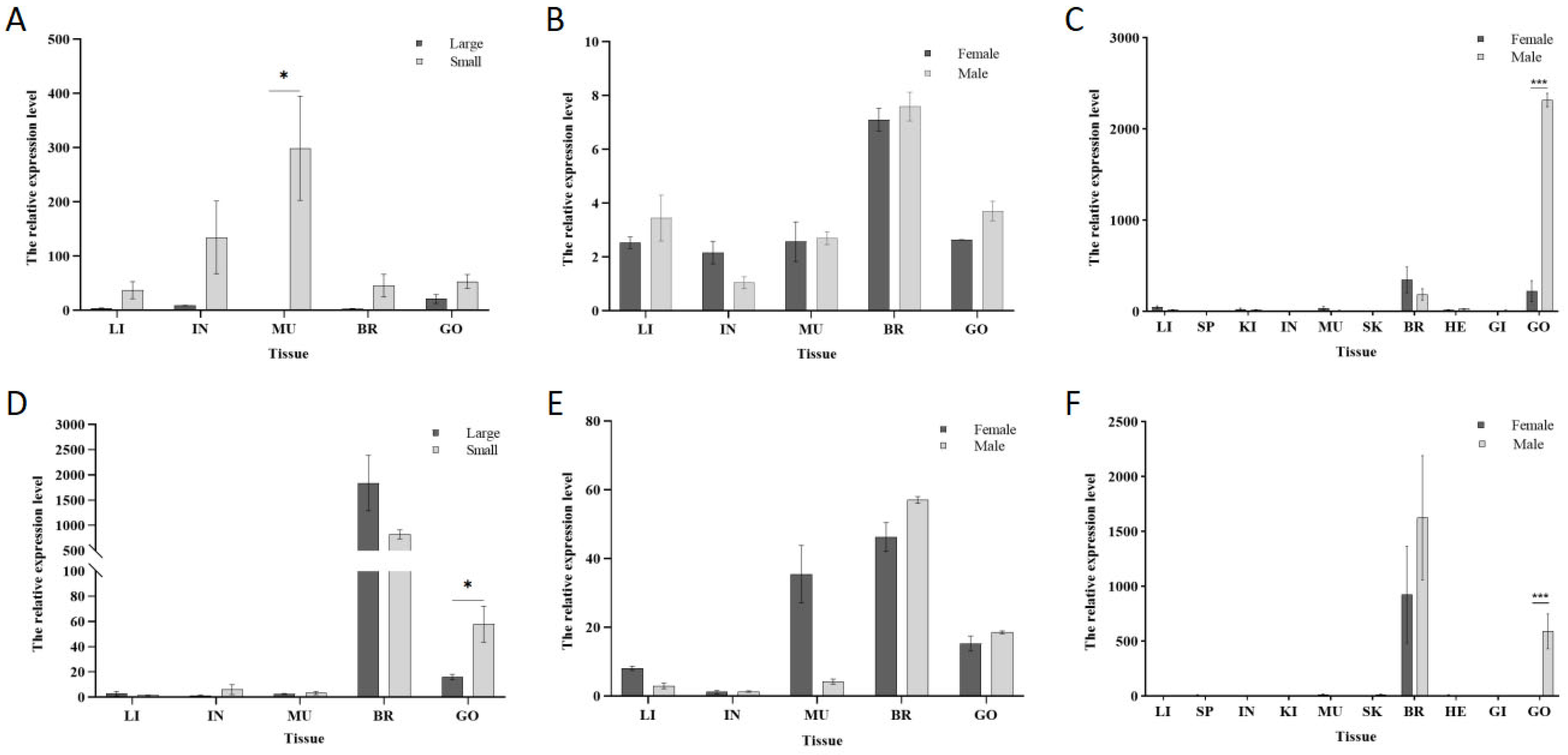
3.3. Differential Expression of the Grid Gene Family Across Tissues
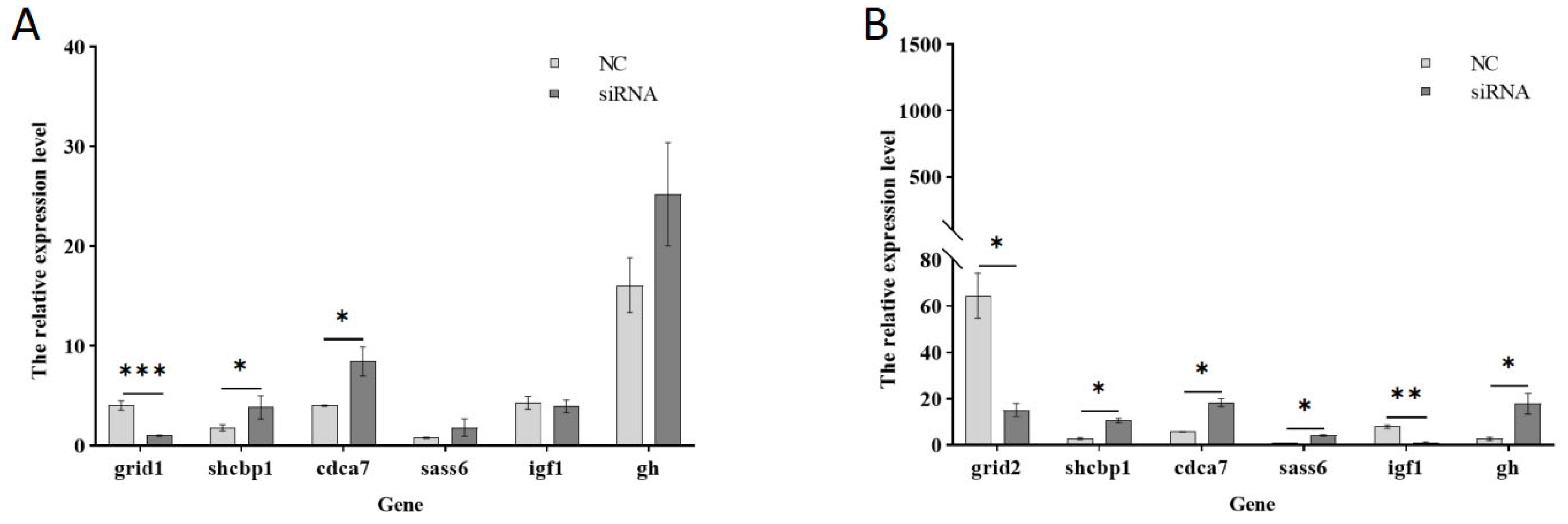
3.4. Detection of the Effect of siRNA-Mediated Interference with the Grid Gene Family
3.5. Comparison of the Grid Gene Family RNAi Transcriptome Sequencing Data with the Cellular Genome
3.5.1. Results of the Grid Gene Family Sequencing Data Quality Preprocessing
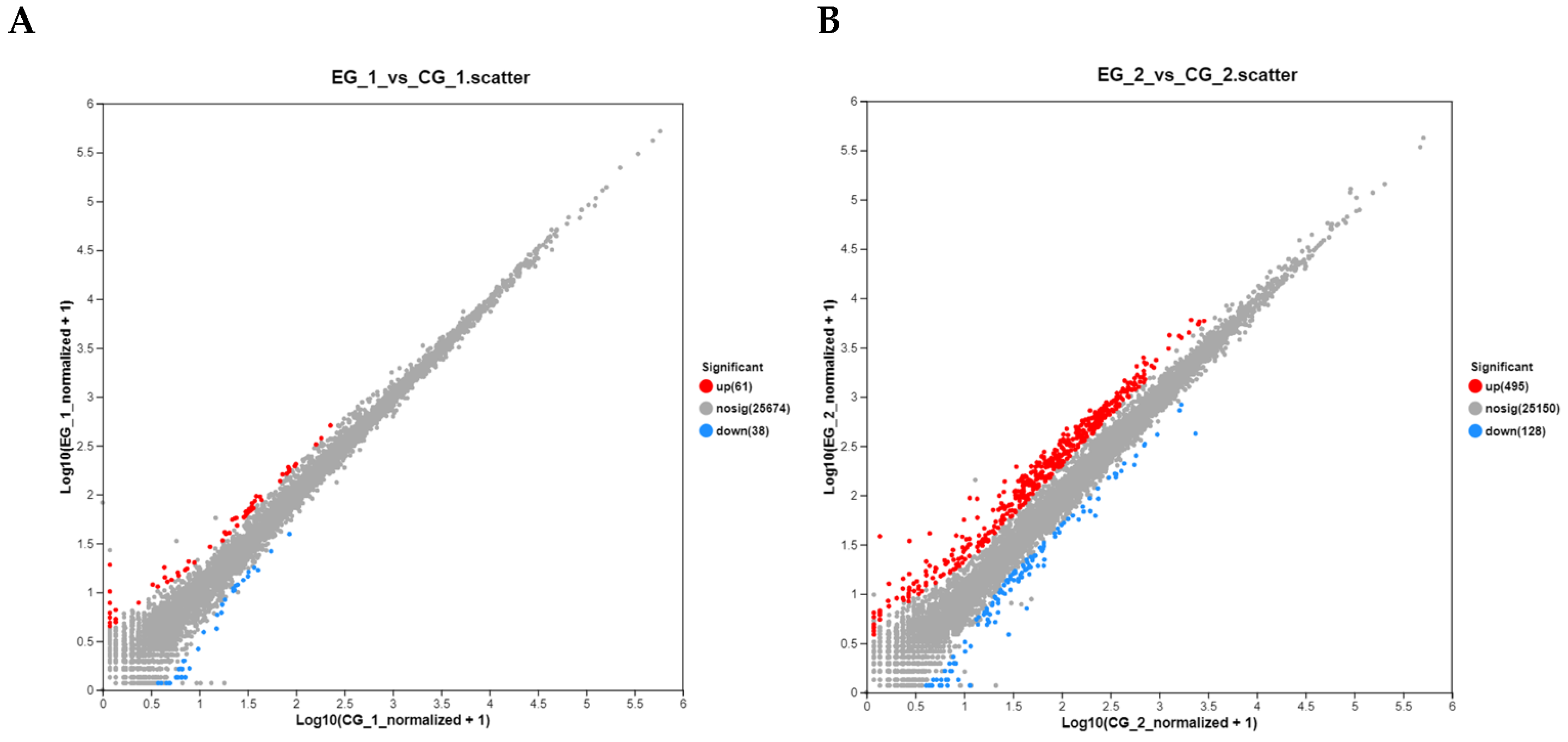
3.5.2. Analysis of DEGs
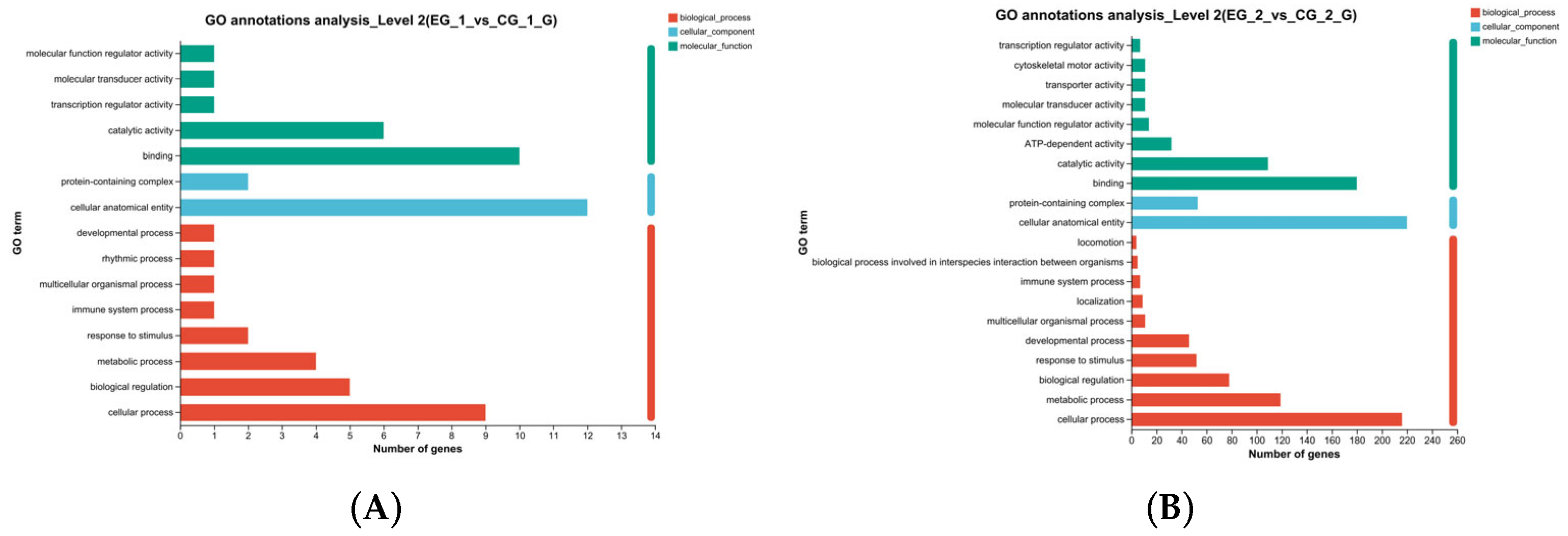
3.5.3. GO Function Annotation, GO Enrichment Analysis of Transcriptomic DEGs in Spermatogonial Cells After Grid1, Grid2 Knockdown
3.5.4. KEGG Signaling Pathway Enrichment Analysis of DEGs in Cynoglossus Semilaevis Testicular Cells
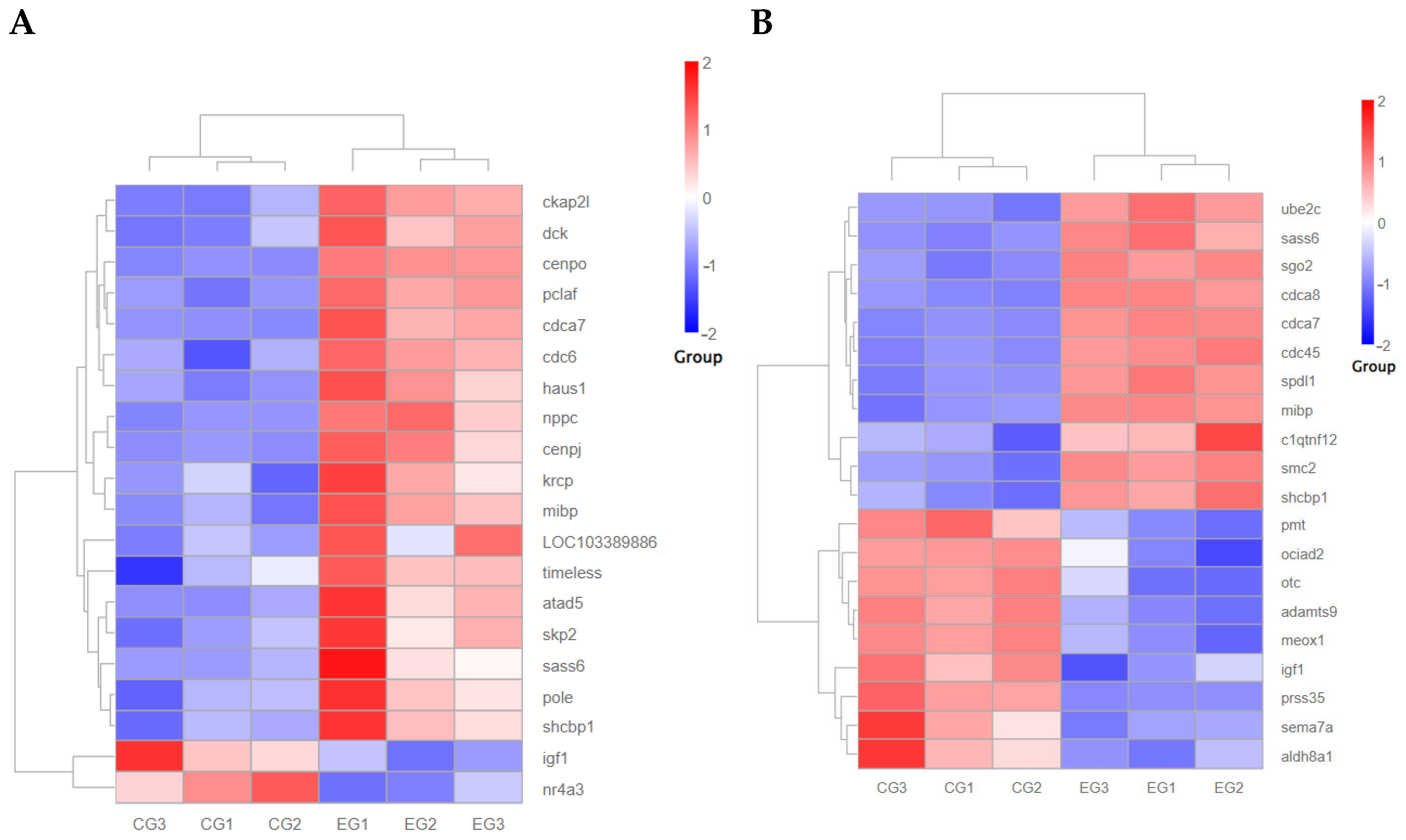
3.5.5. Effect of Expression of Growth-Related Genes After Grid Gene Family Disruption
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bao, B. Flatfish Metamorphosis; Springer Nature: Dordrecht, The Netherlands, 2022. [Google Scholar]
- Bhujel, R.C. Global aquatic food production. In Aquatic Food Security; CABI: Oxfordshire, UK, 2024; pp. 99–126. [Google Scholar]
- Chen, S.-L.; Li, J.; Deng, S.-P.; Tian, Y.-S.; Wang, Q.-Y.; Zhuang, Z.-M.; Sha, Z.-X.; Xu, J.-Y. Isolation of female-specific aflp markers and molecular identification of genetic sex in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. 2007, 9, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-L.; Tian, Y.-S.; Yang, J.-F.; Shao, C.-W.; Ji, X.-S.; Zhai, J.-M.; Liao, X.-L.; Zhuang, Z.-M.; Su, P.-Z.; Xu, J.-Y.; et al. Artificial gynogenesis and sex determination in half-smooth tongue sole (Cynoglossus semilaevis). Mar. Biotechnol. 2008, 11, 243–251. [Google Scholar] [CrossRef]
- Shao, C.; Li, Q.; Chen, S.; Zhang, P.; Lian, J.; Hu, Q.; Sun, B.; Jin, L.; Liu, S.; Wang, Z.; et al. Epigenetic modification and inheritance in sexual reversal of fish. Genome Res. 2014, 24, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhang, G.; Shao, C.; Huang, Q.; Liu, G.; Zhang, P.; Song, W.; An, N.; Chalopin, D.; Volff, J.-N.; et al. Whole-genome sequence of a flatfish provides insights into ZW sex chromosome evolution and adaptation to a benthic lifestyle. Nat. Genet. 2014, 46, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Li, X.; Chen, Y.; Yang, Y.; Wang, N.; Xu, W. Identification and Functional Analysis of Cynoglossus semilaevis Z-Linked E3 Ubiquitin Ligase rnf34. Animals 2024, 14, 311. [Google Scholar] [CrossRef]
- Zhao, N.; Jia, L.; He, X.; Zhang, B. Sex bias miRNAs in Cynoglossus semilaevis could play a role in transgenerational inheritance. Comp. Biochem. Physiol. Part D Genom. Proteom. 2021, 39, 100853. [Google Scholar] [CrossRef]
- Qi, Q.; Dong, Z.; Zhang, N.; Shao, C.; Xu, W. A patched1 gene homologue participates in female differentiation of Cynoglossus semilaevis. Gene Expr. Patterns 2022, 45, 119265. [Google Scholar] [CrossRef]
- Schmid, S.M.; Hollmann, M. To gate or not to gate: Are the delta subunits in the glutamate receptor family functional ion channels? Mol. Neurobiol. 2008, 37, 126–141. [Google Scholar] [CrossRef]
- Ady, V.; Perroy, J.; Tricoire, L.; Piochon, C.; Dadak, S.; Chen, X.; Dusart, I.; Fagni, L.; Lambolez, B.; Levenes, C. Type 1 metabotropic glutamate receptors (mGlu1) trigger the gating of G lu D 2 delta glutamate receptors. EMBO Rep. 2013, 15, 103–109. [Google Scholar] [CrossRef]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef]
- Kumagai, A.; Fujita, A.; Yokoyama, T.; Nonobe, Y.; Hasaba, Y.; Sasaki, T.; Itoh, Y.; Koura, M.; Suzuki, O.; Adachi, S.; et al. Altered Actions of Memantine and NMDA-Induced Currents in a New Grid2-Deleted Mouse Line. Genes 2014, 5, 1095–1114. [Google Scholar] [CrossRef] [PubMed]
- Elegheert, J.; Kakegawa, W.; Clay, J.E.; Shanks, N.F.; Behiels, E.; Matsuda, K.; Kohda, K.; Miura, E.; Rossmann, M.; Mitakidis, N.; et al. Structural basis for integration of GluD receptors within synaptic organizer complexes. Science 2016, 353, 295–299. [Google Scholar] [CrossRef]
- Todman, M.; Han, S.-K.; Herbison, A. Profiling neurotransmitter receptor expression in mouse gonadotropin-releasing hormone neurons using green fluorescent protein-promoter transgenics and microarrays. Neuroscience 2005, 132, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Li, X.; Pan, Z.; Wu, Z.; Zhu, Y.; Zhang, W.; Lu, J.; Xu, S.; Qin, P.; Liu, Y.; et al. Grid1 regulates the onset of puberty in female rats. J. Vet. Med. Sci. 2024, 86, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Behar, T.N.; Scott, C.A.; Greene, C.L.; Wen, X.; Smith, S.V.; Maric, D.; Liu, Q.-Y.; Colton, C.A.; Barker, J.L. Glutamate acting at nmda receptors stimulates embryonic cortical neuronal migration. J. Neurosci. 1999, 19, 4449–4461. [Google Scholar] [CrossRef]
- Rodríguez-Sanz, H.; Moreno-Romero, J.; Solís, M.T.; Köhler, C.; Risueño, M.C.; Testillano, P.S. Changes in histone methylation and acetylation during microspore reprogramming to embryogenesis occur concomitantly with Bn HKMT and Bn HAT expression and are associated with cell totipotency, proliferation, and dif-ferentiation in Brassica napus. Cytogenet. Genome Res. 2014, 143, 209–218. [Google Scholar] [CrossRef]
- Veerapandiyan, A.; Enner, S.; Thulasi, V.; Ming, X. A Rare Syndrome of GRID2 Deletion in 2 Siblings. Child Neurol. Open 2017, 4. [Google Scholar] [CrossRef]
- Wang, N.; Gao, J.; Liu, Y.; Shi, R.; Chen, S. Identification of crucial factors involved in Cynoglossus semilaevis sexual size dimorphism by GWAS and demonstration of zbed1 regulatory network by DAP-seq. Genomics 2022, 114, 110376. [Google Scholar] [CrossRef]
- Zhou, Q.; Su, Z.; Li, Y.; Liu, Y.; Wang, L.; Lu, S.; Wang, S.; Gan, T.; Liu, F.; Zhou, X.; et al. Genome-Wide Association Mapping and Gene Expression Analyses Reveal Genetic Mechanisms of Disease Resistance Variations in Cynoglossus semilaevis. Front. Genet. 2019, 10, 1167. [Google Scholar] [CrossRef]
- Fu, X.; Chen, Y.; Wang, L.; Zhou, Q.; Li, M.; Song, Y.; Li, Y.; Zhao, F.; Chen, S. Identification and functional analysis of the perforin-1 like gene in disease resistance in half smooth tongue sole (Cynoglossus semilaevis). Dev. Comp. Immunol. 2021, 122, 104135. [Google Scholar] [CrossRef]
- Gholizadeh, M.; Esmaeili-Fard, S.M. Meta-analysis of genome-wide association studies for litter size in sheep. Theriogenology 2021, 180, 103–112. [Google Scholar] [CrossRef]
- Nosrati, M.; Asadollahpour Nanaei, H.; Amiri Ghanatsaman, Z.; Esmailizadeh, A. Whole genome sequence analysis to detect signatures of positive selection for high fecundity in sheep. Reprod. Domest. Anim. 2019, 54, 358–364. [Google Scholar] [CrossRef]
- Sánchez-Ramos, R.; Trujano-Chavez, M.Z.; Gallegos-Sánchez, J.; Becerril-Pérez, C.M.; Cadena-Villegas, S.; Cortez-Romero, C. Detection of Candidate Genes Associated with Fecundity through Genome-Wide Selection Signatures of Katahdin Ewes. Animals 2023, 13, 272. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Nanaei, H.A.; Mehrgardi, A.A.; Esmailizadeh, A. Whole-genome sequence analysis reveals candidate genomic footprints and genes associated with reproductive traits in Thoroughbred horse. Reprod. Domest. Anim. 2019, 55, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Martins, T.F.; Magalhães, A.F.B.; Verardo, L.L.; Santos, G.C.; Fernandes, A.A.S.; Vieira, J.I.G.; Irano, N.; dos Santos, D.B. Functional analysis of litter size and number of teats in pigs: From GWAS to post-GWAS. Theriogenology 2022, 193, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Khramtsova, E.A.; Heldman, R.; Derks, E.M.; Yu, D.; Tourette Syndrome/Obsessive-Compulsive Disorder Working Group of the Psychiatric Genomics Consortium; Davis, L.K.; Stranger, B.E. Sex differences in the genetic architecture of obsessive–compulsive disorder. Am. J. Med Genet. Part B Neuropsychiatr. Genet. 2018, 180, 351–364. [Google Scholar] [CrossRef]
- Hu, L.; Zhang, L.; Li, Q.; Liu, H.; Xu, T.; Zhao, N.; Han, X.; Xu, S.; Zhao, X.; Zhang, C. Genome-wide analysis of CNVs in three populations of Tibetan sheep using whole-genome resequencing. Front. Genet. 2022, 13, 971464. [Google Scholar] [CrossRef]
- Sugimoto, M.; Sasaki, S.; Watanabe, T.; Nishimura, S.; Ideta, A.; Yamazaki, M.; Matsuda, K.; Yuzaki, M.; Sakimura, K.; Aoyagi, Y.; et al. Ionotropic glutamate receptor AMPA 1 is associated with ovulation rate. PLoS ONE 2010, 5, e13817. [Google Scholar] [CrossRef]
- Nikpay, M.; Šeda, O.; Tremblay, J.; Petrovich, M.; Gaudet, D.; Kotchen, T.A.; Cowley, A.W.; Hamet, P. Genetic mapping of habitual substance use, obesity-related traits, responses to mental and physical stress, and heart rate and blood pressure measurements reveals shared genes that are overrepresented in the neural synapse. Hypertens Res. 2012, 35, 585–591. [Google Scholar] [CrossRef]
- Locke, A.E.; Kahali, B.; Berndt, S.I.; Justice, A.E.; Pers, T.H.; Day, F.R.; Powell, C.; Vedantam, S.; Buchkovich, M.L.; Yang, J.; et al. Genetic studies of body mass index yield new insights for obesity biology. Nature 2015, 518, 197–206. [Google Scholar] [CrossRef] [PubMed]
- Keele, G.R.; Prokop, J.W.; He, H.; Holl, K.; Littrell, J.; Deal, A.; Francic, S.; Cui, L.; Gatti, D.M.; Broman, K.W.; et al. Genetic Fine-Mapping and Identification of Candidate Genes and Variants for Adiposity Traits in Outbred Rats. Obesity 2017, 26, 213–222. [Google Scholar] [CrossRef]
- Lu, H.; Yin, M.; Wang, L.; Cheng, J.; Cheng, W.; An, H.; Zhang, T. FGF13 interaction with SHCBP1 activates AKT-GSK3α/β signaling and promotes the proliferation of A549 cells. Cancer Biol. Ther. 2020, 21, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Leidel, S.; Delattre, M.; Cerutti, L.; Baumer, K.; Gönczy, P. SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat. Cell Biol. 2005, 7, 115–125. [Google Scholar] [CrossRef]
- Nigg, E.A.; Holland, A.J. Once and only once: Mechanisms of centriole duplication and their deregulation in disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 297–312. [Google Scholar] [CrossRef]
- Kitagawa, D.; Vakonakis, I.; Olieric, N.; Hilbert, M.; Keller, D.; Olieric, V.; Bortfeld, M.; Erat, M.C.; Flückiger, I.; Gönczy, P.; et al. Structural Basis of the 9-Fold Symmetry of Centrioles. Cell 2011, 144, 364–375. [Google Scholar] [CrossRef]
- Grzonka, M.; Bazzi, H. Mouse SAS-6 is required for centriole formation in embryos and integrity in embryonic stem cells. eLife 2024, 13, e94694. [Google Scholar] [CrossRef] [PubMed]
- Vukic, M.; Chouaref, J.; Della Chiara, V.; Dogan, S.; Ratner, F.; Hogenboom, J.Z.M.; Epp, T.A.; Chawengsaksophak, K.; Vonk, K.K.D.; Breukel, C.; et al. CDCA7-associated global aberrant DNA hypomethylation translates to localized, tissue-specific transcriptional responses. Sci. Adv. 2024, 10, eadk3384. [Google Scholar] [CrossRef] [PubMed]
- Vukic, M.; Daxinger, L. DNA methylation in disease: Immunodeficiency, Centromeric instability, Facial anomalies syndrome. Essays Biochem. 2019, 63, 773–783. [Google Scholar] [CrossRef]
- Hyun, S. Body size regulation and insulin-like growth factor signaling. Cell. Mol. Life Sci. 2013, 70, 2351–2365. [Google Scholar] [CrossRef]
- Reinecke, M.; Björnsson, B.T.; Dickhoff, W.W.; McCormick, S.D.; Navarro, I.; Power, D.M.; Gutiérrez, J. Growth hormone and insulin-like growth factors in fish: Where we are and where to go. Gen. Comp. Endocrinol. 2005, 142, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Zhou, Y.; Liu, S.; Tao, M.; Long, Y.; Liu, Z.; Zhang, C.; Duan, W.; Hu, J.; Song, C.; et al. Elevated expressions of GH/IGF axis genes in triploid crucian carp. Gen. Comp. Endocrinol. 2012, 178, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Silva, M.A.; Dupont-Prinet, A.; Houle, C.; Vagner, M.; Garant, D.; Bernatchez, L.; Audet, C. Growth regulation in brook charr Salvelinus fontinalis. Gen. Comp. Endocrinol. 2022, 331, 114160. [Google Scholar] [CrossRef] [PubMed]
- Horie, Y.; Yonekura, K.; Suzuki, A.; Takahashi, C. Zinc chloride influences embryonic development, growth, and Gh/Igf-1 gene expression during the early life stage in zebrafish (Danio rerio). Comp. Biochem. Physiol. Part C Toxicol. Pharmacol. 2020, 230, 108684. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence (5′~3′) | Purpose |
|---|---|---|
| grid1-F | GCGCTGAAATCCCGAGATGG | ORF verification |
| grid1-R | GTCTGTGGATGAATCTCCTGCT | ORF verification |
| grid2-F | TTTCTTCCATCCAGCCTGTGAG | ORF verification |
| grid2-R | AATCAGCCATATTCCCAGCAAC | ORF verification |
| Q-grid1-F | AACATTCTGGGCCAACCCAA | RT-qPCR |
| Q-grid1-R | AGCTCGCTTTCCGATCACTT | RT-qPCR |
| Q-grid2-F | TGATTTCCGCAACACCCACA | RT-qPCR |
| Q-grid2-R | AGGGCTACGGAAAAAGCCAC | RT-qPCR |
| β-actin-F | TTCCAGCCTTCCTTCCTT | RT-qPCR |
| β-actin-R | TACCTCCAGACAGCACAG | RT-qPCR |
| gh-F | ATCCACGCAGCCGGTTATAG | RT-qPCR |
| gh-R | CTCATGCTTGTTGTCGGGGA | RT-qPCR |
| igf1-F | ATGTCCATCTCTGCTCCGTC | RT-qPCR |
| igf1-R | GAAATAAAAGCCTCTCTCTCCAC | RT-qPCR |
| sex-F | CCTAAATGATGGATGTAGATTCTGTC | Gender detection |
| sex-R | GATCCAGAGAAAATAAACCCAGG | Gender detection |
| Q-cdca7-F | AGGAAATGCTCGCCAAACTG | RT-qPCR |
| Q-cdca7-R | TCTTCCTGCATGATCCCGGT | RT-qPCR |
| Q-shcbp1-F | GGCTACAAAGGAAATGCCAGG | RT-qPCR |
| Q-shcbp1-R | CAGGAGTCTGGTAGTGAGCAG | RT-qPCR |
| Q-sass6-F | GAACGAGTCGTCCATCAGAGA | RT-qPCR |
| Q-sass6-R | CTGACTCACCAAACGCTCCT | RT-qPCR |
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| Y1_1 | 48,615,382 | 7,340,922,682 | 48,102,788 | 7,177,069,845 | 0.0125 | 98.41 | 95.14 | 47.25 |
| Y1_2 | 53,804,658 | 8,124,503,358 | 53,197,338 | 7,933,840,777 | 0.0125 | 98.42 | 95.2 | 46.72 |
| Y1_3 | 47,486,224 | 7,170,419,824 | 46,941,306 | 6,986,664,819 | 0.0124 | 98.44 | 95.27 | 46.59 |
| NC_1_1 | 51,427,878 | 7,765,609,578 | 50,797,030 | 7,578,762,047 | 0.0125 | 98.43 | 95.24 | 47.06 |
| NC_1_2 | 51,475,210 | 7,772,756,710 | 50,868,992 | 7,577,515,371 | 0.0125 | 98.42 | 95.2 | 47.01 |
| NC_1_3 | 45,453,332 | 6,863,453,132 | 44,943,864 | 6,709,376,354 | 0.0125 | 98.41 | 95.18 | 46.48 |
| Sample | Raw Reads | Raw Bases | Clean Reads | Clean Bases | Error Rate (%) | Q20 (%) | Q30 (%) | GC Content (%) |
|---|---|---|---|---|---|---|---|---|
| Y2_1 | 47,141,102 | 7,118,306,402 | 46,616,214 | 6,960,704,393 | 0.0125 | 98.41 | 95.18 | 46.58 |
| Y2_2 | 46,248,072 | 6,983,458,872 | 45,720,266 | 6,842,093,724 | 0.0126 | 98.34 | 94.94 | 46.89 |
| Y2_3 | 48,204,732 | 7,278,914,532 | 47,684,778 | 7,130,821,946 | 0.0126 | 98.37 | 95.06 | 46.88 |
| NC_2_1 | 45,554,842 | 6,878,781,142 | 45,025,372 | 6,721,019,003 | 0.0125 | 98.4 | 95.16 | 46.81 |
| NC_2_2 | 45,094,196 | 6,809,223,596 | 44,599,126 | 6,662,407,768 | 0.0126 | 98.38 | 95.08 | 46.61 |
| NC_2_3 | 49,930,426 | 7,539,494,326 | 49,407,854 | 7,390,924,078 | 0.0126 | 98.37 | 95.04 | 47.16 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Chen, Y.; Liu, Y.; Chen, S. Molecular Mechanism of the Grid Gene Family Regulating Growth Size Heteromorphism in Cynoglossus semilaevis. Animals 2025, 15, 1130. https://doi.org/10.3390/ani15081130
Wang Y, Chen Y, Liu Y, Chen S. Molecular Mechanism of the Grid Gene Family Regulating Growth Size Heteromorphism in Cynoglossus semilaevis. Animals. 2025; 15(8):1130. https://doi.org/10.3390/ani15081130
Chicago/Turabian StyleWang, Yaning, Yadong Chen, Yang Liu, and Songlin Chen. 2025. "Molecular Mechanism of the Grid Gene Family Regulating Growth Size Heteromorphism in Cynoglossus semilaevis" Animals 15, no. 8: 1130. https://doi.org/10.3390/ani15081130
APA StyleWang, Y., Chen, Y., Liu, Y., & Chen, S. (2025). Molecular Mechanism of the Grid Gene Family Regulating Growth Size Heteromorphism in Cynoglossus semilaevis. Animals, 15(8), 1130. https://doi.org/10.3390/ani15081130