
Genomic Dissection of Chinese Yangtze River Delta White Goat Based on Whole Genome Sequencing
 , , ,
, , ,
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection
2.2. Genomic DNA Extraction and Sequencing Libraries Construction
2.3. High-Throughput Sequencing and Other Sequencing Data Downloads
2.4. Mapping and Variant Calling
2.5. Population Genetic Analysis
2.6. Genome-Wide Selective Sweeps Detection
2.7. Enrichment Analysis and PPI Network Construction
3. Results
3.1. Genetic Variant Identification
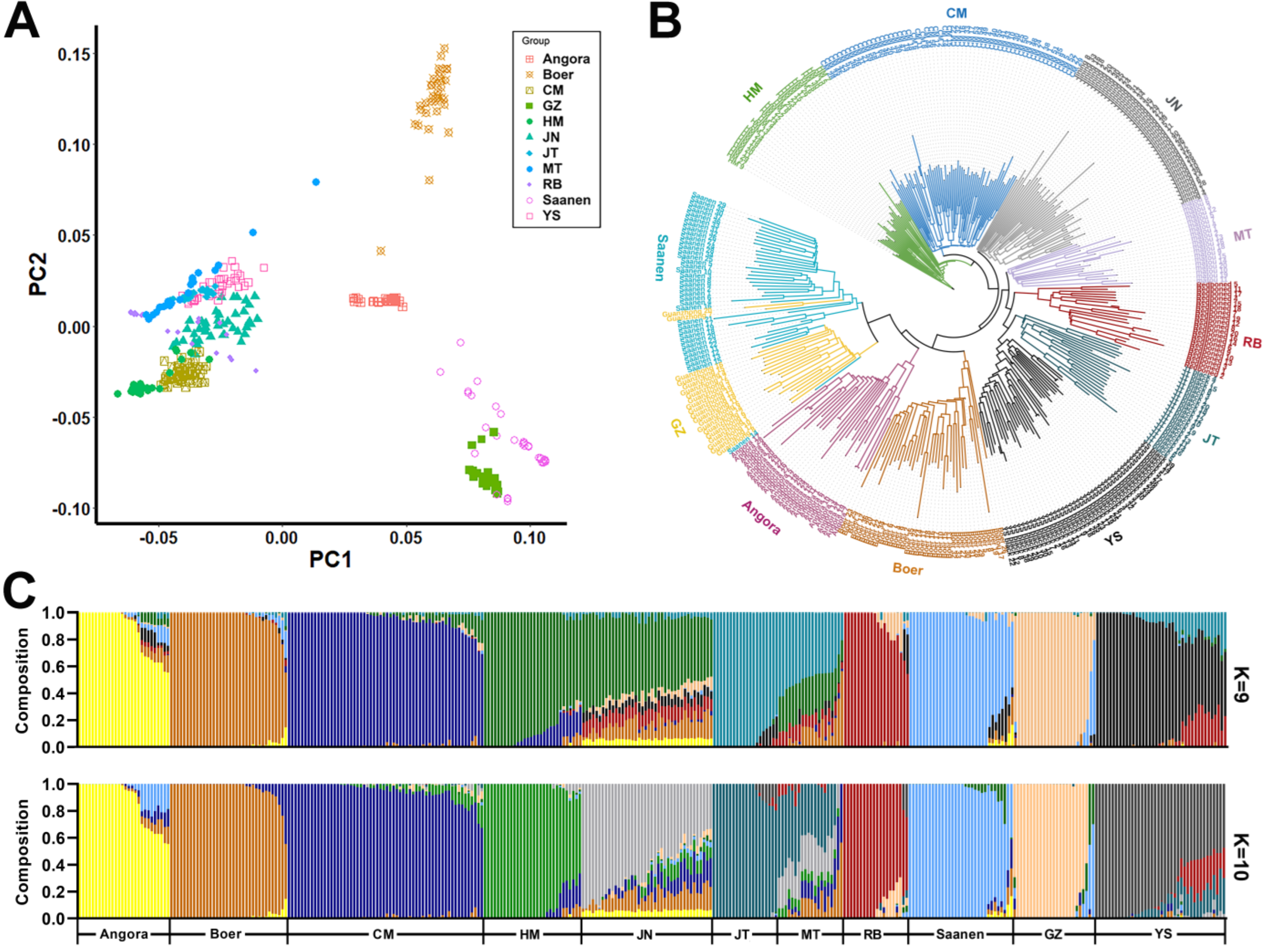
3.2. Population Genetic Structure
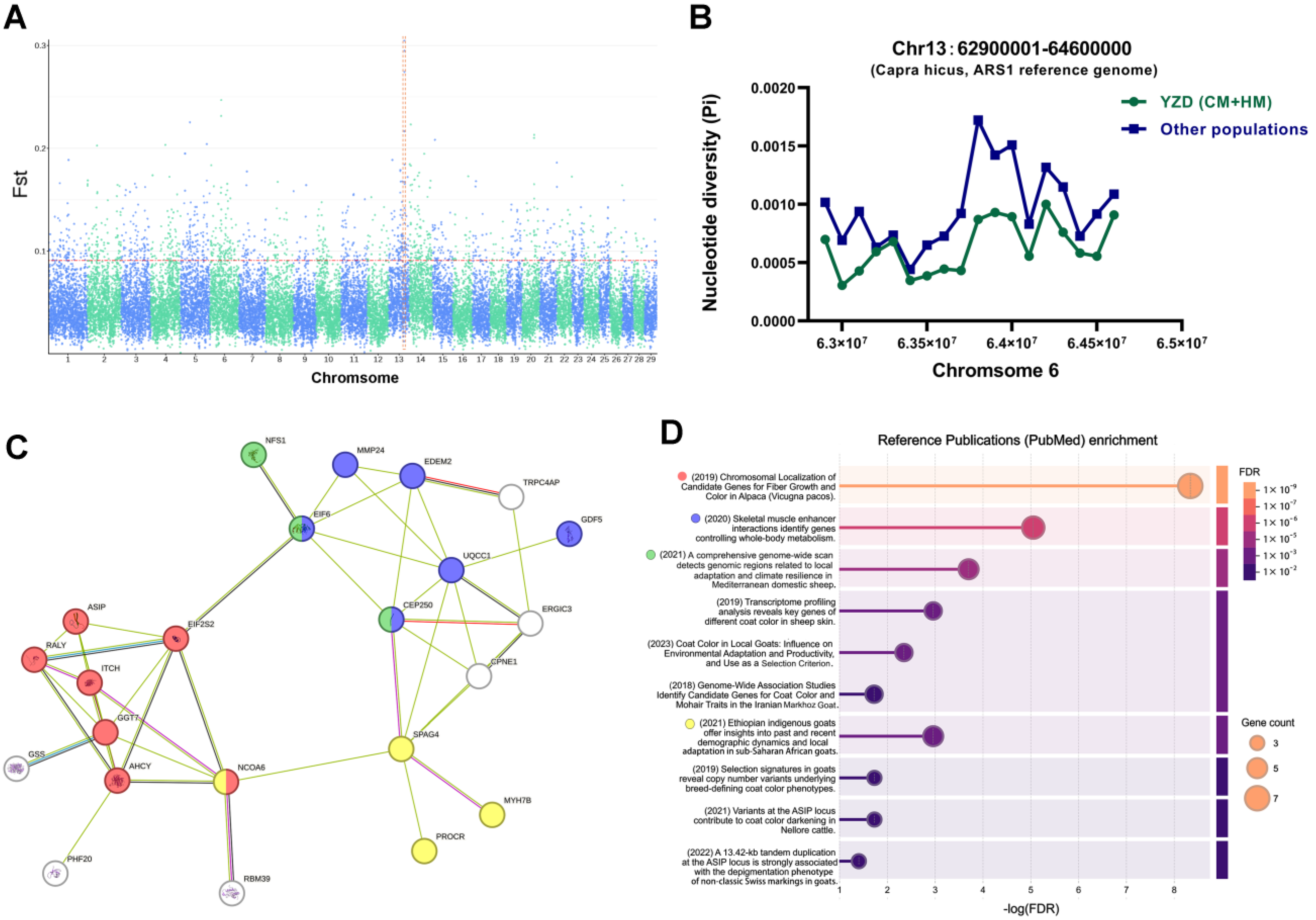
3.3. Selection-Signature Detection
3.4. Functional Analysis of Genes in Selection-Signature Regions
3.5. Polymorphisms of MYH7B Within YRD Population
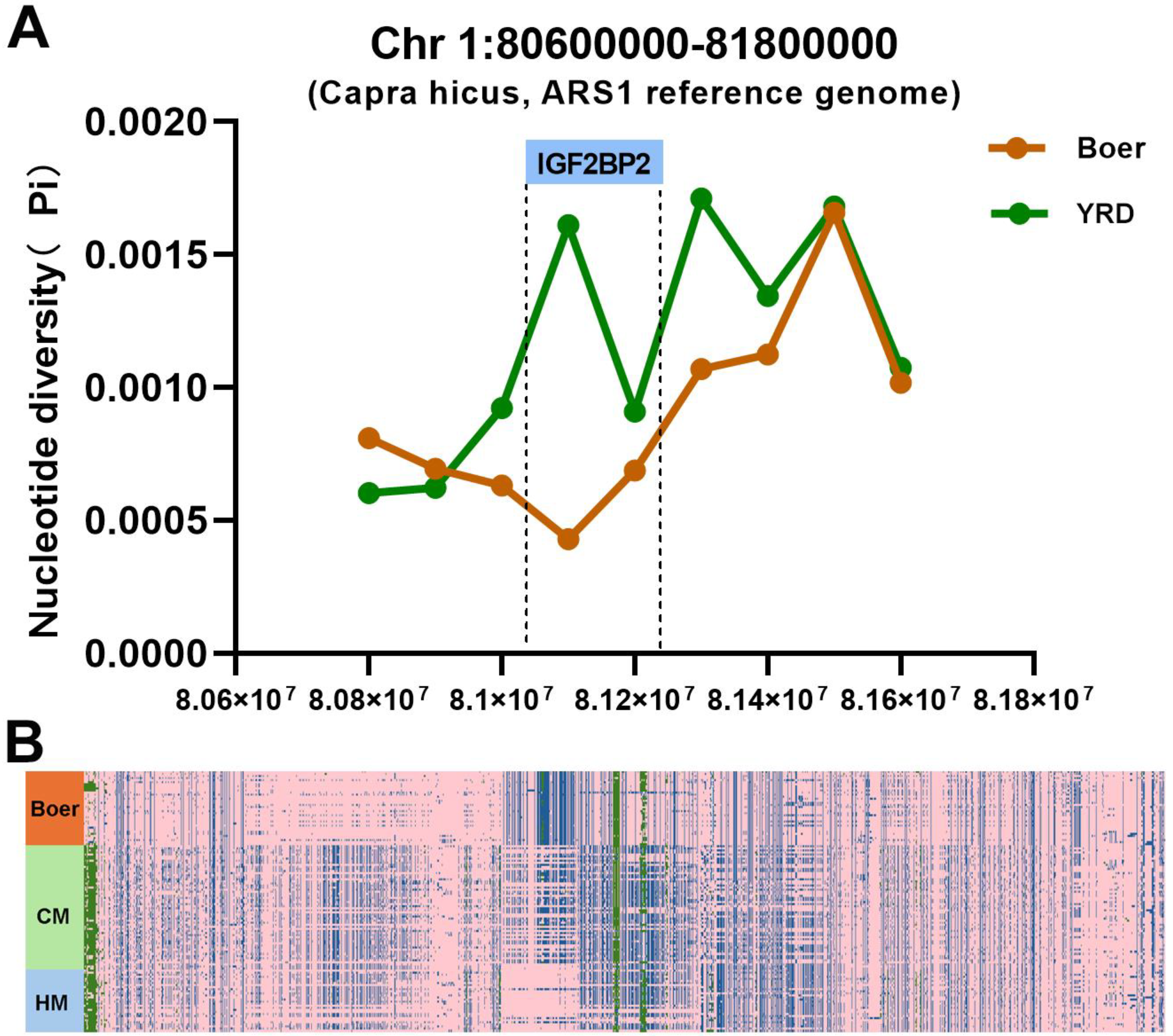
3.6. IGF2BP2 Under Selection in Boer Goat
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nigussie, H.; Legesse, Y.; Pal, S.K. Domestic Animal Diversity Information System (DAD-IS): Its contribution for Animal Genetic Resources (AnGR) management. In Proceedings of the 19th Annual Conference of Ethiopian Society of Animal Production (ESAP), Addis Ababa, Ethiopia, 15–17 December 2011; pp. 15–17. [Google Scholar]
- Gao, J.; Lyu, Y.; Zhang, D.; Reddi, K.K.; Sun, F.; Yi, J.; Liu, C.; Li, H.; Yao, H.; Dai, J.; et al. Genomic characteristics and selection signatures in indigenous chongming white goat (Capra hircus). Front. Genet. 2020, 11, 901. [Google Scholar]
- Huang, X.; Chen, Z.; Wang, K.; He, P.; Li, F.; Zhang, F.; Qi, K.; Song, Q.; Tang, Y.; Tang, Y. A modified goat model of acute spinal cord compression injury from a percutaneous balloon catheter: Method feasibility and preliminary observation. Zhonghua Yi Xue Za Zhi 2013, 93, 2993–2996. [Google Scholar] [PubMed]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Nei, M.; Li, W.-H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 1979, 76, 5269–5273. [Google Scholar]
- Voight, B.F.; Kudaravalli, S.; Wen, X.; Pritchard, J.K. A map of recent positive selection in the human genome. PLoS Biol. 2006, 4, e72. [Google Scholar]
- Chang, L.; Zheng, Y.; Li, S.; Niu, X.; Huang, S.; Long, Q.; Ran, X.; Wang, J. Identification of genomic characteristics and selective signals in Guizhou black goat. BMC Genom. 2024, 25, 164. [Google Scholar]
- Chen, Q.; Chai, Y.; Zhang, W.; Cheng, Y.; Zhang, Z.; An, Q.; Chen, S.; Man, C.; Du, L.; Zhang, W. Whole-genome sequencing reveals the genomic characteristics and selection signatures of hainan black goat. Genes 2022, 13, 1539. [Google Scholar] [CrossRef]
- Russell, D.W.; Sambrook, J. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2001; Volume 1. [Google Scholar]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar]
- Bickhart, D.M.; Rosen, B.D.; Koren, S.; Sayre, B.L.; Hastie, A.R.; Chan, S.; Lee, J.; Lam, E.T.; Liachko, I.; Sullivan, S.T. Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome. Nat. Genet. 2017, 49, 643–650. [Google Scholar]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [PubMed]
- Wickham, H. Ggplot2. In Wiley Interdisciplinary Reviews: Computational Statistics; John Wiley & Sons: Hoboken, NJ, USA, 2011; Volume 3, pp. 180–185. [Google Scholar]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar]
- Wu, T.; Hu, E.; Xu, S.; Chen, M.; Guo, P.; Dai, Z.; Feng, T.; Zhou, L.; Tang, W.; Zhan, L. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation 2021, 2, 100141. [Google Scholar] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar]
- Chen, T.; Chen, X.; Zhang, S.; Zhu, J.; Tang, B.; Wang, A.; Dong, L.; Zhang, Z.; Yu, C.; Sun, Y. The genome sequence archive family: Toward explosive data growth and diverse data types. Genom. Proteom. Bioinform. 2021, 19, 578–583. [Google Scholar]
- Partners, C.-N.M.a. Database resources of the national genomics data center, china national center for bioinformation in 2022. Nucleic Acids Res. 2022, 50, D27–D38. [Google Scholar]
- Xiong, J.; Bao, J.; Hu, W.; Shang, M.; Zhang, L. Whole-genome resequencing reveals genetic diversity and selection characteristics of dairy goat. Front. Genet. 2023, 13, 1044017. [Google Scholar]
- Rossi, A.C.; Mammucari, C.; Argentini, C.; Reggiani, C.; Schiaffino, S. Two novel/ancient myosins in mammalian skeletal muscles: MYH14/7b and MYH15 are expressed in extraocular muscles and muscle spindles. J. Physiol. 2010, 588, 353–364. [Google Scholar] [PubMed]
- Lee, L.A.; Karabina, A.; Broadwell, L.J.; Leinwand, L.A. The ancient sarcomeric myosins found in specialized muscles. Nucleic Acids Res. 2019, 9, 7. [Google Scholar]
- Yeung, F.; Chung, E.; Guess, M.G.; Bell, M.L.; Leinwand, L.A. Myh7b/miR-499 gene expression is transcriptionally regulated by MRFs and Eos. Nucleic Acids Res. 2012, 40, 7303–7318. [Google Scholar]
- Zheng, Q.; Zhang, Y.; Chen, Y.; Yang, N.; Wang, X.-J.; Zhu, D. Systematic identification of genes involved in divergent skeletal muscle growth rates of broiler and layer chickens. BMC Genom. 2009, 10, 87. [Google Scholar]
- Williams, K.; Ingerslev, L.R.; Bork-Jensen, J.; Wohlwend, M.; Hansen, A.N.; Small, L.; Ribel-Madsen, R.; Astrup, A.; Pedersen, O.; Auwerx, J. Skeletal muscle enhancer interactions identify genes controlling whole-body metabolism. Nat. Commun. 2020, 11, 2695. [Google Scholar]
- Mendoza, M.N.; Raudsepp, T.; Alshanbari, F.; Gutiérrez, G.; Ponce de León, F.A. Chromosomal localization of candidate genes for fiber growth and color in alpaca (Vicugna pacos). Front. Genet. 2019, 10, 583. [Google Scholar] [CrossRef]
- Voisey, J.; Van Daal, A. Agouti: From mouse to man, from skin to fat. Pigment Cell Res. 2002, 15, 10–18. [Google Scholar] [PubMed]
- Guo, J.; Tao, H.; Li, P.; Li, L.; Zhong, T.; Wang, L.; Ma, J.; Chen, X.; Song, T.; Zhang, H. Whole-genome sequencing reveals selection signatures associated with important traits in six goat breeds. Sci. Rep. 2018, 8, 10405. [Google Scholar]
- Wang, X.; Liu, J.; Zhou, G.; Guo, J.; Yan, H.; Niu, Y.; Li, Y.; Yuan, C.; Geng, R.; Lan, X. Whole-genome sequencing of eight goat populations for the detection of selection signatures underlying production and adaptive traits. Sci. Rep. 2016, 6, 38932. [Google Scholar]
- Dreger, D.L.; Parker, H.G.; Ostrander, E.A.; Schmutz, S.M. Identification of a mutation that is associated with the saddle tan and black-and-tan phenotypes in Basset Hounds and Pembroke Welsh Corgis. J. Hered. 2013, 104, 399–406. [Google Scholar]
- Zhao, Y.-J.; Huang, Y.-F. Selection signatures of litter size in Dazu black goats based on a whole genome sequencing mixed pools strategy. Mol. Biol. Rep. 2019, 46, 5517–5523. [Google Scholar]
- Dongyun, X.; Yangyang, B.; Yi, B.; Libang, H.; Yuxin, K.; Chuanying, P.; Haijing, Z.; Hong, C.; Lei, Q.; Xianyong, L. Insertion/deletion variants within the IGF2BP2 gene identified in reported genome-wide selective sweep analysis reveal a correlation with goat litter size. J. Zhejiang Univ. Sci. B 2021, 22, 757. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed (Individuals) | Clean Reads Number | Clean Bases (bp) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|
| CM (60) | 182,979,278 | 27,392,763,250 | 97.93 | 93.30 |
| HM (30) | 181,124,770 | 27,126,731,221 | 97.48 | 91.91 |
| Angora (28) | 248,429,018 | 37,297,824,732 | 97.00 | 92.12 |
| Boer (36) | 463,540,265 | 64,152,217,674 | 93.85 | 86.17 |
| Saanen (38) | 413,572,169 | 53,331,587,696 | 95.83 | 89.36 |
| Guanzhong (20) | 204,353,917 | 30,555,062,219 | 97.45 | 92.74 |
| Jining (40) | 226,665,695 | 33,837,615,989 | 97.44 | 95.07 |
| Jingtang (20) | 139,666,239 | 20,923,429,866 | 97.31 | 92.34 |
| Matou (20) | 239,152,704 | 35,849,687,942 | 97.36 | 92.88 |
| Redbone (20) | 356,169,599 | 35,597,422,417 | 97.97 | 91.12 |
| Yunshang(40) | 405,412,921 | 60,633,301,251 | 98.03 | 94.20 |
| Annotation | SNP | InDel |
|---|---|---|
| intergenic | 34,911,798 | 6,701,733 |
| intronic | 14,940,079 | 3,092,147 |
| ncRNA_intronic | 2,177,750 | 425,960 |
| upstream | 773,104 | 181,920 |
| downstream | 749,689 | 179,338 |
| exonic | 497,326 | 43,198 |
| UTR3 | 178,363 | 45,921 |
| ncRNA_exonic | 84,879 | 15,518 |
| UTR5 | 44,842 | 12,067 |
| upstream;downstream | 30,045 | 8448 |
| splicing | 3037 | 4348 |
| ncRNA_splicing | 409 | 58 |
| exonic;splicing | 328 | 159 |
| UTR5;UTR3 | 132 | 28 |
| ncRNA_exonic;splicing | 28 | 9 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Sun, L.; Liao, R.; Lyu, Y.; Zhang, S.; Xu, J.; He, M.; Wu, C.; Zhang, D.; Lin, Y.; et al. Genomic Dissection of Chinese Yangtze River Delta White Goat Based on Whole Genome Sequencing. Animals 2025, 15, 979. https://doi.org/10.3390/ani15070979
Gao J, Sun L, Liao R, Lyu Y, Zhang S, Xu J, He M, Wu C, Zhang D, Lin Y, et al. Genomic Dissection of Chinese Yangtze River Delta White Goat Based on Whole Genome Sequencing. Animals. 2025; 15(7):979. https://doi.org/10.3390/ani15070979
Chicago/Turabian StyleGao, Jun, Lingwei Sun, Rongrong Liao, Yuhua Lyu, Shushan Zhang, Jiehuan Xu, Mengqian He, Caifeng Wu, Defu Zhang, Yuexia Lin, and et al. 2025. "Genomic Dissection of Chinese Yangtze River Delta White Goat Based on Whole Genome Sequencing" Animals 15, no. 7: 979. https://doi.org/10.3390/ani15070979
APA StyleGao, J., Sun, L., Liao, R., Lyu, Y., Zhang, S., Xu, J., He, M., Wu, C., Zhang, D., Lin, Y., & Dai, J. (2025). Genomic Dissection of Chinese Yangtze River Delta White Goat Based on Whole Genome Sequencing. Animals, 15(7), 979. https://doi.org/10.3390/ani15070979