
Transcriptomics Reveals the Differences in mRNA Expression Patterns in Yak Uterus of Follicular, Luteal, and Pregnant Phases

 ,
,
Simple Summary
Abstract
1. Introduction
2. Methods and Materials
2.1. Sample Collections
2.2. Real-Time Quantitative Polymerase Reaction (RT-qPCR)
2.3. RNA Sequencing
2.3.1. Construction of cDNA Library and Transcriptome Sequencing
2.3.2. RNA-Seq and Data Processing
2.3.3. Enrichment Analysis of Differentially Expressed Genes (DEGs)
3. Results
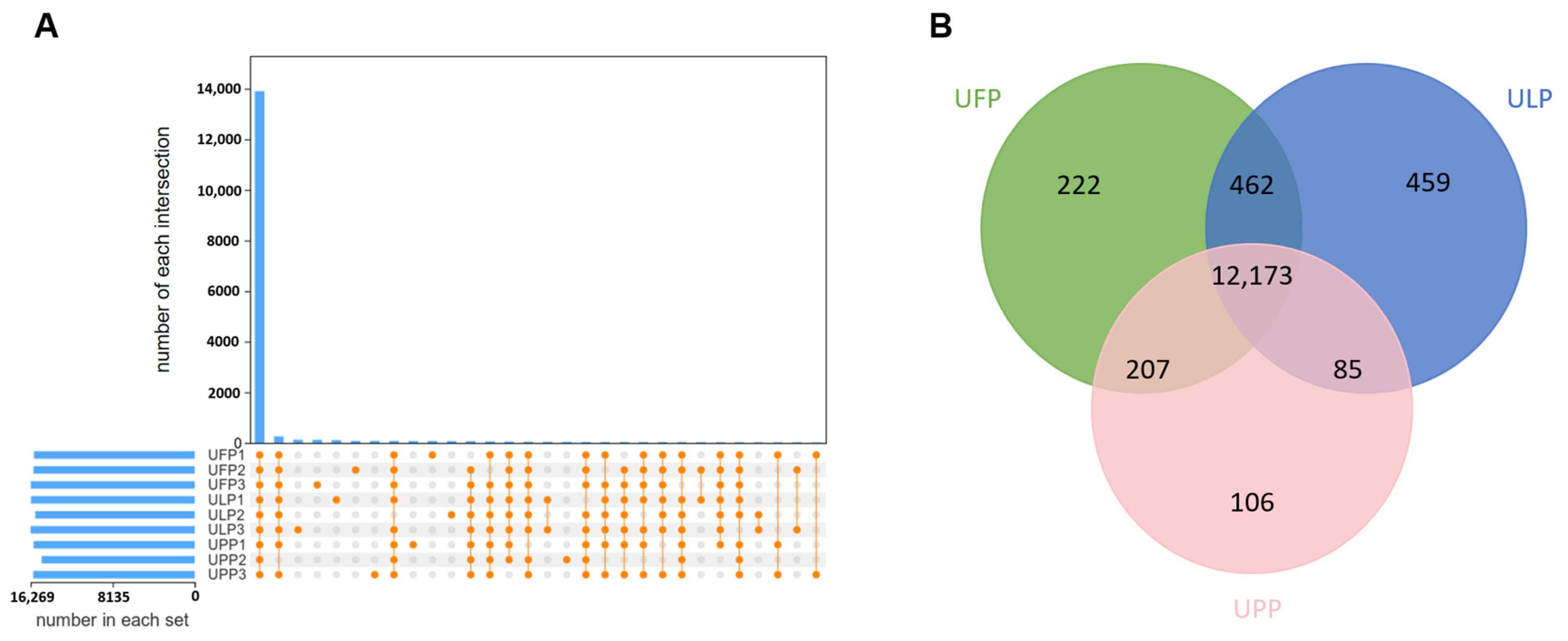
3.1. Transcriptome Reveals the Gene Expression Patterns in Yaks Among the Uterus of Follicular Phase (UFP), the Uterus of Luteal Phase (ULP), and the Uterus of Pregnant Phase (UPP)
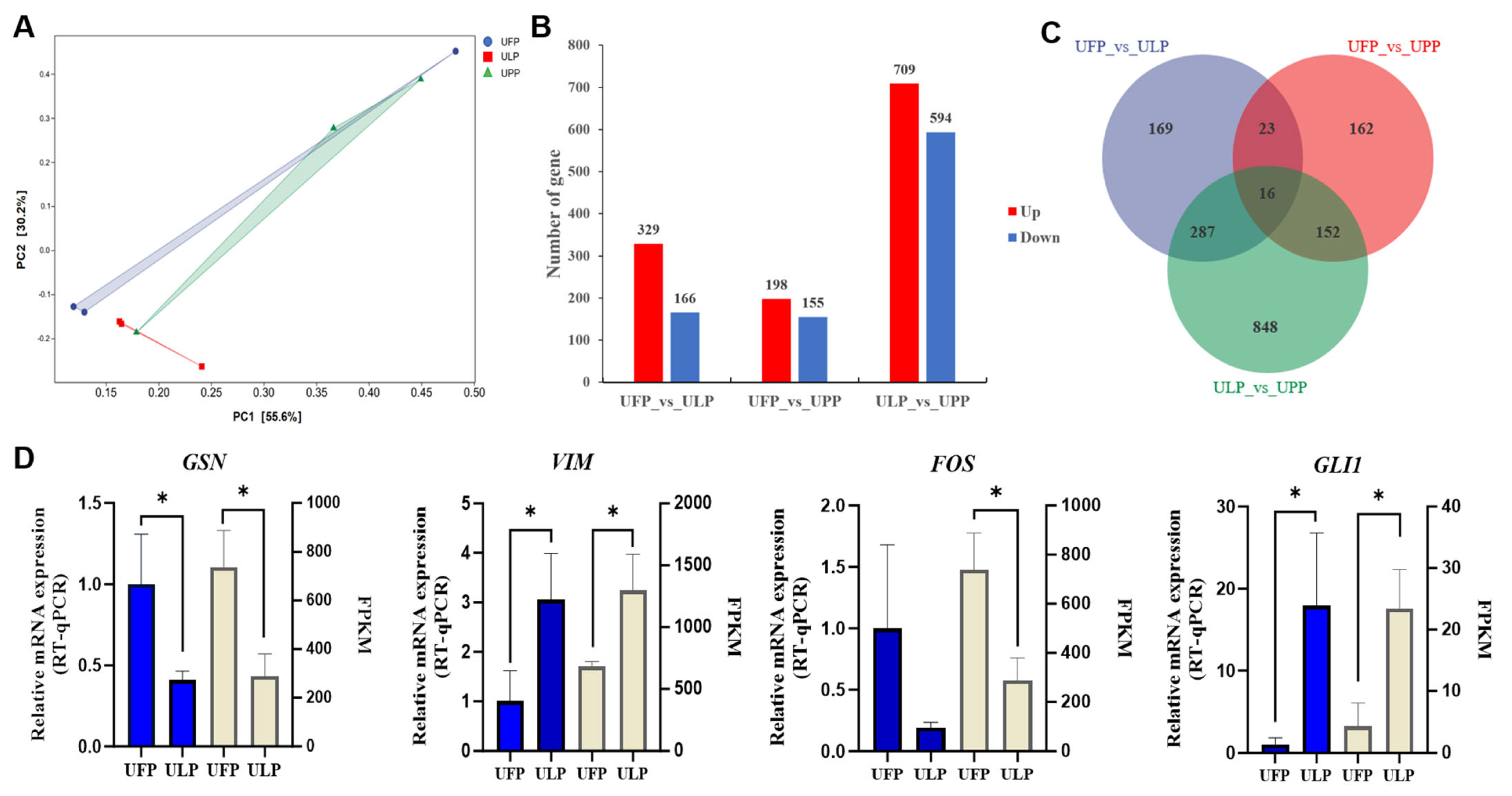
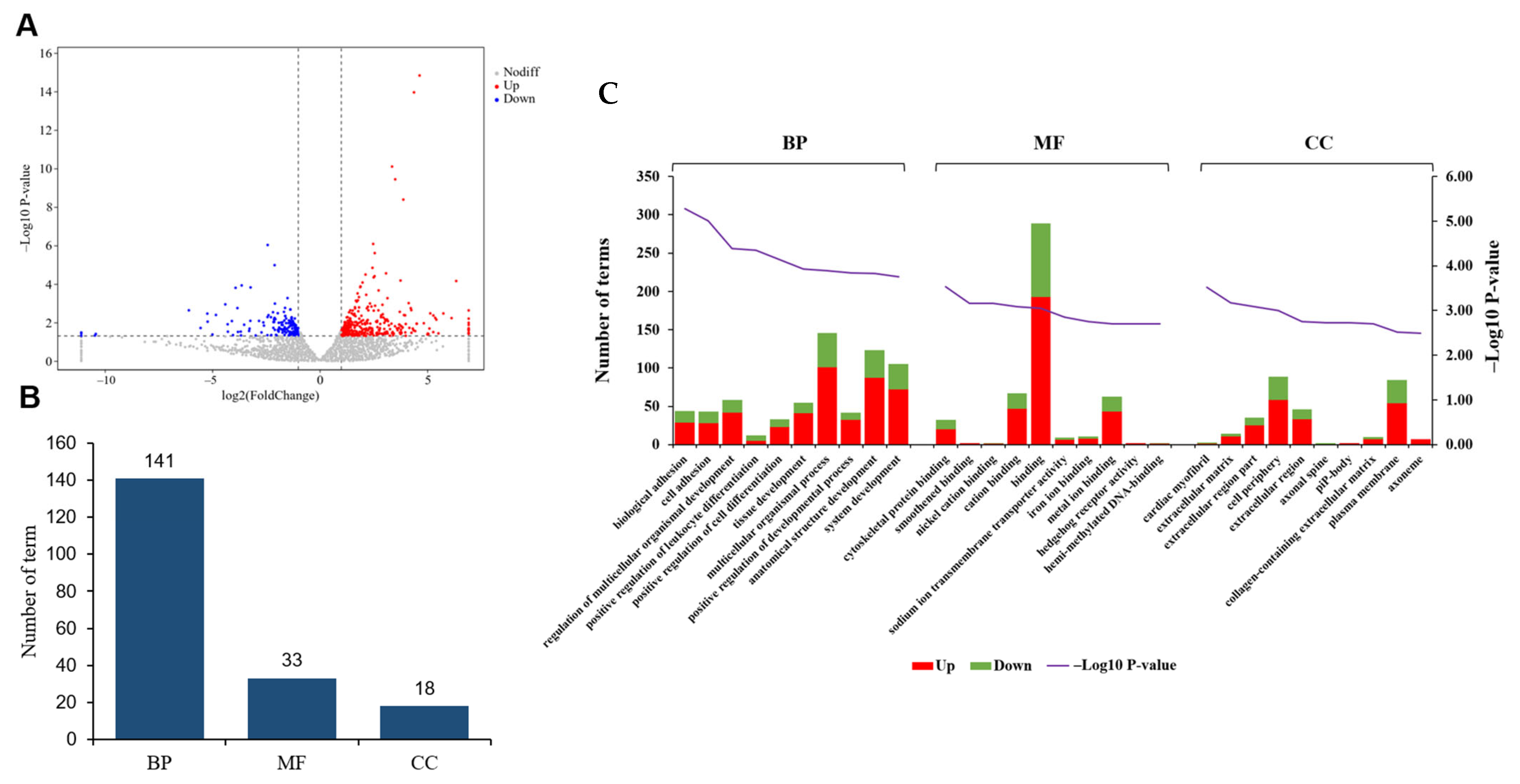
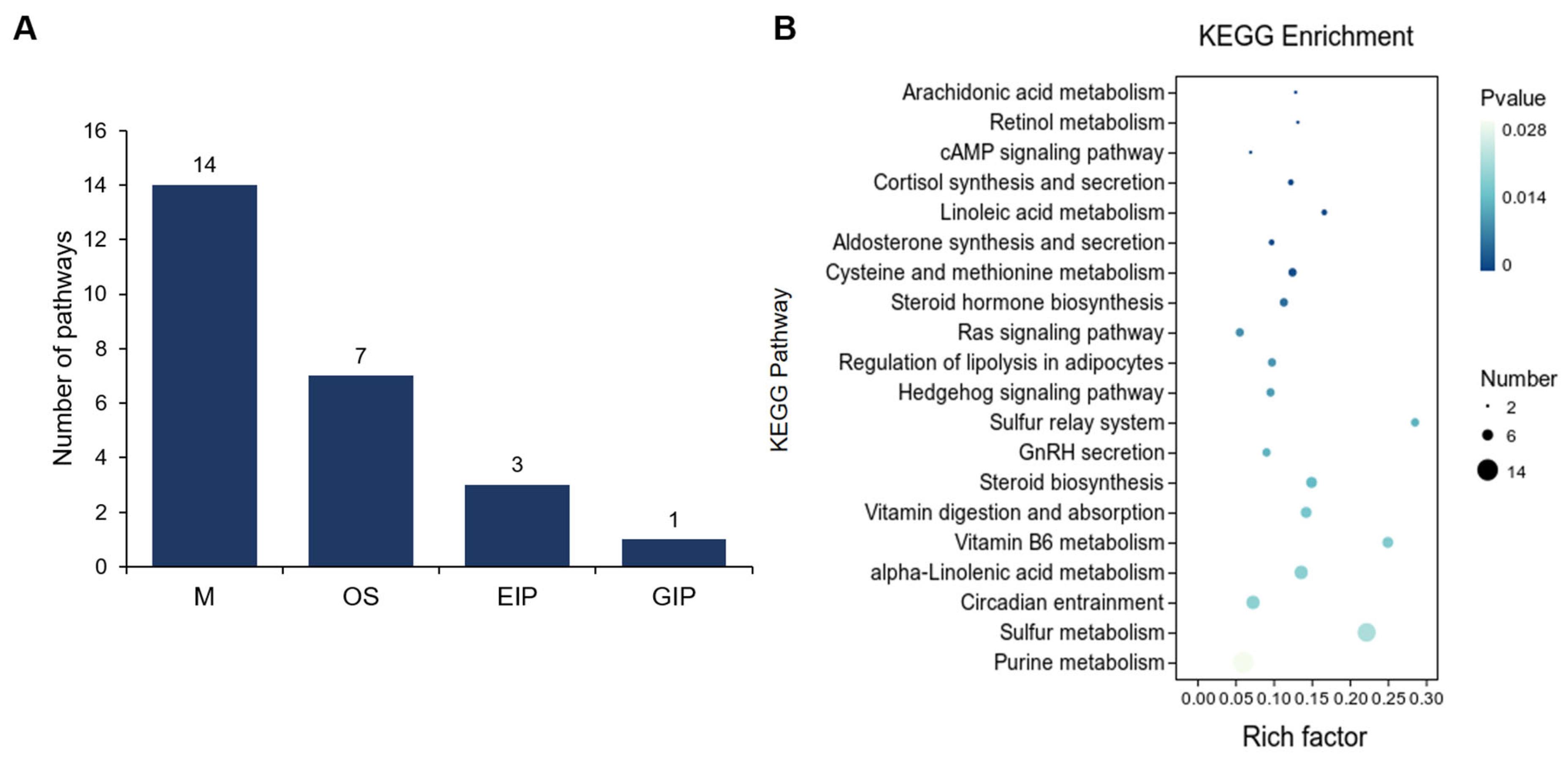
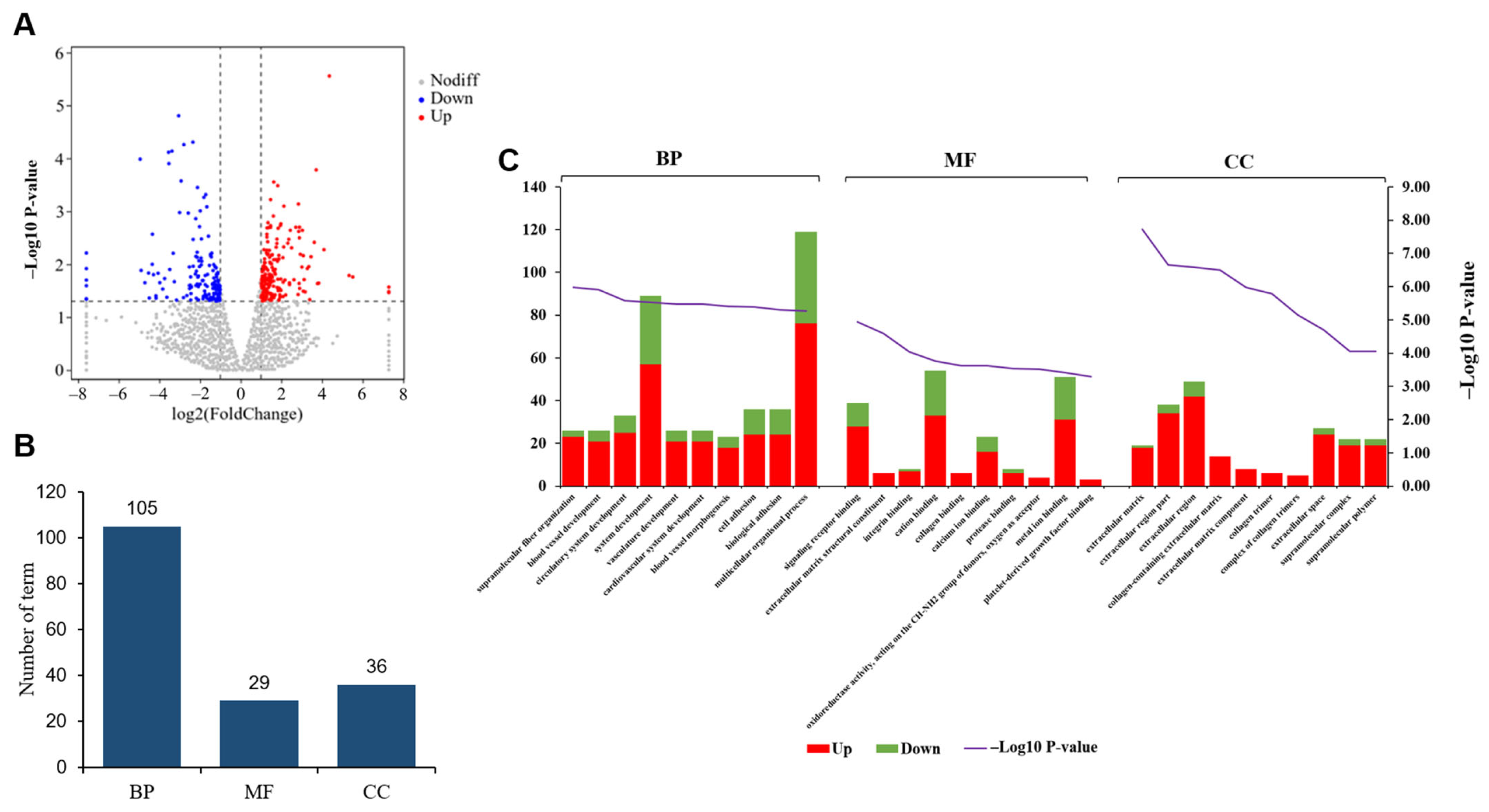
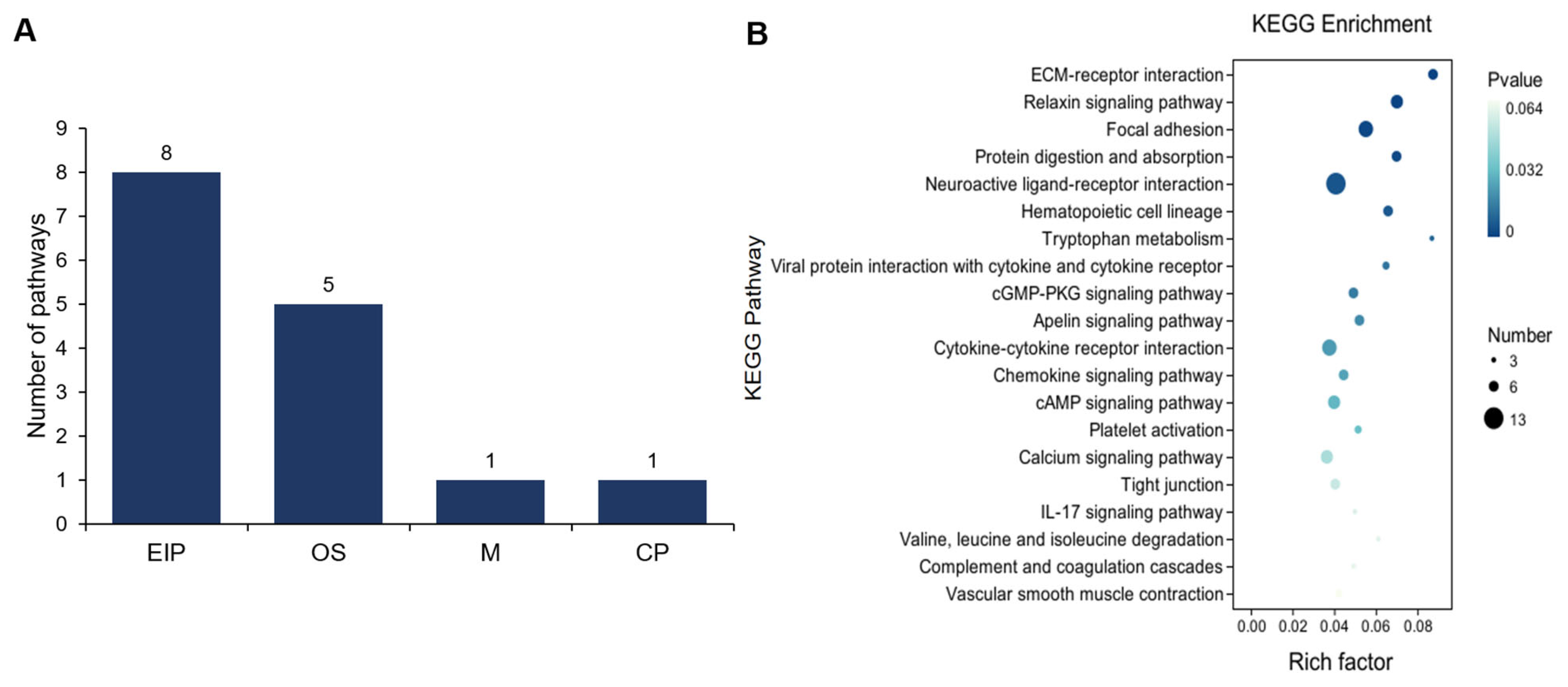
3.2. Comparative Analysis of Gene Expression Patterns in Yak Uterus During the Follicular Phase (UFP) and Luteal Phase (ULP)
3.3. Comparative Analysis of Gene Expression Patterns in Yak Uterus During the Follicular Phase (UFP) and Pregnant Phase (UPP)
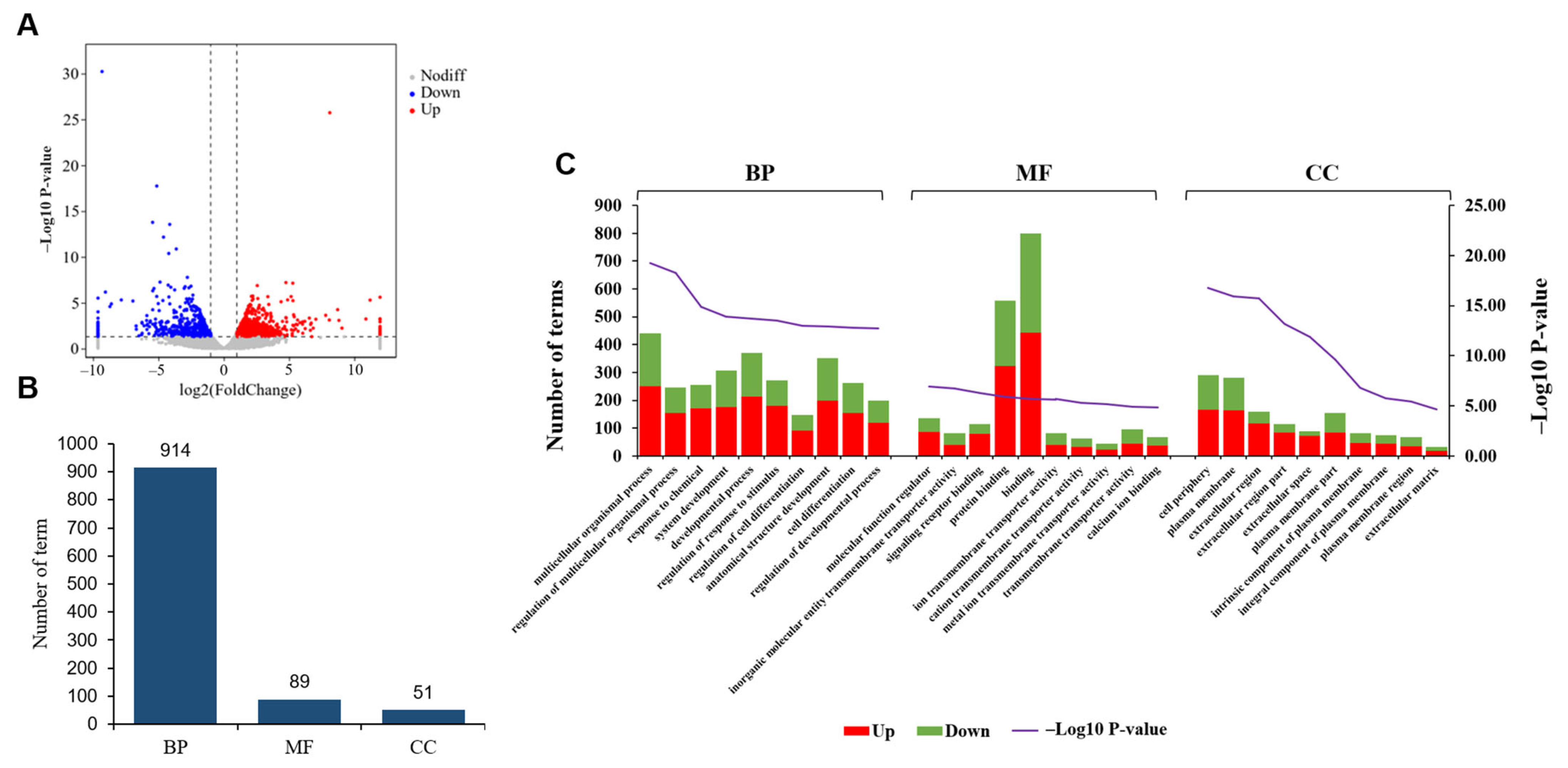
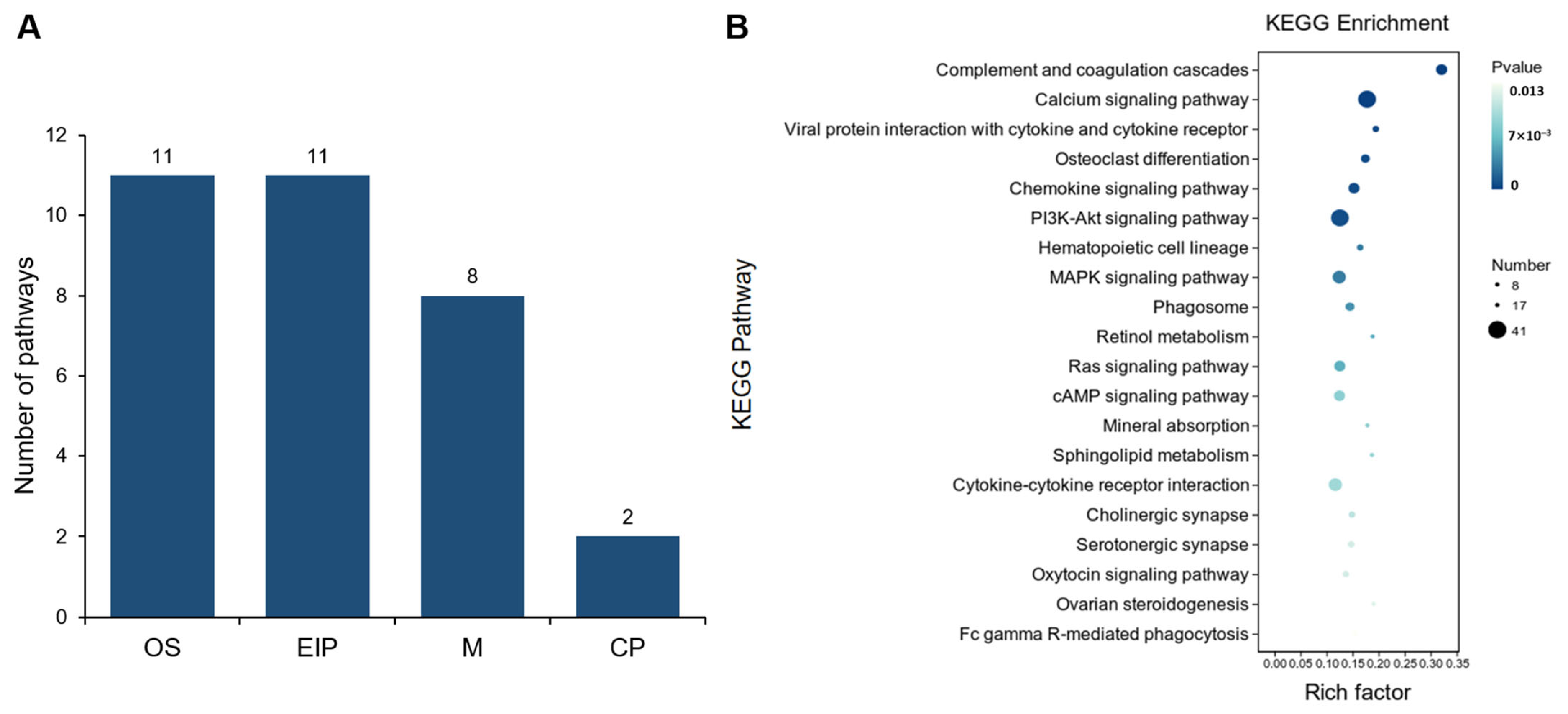
3.4. Comparative Analysis of Gene Expression Patterns in Yak Uterus During the Luteal Phase (ULP)and Pregnant Phase (UPP)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Qiu, Q.; Zhang, G.; Ma, T.; Qian, W.; Wang, J.; Ye, Z.; Cao, C.; Hu, Q.; Kim, J.; Larkin, D.M.; et al. The yak genome and adaptation to life at high altitude. Nat. Genet. 2012, 44, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.X.; Xin, J.W.; Zhang, C.F.; Dawayangla; Luosang; Zhang, Q.; Pingcuozhandui; Li, C.; Zhu, Y.; Cao, H.W.; et al. Whole-genome resequencing provides insights into the evolution and divergence of the native domestic yaks of the Qinghai-Tibet Plateau. BMC Evol. Biol. 2020, 20, 137. [Google Scholar] [CrossRef]
- Jing, X.; Ding, L.; Zhou, J.; Huang, X.; Degen, A.; Long, R. The adaptive strategies of yaks to live in the Asian highlands. Anim. Nutr. 2022, 9, 249–258. [Google Scholar] [CrossRef]
- Yin, S.; Li, Z.; Yang, F.; Guo, H.; Zhao, Q.; Zhang, Y.; Yin, Y.; Wu, X.; He, J. A Comprehensive Genomic Analysis of Chinese Indigenous Ningxiang Pigs: Genomic Breed Compositions, Runs of Homozygosity, and Beyond. Int. J. Mol. Sci. 2023, 24, 14550. [Google Scholar] [CrossRef]
- Yu, S.J.; Huang, Y.M.; Chen, B.X. Reproductive patterns of the yak. I. Reproductive phenomena of the female yak. Br. Vet. J. 1993, 149, 579–583. [Google Scholar] [CrossRef]
- Zi, X.D. Reproduction in female yaks (Bos grunniens) and opportunities for improvement. Theriogenology 2003, 59, 1303–1312. [Google Scholar] [CrossRef]
- Prakash, B.S.; Sarkar, M.; Mondal, M. An update on reproduction in yak and mithun. Reprod. Domest. Anim. 2008, 43 (Suppl. 2), 217–223. [Google Scholar] [CrossRef]
- Xu, S.R.; Wei, P.; Yang, Q.L.; Jia, G.X.; Ma, S.K.; Yang, Q.E.; Jun, Z.; Zhang, R.N. Transcriptome analysis revealed key signaling networks regulating ovarian activities in the domestic yak. Theriogenology 2020, 147, 50–56. [Google Scholar] [CrossRef]
- Boutelis, S.; Laanani, I.; Yahia, M.; Massinissa, Y. Evaluation of the Cytology and Histology of Uterus and Cervix As Predictors of Estrous Stages in Ewes and Dairy Cows. Int. J. Syst. Appl. Eng. Dev. 2022, 16, 38–40. [Google Scholar] [CrossRef]
- Acharya, R.Y.; Hemsworth, P.H.; Coleman, G.J.; Kinder, J.E. The Animal-Human Interface in Farm Animal Production: Animal Fear, Stress, Reproduction and Welfare. Animals 2022, 12, 487. [Google Scholar] [CrossRef]
- Ortega, M.A.; Fraile-Martínez, O.; García-Montero, C.; Paradela, A.; Asunción Sánchez-Gil, M.; Rodriguez-Martin, S.; De León-Luis, J.A.; Pereda-Cerquella, C.; Bujan, J.; Guijarro, L.G.; et al. Unfolding the role of placental-derived Extracellular Vesicles in Pregnancy: From homeostasis to pathophysiology. Front. Cell Dev. Biol. 2022, 10, 1060850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, L.; Gu, Y.; Hu, B.; Zhou, J. Observation on Aging Changes of Uterine Tissue Structure of Huanghuai White Goats with Different Days of Age. J. Anhui Sci. Technol. Univ. 2022, 36, 5–11. [Google Scholar] [CrossRef]
- Mendelson, C.R.; Gao, L.; Montalbano, A.P. Multifactorial Regulation of Myometrial Contractility During Pregnancy and Parturition. Front. Endocrinol. 2019, 10, 714. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, S.K.; Lee, J.R.; Woodruff, T.K. Ovary is necessary to the health of uterus. J. Gynecol. Oncol. 2016, 27, e35. [Google Scholar] [CrossRef]
- Dobrowolski, W.; Hafez, E.S. The uterus and control of ovarian function. Acta Obs. Gynecol. Scand. Suppl. 1971, 12, 1–26. [Google Scholar] [CrossRef]
- Levin, R.J. The Involvement of the Human Cervix in Reproduction and Sex. Sex. Relatsh. Ther. 2005, 20, 251–260. [Google Scholar] [CrossRef]
- Muter, J.; Lynch, V.J.; McCoy, R.C.; Brosens, J.J. Human embryo implantation. Development 2023, 150, dev201507. [Google Scholar] [CrossRef]
- Spencer, T.E. Biological roles of uterine glands in pregnancy. Semin. Reprod. Med. 2014, 32, 346–357. [Google Scholar] [CrossRef]
- Dunlap, K.A.; Brown, J.D.; Keith, A.B.; Satterfield, M.C. Factors controlling nutrient availability to the developing fetus in ruminants. J. Anim. Sci. Biotechnol. 2015, 6, 16. [Google Scholar] [CrossRef]
- Bauersachs, S.; Almiñana, C. Embryo-Maternal Interactions Underlying Reproduction in Mammals. Int. J. Mol. Sci. 2020, 21, 4872. [Google Scholar] [CrossRef]
- Yu, J.; Liu, C.; Chen, H.; Xiang, M.; Hu, X.; Zhong, Z.; Liu, Q.; Wang, D.; Cheng, L. Transcriptomic analysis of bovine endometrial epithelial cells in response to interferon tau and hormone stimulation. Front. Vet. Sci. 2024, 11, 1344259. [Google Scholar] [CrossRef] [PubMed]
- Mathew, D.J.; Sánchez, J.M.; Passaro, C.; Charpigny, G.; Behura, S.K.; Spencer, T.E.; Lonergan, P. Interferon tau-dependent and independent effects of the bovine conceptus on the endometrial transcriptome†. Biol. Reprod. 2019, 100, 365–380. [Google Scholar] [CrossRef] [PubMed]
- Binelli, M.; Scolari, S.C.; Pugliesi, G.; Van Hoeck, V.; Gonella-Diaza, A.M.; Andrade, S.C.; Gasparin, G.R.; Coutinho, L.L. The transcriptome signature of the receptive bovine uterus determined at early gestation. PLoS ONE 2015, 10, e0122874. [Google Scholar] [CrossRef]
- Ding, X.; Lv, H.; Deng, L.; Hu, W.; Peng, Z.; Yan, C.; Yang, D.; Tong, C.; Wang, X. Analysis of Transcriptomic Changes in Bovine Endometrial Stromal Cells Treated With Lipopolysaccharide. Front. Vet. Sci. 2020, 7, 575865. [Google Scholar] [CrossRef]
- Fazleabas, A.T.; Bell, S.C.; Fleming, S.; Sun, J.; Lessey, B.A. Distribution of integrins and the extracellular matrix proteins in the baboon endometrium during the menstrual cycle and early pregnancy. Biol. Reprod. 1997, 56, 348–356. [Google Scholar] [CrossRef]
- Genbacev, O.D.; Prakobphol, A.; Foulk, R.A.; Krtolica, A.R.; Ilic, D.; Singer, M.S.; Yang, Z.Q.; Kiessling, L.L.; Rosen, S.D.; Fisher, S.J. Trophoblast L-selectin-mediated adhesion at the maternal-fetal interface. Science 2003, 299, 405–408. [Google Scholar] [CrossRef]
- Bazer, F.W.; Spencer, T.E.; Johnson, G.A.; Burghardt, R.C. Uterine receptivity to implantation of blastocysts in mammals. Front. Biosci. Sch. Ed. 2011, 3, 745–767. [Google Scholar] [CrossRef]
- Moorey, S.E.; Hessock, E.A.; Edwards, J.L. Preovulatory follicle contributions to oocyte competence in cattle: Importance of the ever-evolving intrafollicular environment leading up to the luteinizing hormone surge. J. Anim. Sci. 2022, 100, skac153. [Google Scholar] [CrossRef]
- Chen, H.; Yu, S.; Cui, Y. Label-free proteomic analysis reveals an estrous cycle transition between the follicular phase and the luteal phase in yak ovary. Anim. Sci. J. 2023, 94, e13855. [Google Scholar] [CrossRef]
- McCracken, J.A.; Custer, E.E.; Lamsa, J.C. Luteolysis: A neuroendocrine-mediated event. Physiol. Rev. 1999, 79, 263–323. [Google Scholar] [CrossRef]
- Arosh, J.A.; Banu, S.K.; Chapdelaine, P.; Fortier, M.A. Temporal and tissue-specific expression of prostaglandin receptors EP2, EP3, EP4, FP, and cyclooxygenases 1 and 2 in uterus and fetal membranes during bovine pregnancy. Endocrinology 2004, 145, 407–417. [Google Scholar] [CrossRef] [PubMed]
- Roumeliotis, S.; Roumeliotis, A.; Stamou, A.; Panagoutsos, S.; Manolopoulos, V.G.; Tsetsos, F.; Georgitsi, M.; Liakopoulos, V. Association of rs11780592 Polymorphism in the Human Soluble Epoxide Hydrolase Gene (EPHX2) with Oxidized LDL and Mortality in Patients with Diabetic Chronic Kidney Disease. Oxidative Med. Cell. Longev. 2021, 2021, 8817502. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yang, J.; Wang, W.; Sanidad, K.Z.; Cinelli, M.A.; Wan, D.; Hwang, S.H.; Kim, D.; Lee, K.S.S.; Xiao, H.; et al. Soluble epoxide hydrolase is an endogenous regulator of obesity-induced intestinal barrier dysfunction and bacterial translocation. Proc. Natl. Acad. Sci. USA 2020, 117, 8431–8436. [Google Scholar] [CrossRef] [PubMed]
- Utomo, A.; Jiang, X.; Furuta, S.; Yun, J.; Levin, D.S.; Wang, Y.C.; Desai, K.V.; Green, J.E.; Chen, P.L.; Lee, W.H. Identification of a novel putative non-selenocysteine containing phospholipid hydroperoxide glutathione peroxidase (NPGPx) essential for alleviating oxidative stress generated from polyunsaturated fatty acids in breast cancer cells. J. Biol. Chem. 2004, 279, 43522–43529. [Google Scholar] [CrossRef]
- Peng, D.; Belkhiri, A.; Hu, T.; Chaturvedi, R.; Asim, M.; Wilson, K.T.; Zaika, A.; El-Rifai, W. Glutathione peroxidase 7 protects against oxidative DNA damage in oesophageal cells. Gut 2012, 61, 1250–1260. [Google Scholar] [CrossRef]
- Starkl, P.; Marichal, T.; Galli, S.J. PLA2G3 promotes mast cell maturation and function. Nat. Immunol. 2013, 14, 527–529. [Google Scholar] [CrossRef]
- Shahrooz, R.; Ayan, E.; Rustami, B. Histomorphometrical Study of the Cervix During the Oestrous Cycle in Adult Azarbaijan Buffalo. Iran. J. Vet. Res. 2009, 10, 75–80. [Google Scholar] [CrossRef]
- Takemura, Y.; Osuga, Y.; Yamauchi, T.; Kobayashi, M.; Harada, M.; Hirata, T.; Morimoto, C.; Hirota, Y.; Yoshino, O.; Koga, K.; et al. Expression of adiponectin receptors and its possible implication in the human endometrium. Endocrinology 2006, 147, 3203–3210. [Google Scholar] [CrossRef]
- Palin, M.F.; Bordignon, V.V.; Murphy, B.D. Adiponectin and the control of female reproductive functions. Vitam. Horm. 2012, 90, 239–287. [Google Scholar] [CrossRef]
- Jousan, F.D.; Oliveira, L.J.; Hansen, P.J. Short-Term culture of in vitro produced bovine preimplantation embryos with insulin-like growth factor-i prevents heat shock-induced apoptosis through activation of the Phosphatidylinositol 3-Kinase/Akt pathway. Mol. Reprod. Dev. 2008, 75, 681–688. [Google Scholar] [CrossRef]
- Jousan, F.D.; Hansen, P.J. Insulin-like growth factor-I promotes resistance of bovine preimplantation embryos to heat shock through actions independent of its anti-apoptotic actions requiring PI3K signaling. Mol. Reprod. Dev. 2007, 74, 189–196. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Liu, Z.; Jia, J. Mechanisms of Smoothened Regulation in Hedgehog Signaling. Cells 2021, 10, 2138. [Google Scholar] [CrossRef]
- Sigafoos, A.N.; Paradise, B.D.; Fernandez-Zapico, M.E. Hedgehog/GLI Signaling Pathway: Transduction, Regulation, and Implications for Disease. Cancers 2021, 13, 3410. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Levens, E.D.; Stefansson, L.; Nieman, L.K. Indian Hedgehog and its targets in human endometrium: Menstrual cycle expression and response to CDB-2914. J. Clin. Endocrinol. Metab. 2010, 95, 5330–5337. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Zhao, J.; Liu, Y.; Ba, H.; Wei, H.; Zhang, Y.; Wang, G.; Murphy, B.D.; Xing, X. Transcriptome Changes in the Mink Uterus during Blastocyst Dormancy and Reactivation. Int. J. Mol. Sci. 2019, 20, 2099. [Google Scholar] [CrossRef]
- Alcorta-Sevillano, N.; Macías, I.; Infante, A.; Rodríguez, C.I. Deciphering the Relevance of Bone ECM Signaling. Cells 2020, 9, 2630. [Google Scholar] [CrossRef]
- Gresham, R.C.H.; Bahney, C.S.; Leach, J.K. Growth factor delivery using extracellular matrix-mimicking substrates for musculoskeletal tissue engineering and repair. Bioact. Mater. 2021, 6, 1945–1956. [Google Scholar] [CrossRef]
- Kim, M.S.; Yu, J.H.; Lee, M.Y.; Kim, A.L.; Jo, M.H.; Kim, M.; Cho, S.R.; Kim, Y.H. Differential Expression of Extracellular Matrix and Adhesion Molecules in Fetal-Origin Amniotic Epithelial Cells of Preeclamptic Pregnancy. PLoS ONE 2016, 11, e0156038. [Google Scholar] [CrossRef]
- Kong, S.; Han, X.; Cui, T.; Zhou, C.; Jiang, Y.; Zhang, H.; Wang, B.; Wang, H.; Zhang, S. MCM2 mediates progesterone-induced endometrial stromal cell proliferation and differentiation in mice. Endocrine 2016, 53, 595–606. [Google Scholar] [CrossRef]
- Spiess, K.; Teodoro, W.R.; Zorn, T.M. Distribution of collagen types I, III, and V in pregnant mouse endometrium. Connect. Tissue Res. 2007, 48, 99–108. [Google Scholar] [CrossRef]
- Goossens, K.; Van Soom, A.; Van Zeveren, A.; Favoreel, H.; Peelman, L.J. Quantification of fibronectin 1 (FN1) splice variants, including two novel ones, and analysis of integrins as candidate FN1 receptors in bovine preimplantation embryos. BMC Dev. Biol. 2009, 9, 1. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Armant, D.R. Integrin-mediated adhesion and signaling during blastocyst implantation. Cells Tissues Organs. 2002, 172, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Acién, P.; Velasco, I.; Acién, M.; Capello, C.; Vela, P. Epithelial ovarian cancers and endometriosis. Gynecol. Obstet. Investig. 2015, 79, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Prockop, D.J. Mutations that alter the primary structure of type I collagen. The perils of a system for generating large structures by the principle of nucleated growth. J. Biol. Chem. 1990, 265, 15349–15352. [Google Scholar] [CrossRef]
- Gould, D.B.; Phalan, F.C.; Breedveld, G.J.; van Mil, S.E.; Smith, R.S.; Schimenti, J.C.; Aguglia, U.; van der Knaap, M.S.; Heutink, P.; John, S.W. Mutations in Col4a1 cause perinatal cerebral hemorrhage and porencephaly. Science 2005, 308, 1167–1171. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Sequence | Lengths (bp) |
|---|---|---|
| GSN | F: CACTACTGGCTGGGCAATGA R: CCGTTCAGGTAGTCGTCCAG | 86 |
| VIM | F: CCCTGAACCTGAGGGAAACC R: CGTGATGCTGGGAAGTTTCG | 129 |
| FOS | F: CGTCAATGCGCAGGACTACT R: GGAGACTAGGGTGGGCTGTA | 121 |
| GLI1 | F: CGCCCAGACAGAGTGCC R: GCGGATAACTGTCTGCAGGT | 120 |
| β-ACTIN | F: GCAATGAGCGGTTCC R: CCGTGTTGGCGTAGAG | 141 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beng, S.; Lan, D.; Li, Y.; Li, D.; Zhang, Y.; Ma, Z.; Zhu, J.; Liu, S.; Chen, K.; Li, J.; et al. Transcriptomics Reveals the Differences in mRNA Expression Patterns in Yak Uterus of Follicular, Luteal, and Pregnant Phases. Animals 2025, 15, 837. https://doi.org/10.3390/ani15060837
Beng S, Lan D, Li Y, Li D, Zhang Y, Ma Z, Zhu J, Liu S, Chen K, Li J, et al. Transcriptomics Reveals the Differences in mRNA Expression Patterns in Yak Uterus of Follicular, Luteal, and Pregnant Phases. Animals. 2025; 15(6):837. https://doi.org/10.3390/ani15060837
Chicago/Turabian StyleBeng, Shaohui, Daoliang Lan, Yueyue Li, Deping Li, Yuehuan Zhang, Zelang Ma, Jianbo Zhu, Shunyang Liu, Kechao Chen, Jian Li, and et al. 2025. "Transcriptomics Reveals the Differences in mRNA Expression Patterns in Yak Uterus of Follicular, Luteal, and Pregnant Phases" Animals 15, no. 6: 837. https://doi.org/10.3390/ani15060837
APA StyleBeng, S., Lan, D., Li, Y., Li, D., Zhang, Y., Ma, Z., Zhu, J., Liu, S., Chen, K., Li, J., Wang, P., & Fu, W. (2025). Transcriptomics Reveals the Differences in mRNA Expression Patterns in Yak Uterus of Follicular, Luteal, and Pregnant Phases. Animals, 15(6), 837. https://doi.org/10.3390/ani15060837