
Whole-Genome Sequencing of Hexagrammos otakii Provides Insights into Its Genomic Characteristics and Population Dynamics
Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Specimen Collection and DNA Extraction
2.2. Library Construction, Whole-Genome Sequencing, and Raw Read Quality Control
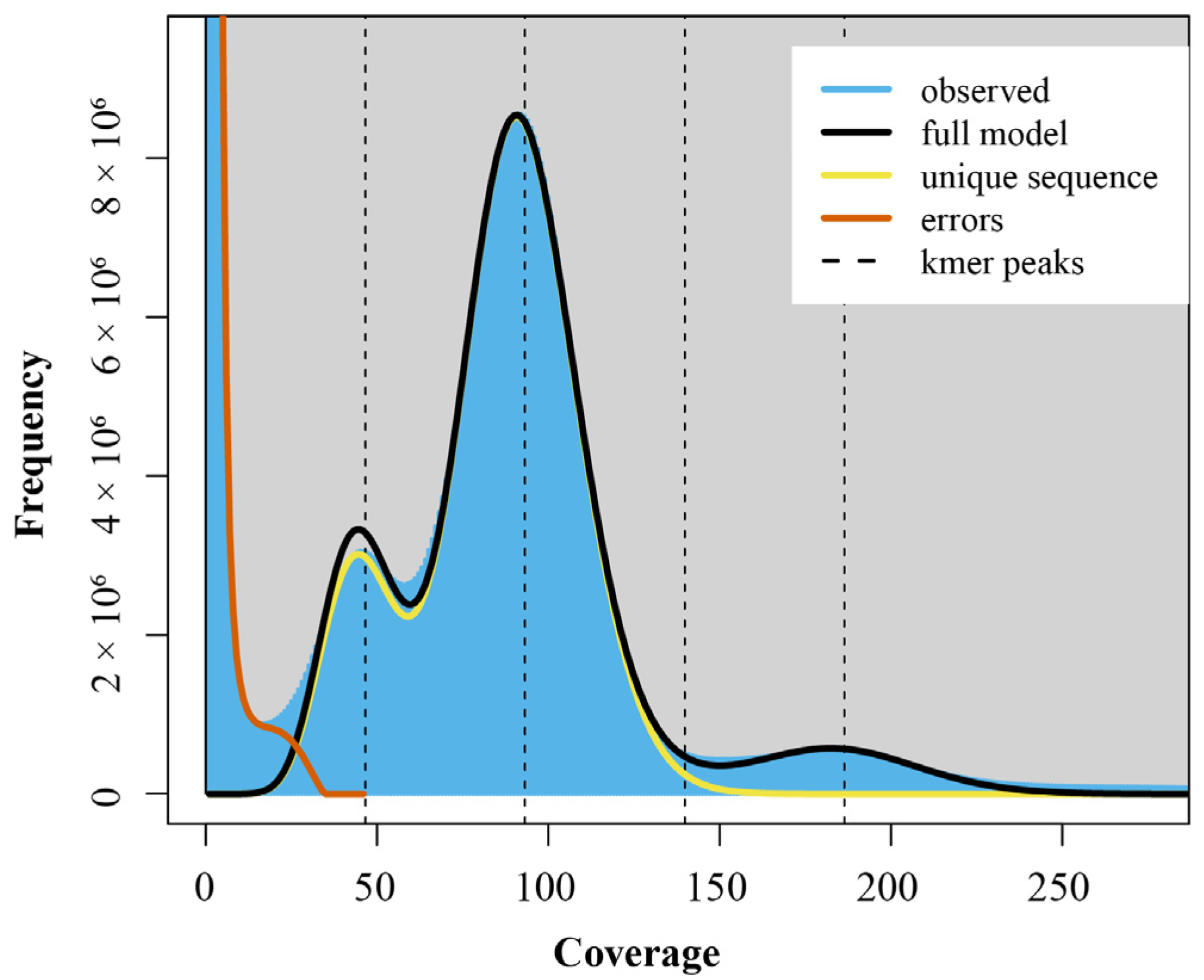
2.3. Estimation of Genome Size, Heterozygosity Ratio, and Repeat Ratio for H. otakii
2.4. Sequence Assembly and Analysis of Guanine and Cytosine (GC) Content
2.5. Identification of Simple Sequence Repeats (SSRs)
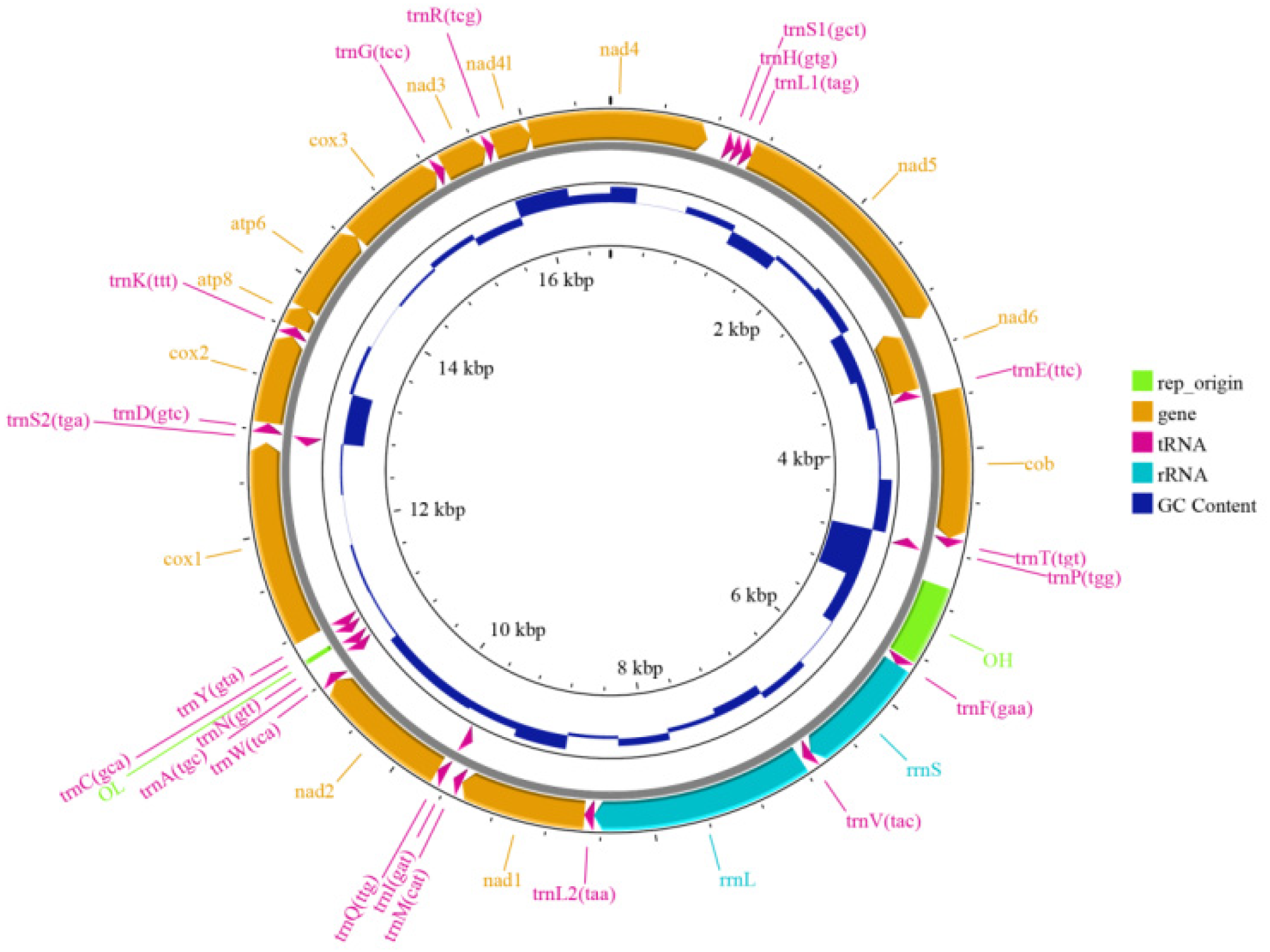
2.6. Mitochondrial Genome Assembly and Annotation
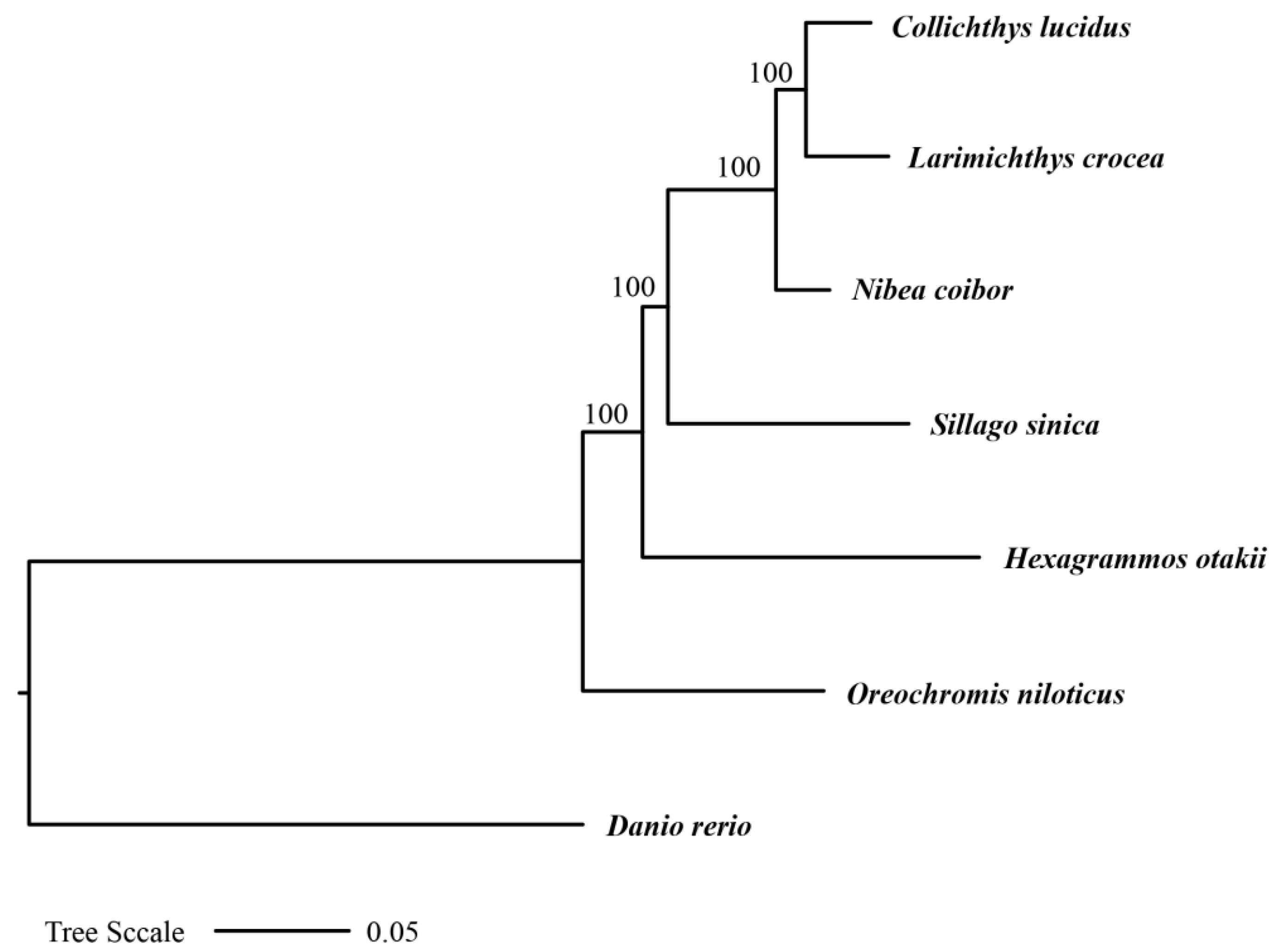
2.7. Construction of a Phylogenetic Tree Based on Orthologous Single-Copy Genes
2.8. Inference of Population Size Dynamics for H. otakii
3. Results
3.1. Genome Survey Analysis of H. otakii
3.2. Genome Assembly and Mitochondrial Genome Assembly of H. otakii
3.3. Profile of Satellite DNA Sequences
3.4. Phylogenetic Relationship of H. otakii with Six Other Species Based on Orthologous Genes
3.5. The Population Size Dynamics of H. otakii
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Habib, K.A.; Jeong, D.; Myoung, J.-G.; Kim, M.S.; Jang, Y.S.; Shim, J.S.; Lee, Y.-H. Population genetic structure and demographic history of the fat greenling Hexagrammos otakii. Genes Genomics 2011, 33, 413–423. [Google Scholar] [CrossRef]
- Kwak, S.N.; Baeck, G.W.; Klumpp, D.W. Comparative feeding ecology of two sympatric greenling species, Hexagrammos otakii and Hexagrammos agrammus in eelgrass Zostera marina beds. Environ. Biol. Fishes 2005, 74, 129–140. [Google Scholar] [CrossRef]
- Cho, G.; Heath, D. Comparison of tricaine methanesulphonate (MS222) and clove oil anaesthesia effects on the physiology of juvenile chinook salmon Oncorhynchus tshawytscha (Walbaum). Aquac. Res. 2000, 31, 537–546. [Google Scholar] [CrossRef]
- Li, L.; Hu, F.; Liu, D.; Wang, X.; Diao, J.; Zhu, Y.; Gao, F.; Fan, Y.; Jian, Y.; Wang, X. A Chromosomal-level genome assembly and annotation of fat greenling (Hexagrammos otakii). Sci. Data 2025, 12, 78. [Google Scholar] [CrossRef]
- Matsumiya, M.; Arakane, Y.; Haga, A.; Muthukrishnan, S.; Kramer, K.J. Substrate specificity of chitinases from two species of fish, greenling, Hexagrammos otakii, and common mackerel, Scomber japonicus, and the insect, tobacco hornworm, Manduca sexta. Biosci. Biotechnol. Biochem. 2006, 70, 971–979. [Google Scholar] [CrossRef]
- Wen, H.; Wang, L.; Mou, X.; Chen, C.; Yao, J.; Chen, S. Study on the annual variation of testis development of Hexagrammos otakii jordan and Starks. J. Ocean Univ. China 2007, 37, 581–585. [Google Scholar]
- Natural Resources Species Survival Commission. IUCN Red List Categories and Criteria; IUCN: Gland, Switzerland, 2001. [Google Scholar]
- Rodrigues, A.S.; Pilgrim, J.D.; Lamoreux, J.F.; Hoffmann, M.; Brooks, T.M. The value of the IUCN Red List for conservation. Trends Ecol. Evol. 2006, 21, 71–76. [Google Scholar] [CrossRef]
- Zhang, R.; Liu, H.; Zhang, Q.; Zhang, H.; Zhao, J. Trophic interactions of reef-associated predatory fishes (Hexagrammos otakii and Sebastes schlegelii) in natural and artificial reefs along the coast of North Yellow Sea, China. Sci. Total Environ. 2021, 791, 148250. [Google Scholar] [CrossRef]
- Yue, G.; Wang, L. Current status of genome sequencing and its applications in aquaculture. Aquaculture 2017, 468, 337–347. [Google Scholar] [CrossRef]
- Ahmad, S.F.; Jehangir, M.; Srikulnath, K.; Martins, C. Fish genomics and its impact on fundamental and applied research of vertebrate biology. Rev. Fish Biol. Fish. 2021, 32, 357–385. [Google Scholar] [CrossRef]
- Huang, Y.; Li, Z.; Li, M.; Zhang, X.; Shi, Q.; Xu, Z. Fish Genomics and Its Application in Disease-Resistance Breeding. Rev. Aquac. 2024, 17, e12973. [Google Scholar] [CrossRef]
- Zeng, J.; Zhao, J.; Wang, J.; Bai, Y.; Long, F.; Deng, Y.; Jiang, P.; Xiao, J.; Qu, A.; Tong, B. Genetic linkage between swimming performance and disease resistance enables multitrait breeding strategies in large yellow croaker. Agric. Commun. 2023, 1, 100019. [Google Scholar] [CrossRef]
- Zhang, C.; Wen, H.; Zhang, Y.; Zhang, K.; Qi, X.; Li, Y. First genome-wide association study and genomic prediction for growth traits in spotted sea bass (Lateolabrax maculatus) using whole-genome resequencing. Aquaculture 2023, 566, 739194. [Google Scholar] [CrossRef]
- Li, L.; Gao, F.; Jian, Y.; Wang, X.; Wang, X.; Pan, L.; Guo, W.; Liu, D.; Hu, F. Transcriptomic analysis of liver tissue in fat greenling (Hexagrammos otakii) exposed to elevated ambient ammonia. Front. Mar. Sci. 2020, 7, 418. [Google Scholar] [CrossRef]
- Hu, F.; Sun, M.; Li, L.; Gao, F.; Jian, Y.; Wang, X.; Wang, X.; Guo, W. Effects of environmental cadmium on cadmium accumulation, oxidative response, and microelements regulation in the liver and kidney of Hexagrammos otakii. J. Ocean. Univ. China 2022, 21, 479–485. [Google Scholar] [CrossRef]
- Hitte, C.; Madeoy, J.; Kirkness, E.F.; Priat, C.; Lorentzen, T.D.; Senger, F.; Thomas, D.; Derrien, T.; Ramirez, C.; Scott, C. Facilitating genome navigation: Survey sequencing and dense radiation-hybrid gene mapping. Nat. Rev. Genet. 2005, 6, 643–648. [Google Scholar] [CrossRef]
- Lei, Y.; Zhou, Y.; Price, M.; Song, Z. Genome-wide characterization of microsatellite DNA in fishes: Survey and analysis of their abundance and frequency in genome-specific regions. BMC Genom. 2021, 22, 421. [Google Scholar] [CrossRef]
- Song, N.; Zhao, X.; Cai, C.; Gao, T. Profile of the genomic characteristics and comparative studies of five Trichiuridae species by genome survey sequencing. Front. Mar. Sci. 2022, 9, 962307. [Google Scholar] [CrossRef]
- Venkatesh, B.; Kirkness, E.F.; Loh, Y.-H.; Halpern, A.L.; Lee, A.P.; Johnson, J.; Dandona, N.; Viswanathan, L.D.; Tay, A.; Venter, J.C. Survey sequencing and comparative analysis of the elephant shark (Callorhinchus milii) genome. PLoS Biol. 2007, 5, e101. [Google Scholar] [CrossRef]
- Zhao, X.; Zheng, T.; Song, N.; Qu, Y.; Gao, T. Whole-genome survey reveals interspecific differences in genomic characteristics and evolution of Pampus fish. Front. Mar. Sci. 2024, 10, 1332250. [Google Scholar] [CrossRef]
- Luo, R.; Liu, B.; Xie, Y.; Li, Z.; Huang, W.; Yuan, J.; He, G.; Chen, Y.; Pan, Q.; Liu, Y. SOAPdenovo2: An empirically improved memory-efficient short-read de novo assembler. Gigascience 2012, 1, 2047-2217X-2041-2018. [Google Scholar] [CrossRef] [PubMed]
- Marcionetti, A.; Rossier, V.; Bertrand, J.A.; Litsios, G.; Salamin, N. First draft genome of an iconic clownfish species (Amphiprion frenatus). Mol. Ecol. Resour. 2018, 18, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Surachat, K.; Deachamag, P.; Wonglapsuwan, M. The first de novo genome assembly and sex marker identification of Pluang Chomphu fish (Tor tambra) from Southern Thailand. Comput. Struct. Biotechnol. J. 2022, 20, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Bargelloni, L.; Babbucci, M.; Ferraresso, S.; Papetti, C.; Vitulo, N.; Carraro, R.; Pauletto, M.; Santovito, G.; Lucassen, M.; Mark, F.C. Draft genome assembly and transcriptome data of the icefish Chionodraco myersi reveal the key role of mitochondria for a life without hemoglobin at subzero temperatures. Commun. Biol. 2019, 2, 443. [Google Scholar] [CrossRef]
- Kasahara, M.; Naruse, K.; Sasaki, S.; Nakatani, Y.; Qu, W.; Ahsan, B.; Yamada, T.; Nagayasu, Y.; Doi, K.; Kasai, Y. The medaka draft genome and insights into vertebrate genome evolution. Nature 2007, 447, 714–719. [Google Scholar] [CrossRef]
- Halley, Y.A.; Dowd, S.E.; Decker, J.E.; Seabury, P.M.; Bhattarai, E.; Johnson, C.D.; Rollins, D.; Tizard, I.R.; Brightsmith, D.J.; Peterson, M.J. A draft de novo genome assembly for the northern bobwhite (Colinus virginianus) reveals evidence for a rapid decline in effective population size beginning in the Late Pleistocene. PLoS ONE 2014, 9, e90240. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Münch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Allio, R.; Schomaker-Bastos, A.; Romiguier, J.; Prosdocimi, F.; Nabholz, B.; Delsuc, F. MitoFinder: Efficient automated large-scale extraction of mitogenomic data in target enrichment phylogenomics. Mol. Ecol. Resour. 2020, 20, 892–905. [Google Scholar] [CrossRef]
- Grant, J.R.; Enns, E.; Marinier, E.; Mandal, A.; Herman, E.K.; Chen, C.-Y.; Graham, M.; Van Domselaar, G.; Stothard, P. Proksee: In-depth characterization and visualization of bacterial genomes. Nucleic Acids Res. 2023, 51, W484–W492. [Google Scholar] [CrossRef]
- Xu, S.; Xiao, S.; Zhu, S.; Zeng, X.; Luo, J.; Liu, J.; Gao, T.; Chen, N. A draft genome assembly of the Chinese sillago (Sillago sinica), the first reference genome for Sillaginidae fishes. GigaScience 2018, 7, giy108. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.i.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Inference of human population history from individual whole-genome sequences. Nature 2011, 475, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Bista, I.; Wood, J.M.; Desvignes, T.; McCarthy, S.A.; Matschiner, M.; Ning, Z.; Tracey, A.; Torrance, J.; Sims, Y.; Chow, W. Genomics of cold adaptations in the Antarctic notothenioid fish radiation. Nat. Commun. 2023, 14, 3412. [Google Scholar] [CrossRef]
- Guan, D.; McCarthy, S.A.; Wood, J.; Howe, K.; Wang, Y.; Durbin, R. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics 2020, 36, 2896–2898. [Google Scholar] [CrossRef]
- Dudchenko, O.; Batra, S.S.; Omer, A.D.; Nyquist, S.K.; Hoeger, M.; Durand, N.C.; Shamim, M.S.; Machol, I.; Lander, E.S.; Aiden, A.P. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science 2017, 356, 92–95. [Google Scholar] [CrossRef]
- Wang, C.; Chen, Q.; Lu, G.; Xu, J.; Yang, Q.; Li, S. Complete mitochondrial genome of the grass carp (Ctenopharyngodon idella, Teleostei): Insight into its phylogenic position within Cyprinidae. Gene 2008, 424, 96–101. [Google Scholar] [CrossRef]
- Wang, Q.; Zheng, F.; Wang, B.; Luo, J.; Chen, G. The complete mitochondrial genome of the Hexagrammos otakii (Scorpaeniformes: Hexagrammidae). Mitochondrial DNA Part A 2016, 27, 2915–2916. [Google Scholar] [CrossRef]
- Finney, B.P.; Alheit, J.; Emeis, K.-C.; Field, D.B.; Gutiérrez, D.; Struck, U. Paleoecological studies on variability in marine fish populations: A long-term perspective on the impacts of climatic change on marine ecosystems. J. Mar. Syst. 2010, 79, 316–326. [Google Scholar] [CrossRef]
- Ruzzante, D.E.; Walde, S.J.; Gosse, J.C.; Cussac, V.E.; Habit, E.; Zemlak, T.S.; Adams, E.D. Climate control on ancestral population dynamics: Insight from Patagonian fish phylogeography. Mol. Ecol. 2008, 17, 2234–2244. [Google Scholar] [CrossRef]
- Sims, D.; Sudbery, I.; Ilott, N.E.; Heger, A.; Ponting, C.P. Sequencing depth and coverage: Key considerations in genomic analyses. Nat. Rev. Genet. 2014, 15, 121–132. [Google Scholar] [CrossRef]
- Guo, Y.; Ye, F.; Sheng, Q.; Clark, T.; Samuels, D.C. Three-stage quality control strategies for DNA re-sequencing data. Brief. Bioinform. 2014, 15, 879–889. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Shao, Y.; Tian, L.; Flasch, D.A.; Mulder, H.L.; Edmonson, M.N.; Liu, Y.; Chen, X.; Newman, S.; Nakitandwe, J. Analysis of error profiles in deep next-generation sequencing data. Genome Biol. 2019, 20, 50. [Google Scholar] [CrossRef]
- Kajitani, R.; Toshimoto, K.; Noguchi, H.; Toyoda, A.; Ogura, Y.; Okuno, M.; Yabana, M.; Harada, M.; Nagayasu, E.; Maruyama, H. Efficient de novo assembly of highly heterozygous genomes from whole-genome shotgun short reads. Genome Res. 2014, 24, 1384–1395. [Google Scholar] [CrossRef] [PubMed]
- Pryszcz, L.P.; Gabaldón, T. Redundans: An assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 2016, 44, e113. [Google Scholar] [CrossRef] [PubMed]
- Brown, K. Fish mitochondrial genomics: Sequence, inheritance and functional variation. J. Fish Biol. 2008, 72, 355–374. [Google Scholar] [CrossRef]
- Iwasaki, W.; Fukunaga, T.; Isagozawa, R.; Yamada, K.; Maeda, Y.; Satoh, T.P.; Sado, T.; Mabuchi, K.; Takeshima, H.; Miya, M. MitoFish and MitoAnnotator: A mitochondrial genome database of fish with an accurate and automatic annotation pipeline. Mol. Biol. Evol. 2013, 30, 2531–2540. [Google Scholar] [CrossRef]
- Hohenlohe, P.A.; Hand, B.K.; Andrews, K.R.; Luikart, G. Population genomics provides key insights in ecology and evolution. In Population Genomics: Concepts, Approaches and Applications; Springer: Cham, Switzerland, 2019; pp. 483–510. [Google Scholar]
- Breed, M.F.; Harrison, P.A.; Blyth, C.; Byrne, M.; Gaget, V.; Gellie, N.J.; Groom, S.V.; Hodgson, R.; Mills, J.G.; Prowse, T.A. The potential of genomics for restoring ecosystems and biodiversity. Nat. Rev. Genet. 2019, 20, 615–628. [Google Scholar] [CrossRef]
- Harley, C.D. Climate change, keystone predation, and biodiversity loss. Science 2011, 334, 1124–1127. [Google Scholar] [CrossRef]
- Selwood, K.E.; McGeoch, M.A.; Mac Nally, R. The effects of climate change and land-use change on demographic rates and population viability. Biol. Rev. 2015, 90, 837–853. [Google Scholar] [CrossRef]
- Logsdon, G.A.; Vollger, M.R.; Eichler, E.E. Long-read human genome sequencing and its applications. Nat. Rev. Genet. 2020, 21, 597–614. [Google Scholar] [CrossRef]
- Garg, V.; Bohra, A.; Mascher, M.; Spannagl, M.; Xu, X.; Bevan, M.W.; Bennetzen, J.L.; Varshney, R.K. Unlocking plant genetics with telomere-to-telomere genome assemblies. Nat. Genet. 2024, 56, 1788–1799. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Chen, C.; Lin, D.; Hua, Z.; Ying, C.; Zhang, J.; Zhao, C.; Liu, Y.; Cao, Z.; Zhang, H. Telomere-to-telomere gap-free genome assembly of the endangered Yangtze finless porpoise and East Asian finless porpoise. GigaScience 2024, 13, giae067. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Read Number | Base Count (bp) | Read Length (bp) | Q20 (%) | Q30 (%) |
|---|---|---|---|---|---|
| H. otakii | 494,440,704 | 73,193,542,644 | 150 | 97.50 | 93.23 |
| Metrics | Value |
|---|---|
| K-mer size | 17 |
| K-mer depth | 97 |
| Genome size (bp) | 679,232,407 |
| Genome repeat length (bp) | 296,145,329 |
| Genome unique length (bp) | 383,087,078 |
| Repeat (%) | 43.60% |
| Heterozygous ratio (%) | 0.68% |
| Assembly Level | Total Length (bp) | Total Number | Max Length (bp) | N50 Length (bp) | N90 Length (bp) | GC Content (%) |
|---|---|---|---|---|---|---|
| Contig | 716,659,970 | 1,484,256 | 29,264 | 695 | 230 | 42.79 |
| Scaffold | 723,306,185 | 1,224,914 | 86,243 | 1183 | 246 | 42.40 |
| Assembly Level | Total Length (bp) | GC Content (%) |
|---|---|---|
| Contig | 16,513 | 47.20 |
| Type | Total Number | Total Length (bp) | Mean Length (bp) |
|---|---|---|---|
| Gene | 13 | 11,427 | 879 |
| tRNA | 22 | 1555 | 71 |
| rRNA | 2 | 2614 | 1307 |
| Type | Definition | Total Number | Total Length (bp) | Rate (%) |
|---|---|---|---|---|
| Microsatellite | 1–9 bp units | 192,961 | 6,157,332 | 0.85 |
| Minisatellite | 10–99 bp units | 338,654 | 48,903,386 | 6.76 |
| Satellite | ≥100 bp units | 12,649 | 3,999,780 | 0.55 |
| Total | 544,264 | 59,060,498 | 8.17 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.; Wang, X.; Lü, J.; Zhu, Y.; Jian, Y.; Wang, X.; Gao, F.; Li, L.; Hu, F. Whole-Genome Sequencing of Hexagrammos otakii Provides Insights into Its Genomic Characteristics and Population Dynamics. Animals 2025, 15, 782. https://doi.org/10.3390/ani15060782
Liu D, Wang X, Lü J, Zhu Y, Jian Y, Wang X, Gao F, Li L, Hu F. Whole-Genome Sequencing of Hexagrammos otakii Provides Insights into Its Genomic Characteristics and Population Dynamics. Animals. 2025; 15(6):782. https://doi.org/10.3390/ani15060782
Chicago/Turabian StyleLiu, Dong, Xiaolong Wang, Jifa Lü, Yijing Zhu, Yuxia Jian, Xue Wang, Fengxiang Gao, Li Li, and Fawen Hu. 2025. "Whole-Genome Sequencing of Hexagrammos otakii Provides Insights into Its Genomic Characteristics and Population Dynamics" Animals 15, no. 6: 782. https://doi.org/10.3390/ani15060782
APA StyleLiu, D., Wang, X., Lü, J., Zhu, Y., Jian, Y., Wang, X., Gao, F., Li, L., & Hu, F. (2025). Whole-Genome Sequencing of Hexagrammos otakii Provides Insights into Its Genomic Characteristics and Population Dynamics. Animals, 15(6), 782. https://doi.org/10.3390/ani15060782