
Whole-Genome Sequencing-Based Population Genetic Analysis of Wild and Domestic Rabbit Breeds
 , , , , and
, , , , and 
Simple Summary
Abstract

1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Isolation
2.2. Sequencing and Public Data Collection
2.3. Quality Control and Variant Calling
2.4. Detecting Population Structure and Inbreeding
2.5. Selected Genes in Domestic Breeds Based on Wgs Data of Unadmixed Individuals
3. Results
3.1. Sequencing and Data Collection
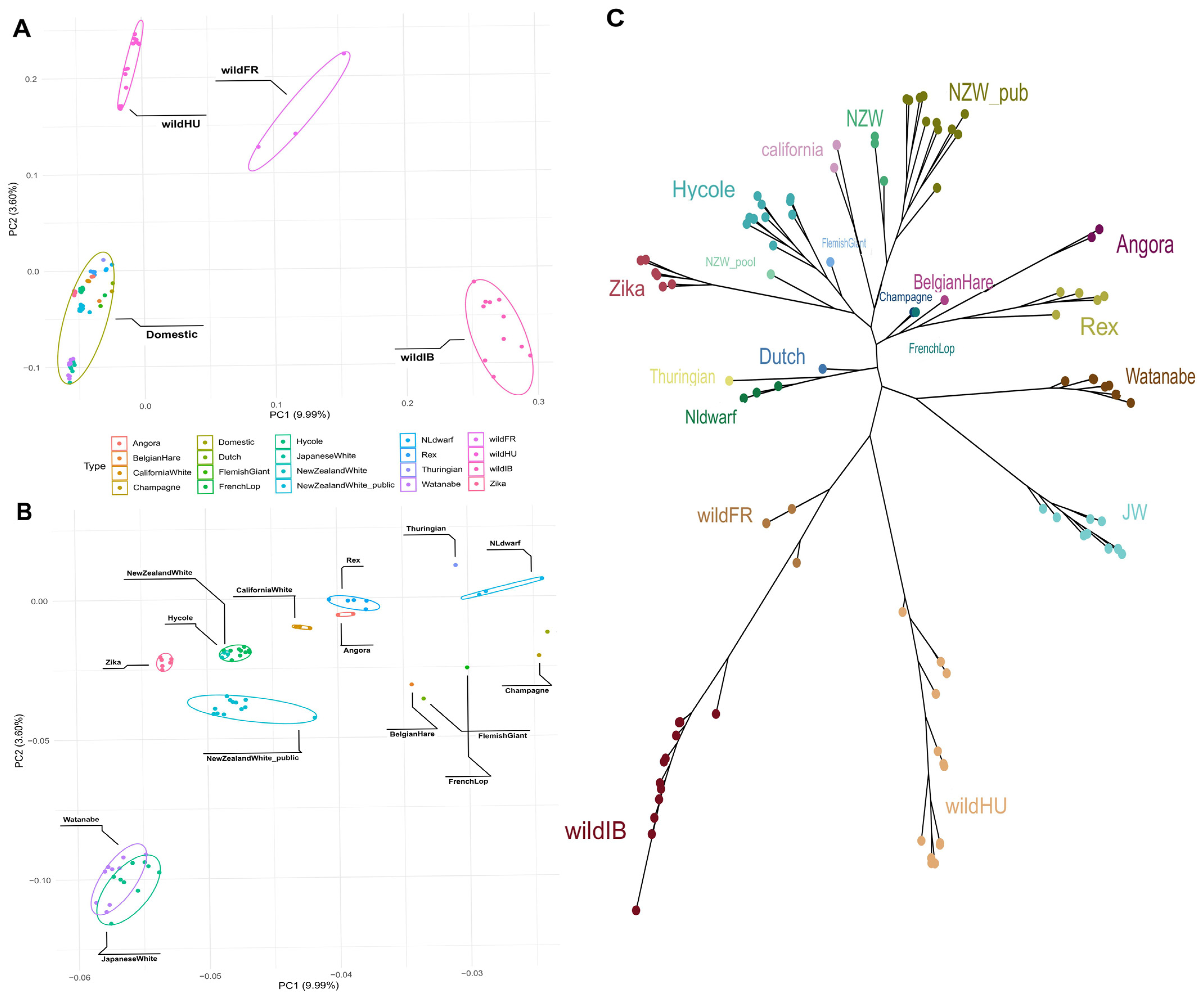
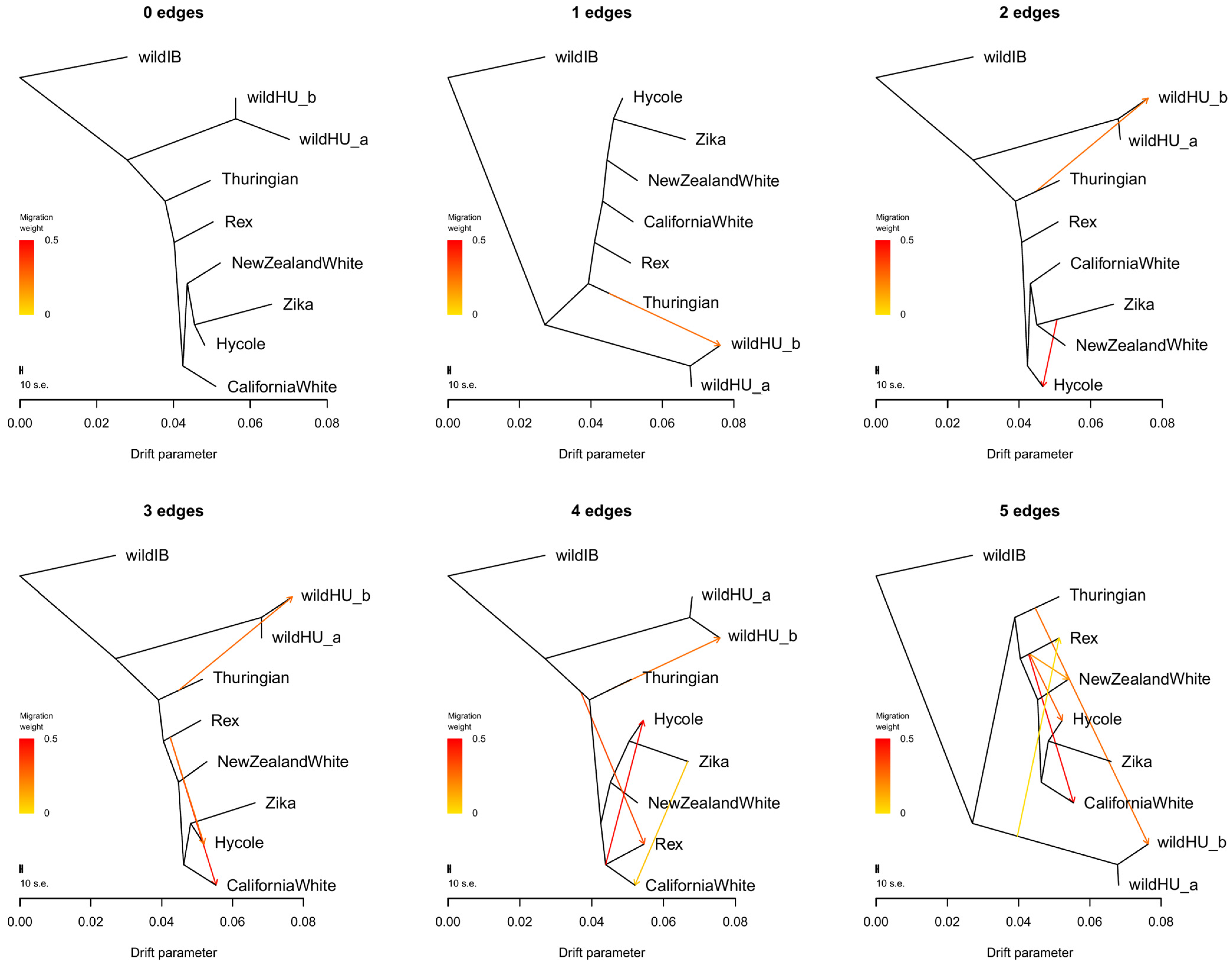
3.2. Population Structure and Phylogenetic Tree
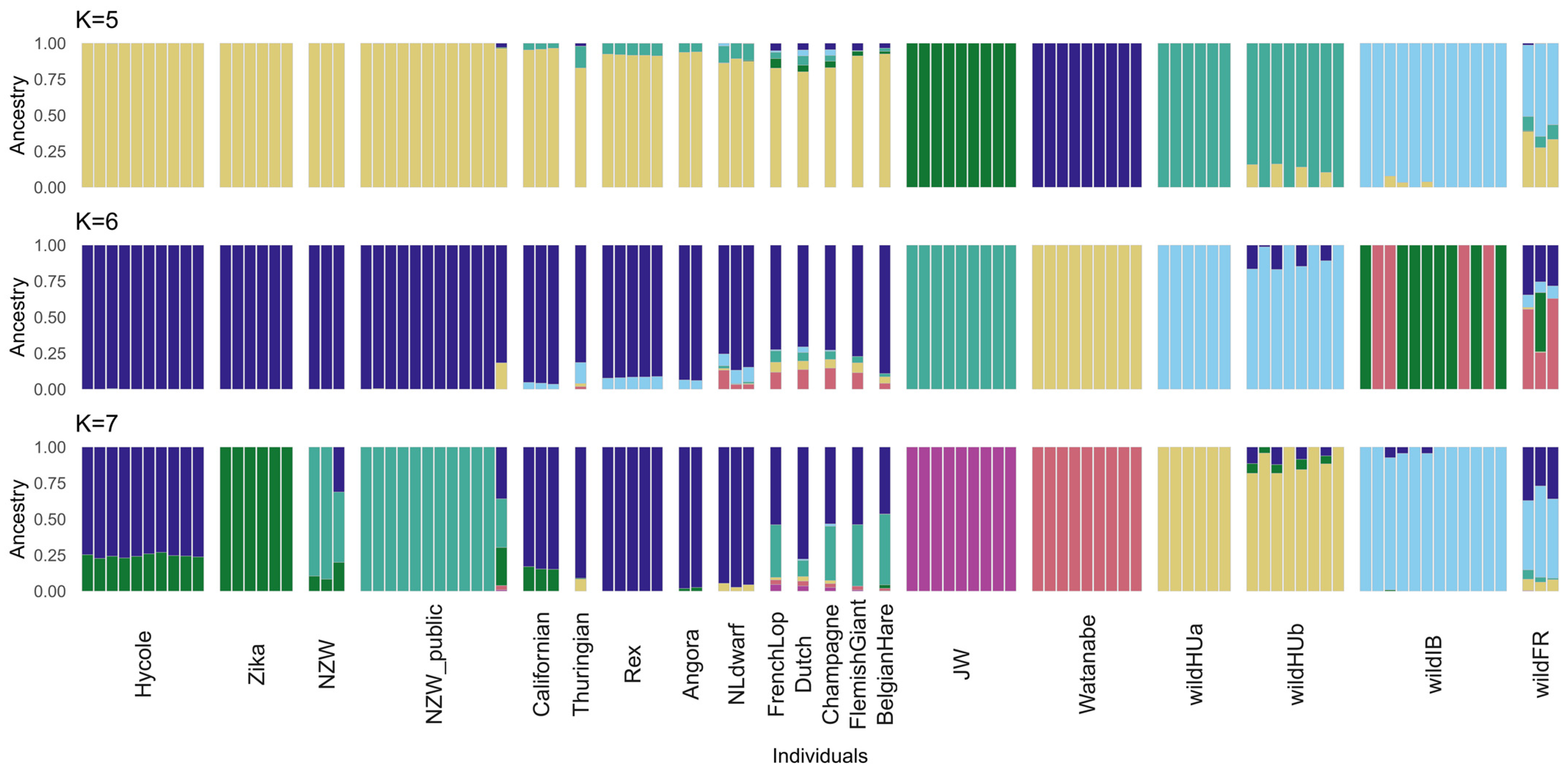
3.3. Admixture of Rabbit Breeds and Populations
3.4. Runs of Homozygosity and Inbreeding
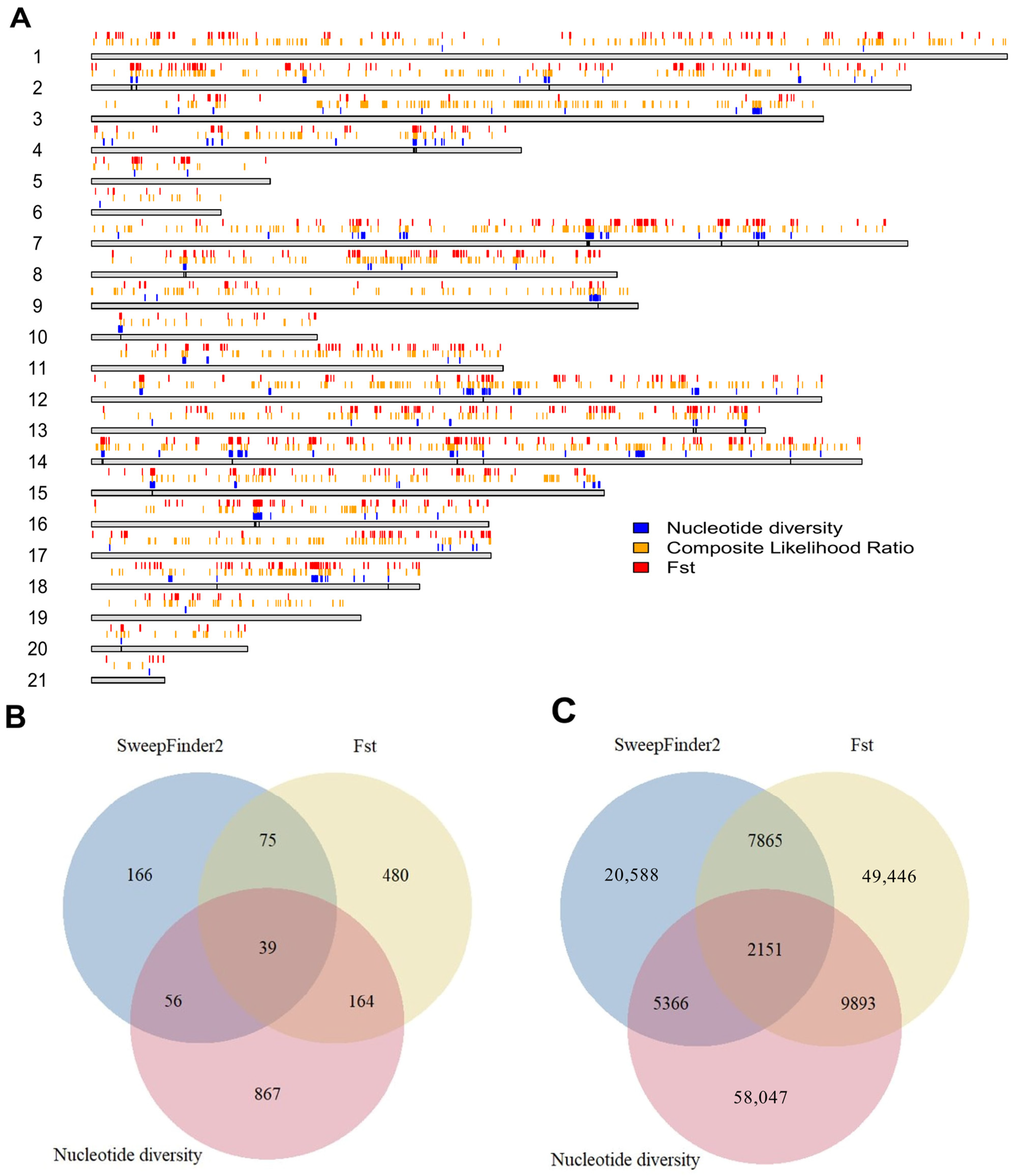
3.5. Selection in Domestic Rabbits
4. Discussion
4.1. Population Structure and Diversity
4.2. Divergence and Selection of Wild and Domestic Rabbits
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Delibes-Mateos, M.; Ferreras, P.; Villafuerte, R. European rabbit population trends and associated factors: A review of the situation in the Iberian Peninsula. Mamm. Rev. 2009, 39, 124–140. [Google Scholar] [CrossRef]
- Delibes-Mateos, M.; Delibes, M.; Ferreras, P.; Villafuerte, R. Key role of European rabbits in the conservation of the Western Mediterranean basin hotspot. Conserv. Biol. 2008, 22, 1106–1117. [Google Scholar] [CrossRef] [PubMed]
- Alves, J.M.; Carneiro, M.; Day, J.P.; Welch, J.J.; Duckworth, J.A.; Cox, T.E.; Letnic, M.; Strive, T.; Ferrand, N.; Jiggins, F.M. A single introduction of wild rabbits triggered the biological invasion of Australia. Proc. Natl. Acad. Sci. USA 2022, 119, e2122734119. [Google Scholar] [CrossRef]
- Delibes, R.; Delibes-Mateos, M. Linking historical ecology and invasion biology: Some lessons from European rabbit introductions into the new world before the nineteenth century. Biol. Invasions 2015, 17, 2505–2515. [Google Scholar] [CrossRef]
- Bello-Rodriguez, V.; Mateo, R.G.; Pellissier, L.; Cubas, J.; Cooke, B.; Gonzalez-Mancebo, J.M. Forecast increase in invasive rabbit spread into ecosystems of an oceanic island (Tenerife) under climate change. Ecol. Appl. 2021, 31, e02206. [Google Scholar] [CrossRef] [PubMed]
- Smyser, T.J.; Tabak, M.A.; Slootmaker, C.; Robeson, M.S.; Miller, R.S.; Bosse, M.; Megens, H.-J.; Groenen, M.A.M.; Paiva, S.R.; de Faria, D.A.; et al. Mixed ancestry from wild and domestic lineages contributes to the rapid expansion of invasive feral swine. Mol. Ecol. 2020, 29, 1103–1119. [Google Scholar] [CrossRef]
- Quilodrán, C.S.; Nussberger, B.; Macdonald, D.W.; Montoya-Burgos, J.I.; Currat, M. Projecting introgression from domestic cats into European wildcats in the Swiss Jura. Evol. Appl. 2020, 13, 2101–2112. [Google Scholar] [CrossRef]
- Wu, M.Y.; Low, G.W.; Forcina, G.; van Grouw, H.; Lee, B.P.Y.-H.; Oh, R.R.Y.; Rheindt, F.E. Historic and modern genomes unveil a domestic introgression gradient in a wild red junglefowl population. Evol. Appl. 2020, 13, 2300–2315. [Google Scholar] [CrossRef]
- Pilot, M.; Moura, A.E.; Okhlopkov, I.M.; Mamaev, N.V.; Manaseryan, N.H.; Hayrapetyan, V.; Kopaliani, N.; Tsingarska, E.; Alagaili, A.N.; Mohammed, O.B.; et al. Human-modified canids in human-modified landscapes: The evolutionary consequences of hybridization for grey wolves and free-ranging domestic dogs. Evol. Appl. 2021, 14, 2433–2456. [Google Scholar] [CrossRef]
- Grossen, C.; Keller, L.; Biebach, I.; Consortium, I.G.G.; Croll, D. Introgression from domestic goat generated variation at the major histocompatibility complex of Alpine ibex. PLoS Genet. 2014, 10, e1004438. [Google Scholar] [CrossRef]
- Alves, J.M.; Carneiro, M.; Afonso, S.; Lopes, S.; Garreau, H.; Boucher, S.; Allain, D.; Queney, G.; Esteves, P.J.; Bolet, G.; et al. Levels and Patterns of Genetic Diversity and Population Structure in Domestic Rabbits. PLoS ONE 2015, 10, e0144687. [Google Scholar] [CrossRef]
- Carneiro, M.; Afonso, S.; Geraldes, A.; Garreau, H.; Bolet, G.; Boucher, S.; Tircazes, A.; Queney, G.; Nachman, M.W.; Ferrand, N. The genetic structure of domestic rabbits. Mol. Biol. Evol. 2011, 28, 1801–1816. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, M.; Rubin, C.-J.; Di Palma, F.; Albert, F.W.; Alföldi, J.; Martinez Barrio, A.; Pielberg, G.; Rafati, N.; Sayyab, S.; Turner-Maier, J.; et al. Rabbit genome analysis reveals a polygenic basis for phenotypic change during domestication. Science 2014, 345, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Wang, X.; Li, M.; Li, Y.; Yang, Z.; Wang, X.; Pan, X.; Gong, M.; Zhang, Y.; Guo, Y.; et al. The origin of domestication genes in goats. Sci. Adv. 2020, 6, eaaz5216. [Google Scholar] [CrossRef]
- Rochus, C.M.; Tortereau, F.; Plisson-Petit, F.; Restoux, G.; Moreno-Romieux, C.; Tosser-Klopp, G.; Servin, B. Revealing the selection history of adaptive loci using genome-wide scans for selection: An example from domestic sheep. BMC Genom. 2018, 19, 71. [Google Scholar] [CrossRef]
- Fatima, N.; Jia, L.; Liu, B.; Li, L.; Bai, L.; Wang, W.; Zhao, S.; Wang, R.; Liu, E. A homozygous missense mutation in the fibroblast growth factor 5 gene is associated with the long-hair trait in Angora rabbits. BMC Genom. 2023, 24, 298. [Google Scholar] [CrossRef] [PubMed]
- Demars, J.; Iannuccelli, N.; Utzeri, V.J.; Auvinet, G.; Riquet, J.; Fontanesi, L.; Allain, D. New Insights into the Melanophilin (MLPH) Gene Affecting Coat Color Dilution in Rabbits. Genes 2018, 9, 430. [Google Scholar] [CrossRef]
- Ballan, M.; Bovo, S.; Schiavo, G.; Schiavitto, M.; Negrini, R.; Fontanesi, L. Genomic diversity and signatures of selection in meat and fancy rabbit breeds based on high-density marker data. Genet. Sel. Evol. 2022, 54, 3. [Google Scholar] [CrossRef]
- Ballan, M.; Bovo, S.; Bertolini, F.; Schiavo, G.; Schiavitto, M.; Negrini, R.; Fontanesi, L. Population genomic structures and signatures of selection define the genetic uniqueness of several fancy and meat rabbit breeds. J. Anim. Breed. Genet. 2023, 140, 663–678. [Google Scholar] [CrossRef]
- Sato, D.X.; Rafati, N.; Ring, H.; Younis, S.; Feng, C.; Blanco-Aguiar, J.A.; Rubin, C.-J.; Villafuerte, R.; Hallböök, F.; Carneiro, M.; et al. Brain Transcriptomics of Wild and Domestic Rabbits Suggests That Changes in Dopamine Signaling and Ciliary Function Contributed to Evolution of Tameness. Genome Biol. Evol. 2020, 12, 1918–1928. [Google Scholar] [CrossRef]
- Brusini, I.; Carneiro, M.; Wang, C.; Rubin, C.J.; Ring, H.; Afonso, S.; Blanco-Aguiar, J.A.; Ferrand, N.; Rafati, N.; Villafuerte, R.; et al. Changes in brain architecture are consistent with altered fear processing in domestic rabbits. Proc. Natl. Acad. Sci. USA 2018, 115, 7380–7385. [Google Scholar] [CrossRef]
- Carneiro, M.; Piorno, V.; Rubin, C.-J.; Alves, J.M.; Ferrand, N.; Alves, P.C.; Andersson, L. Candidate genes underlying heritable differences in reproductive seasonality between wild and domestic rabbits. Anim. Genet. 2015, 46, 418–425. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhang, J.; Li, H.; Li, J.; Niimi, M.; Ding, G.; Chen, H.; Xu, J.; Zhang, H.; Xu, Z.; et al. Hyperlipidemia-associated gene variations and expression patterns revealed by whole-genome and transcriptome sequencing of rabbit models. Sci. Rep. 2016, 6, 26942. [Google Scholar] [CrossRef]
- Carneiro, M.; Hu, D.; Archer, J.; Feng, C.; Afonso, S.; Chen, C.; Blanco-Aguiar, J.A.; Garreau, H.; Boucher, S.; Ferreira, P.G.; et al. Dwarfism and Altered Craniofacial Development in Rabbits Is Caused by a 12.1 kb Deletion at the HMGA2 Locus. Genetics 2017, 205, 955–965. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Poplin, R.; Ruano-Rubio, V.; DePristo, M.A.; Fennell, T.J.; Carneiro, M.O.; Van der Auwera, G.A.; Kling, D.E.; Gauthier, L.D.; Levy-Moonshine, A.; Roazen, D.; et al. Scaling accurate genetic variant discovery to tens of thousands of samples. BioRxiv 2018, 201178. [Google Scholar] [CrossRef]
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ data to high confidence variant calls: The Genome Analysis Toolkit best practices pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.11–11.10.33. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Howe, K.L.; Achuthan, P.; Allen, J.; Allen, J.; Alvarez-Jarreta, J.; Amode, M.R.; Armean, I.M.; Azov, A.G.; Bennett, R.; Bhai, J.; et al. Ensembl 2021. Nucleic Acids Res. 2021, 49, D884–D891. [Google Scholar] [CrossRef] [PubMed]
- Lee, T.-H.; Guo, H.; Wang, X.; Kim, C.; Paterson, A.H. SNPhylo: A pipeline to construct a phylogenetic tree from huge SNP data. BMC Genom. 2014, 15, 162. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Patterson, N.; Moorjani, P.; Luo, Y.; Mallick, S.; Rohland, N.; Zhan, Y.; Genschoreck, T.; Webster, T.; Reich, D. Ancient admixture in human history. Genetics 2012, 192, 1065–1093. [Google Scholar] [CrossRef]
- Petr, M.; Vernot, B.; Kelso, J. admixr-R package for reproducible analyses using ADMIXTOOLS. Bioinformatics 2019, 35, 3194–3195. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, M.; Huber, C.D.; Hubisz, M.J.; Hellmann, I.; Nielsen, R. SweepFinder2: Increased sensitivity, robustness and flexibility. Bioinformatics 2016, 32, 1895–1897. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Hinrichs, A.S.; Karolchik, D.; Baertsch, R.; Barber, G.P.; Bejerano, G.; Clawson, H.; Diekhans, M.; Furey, T.S.; Harte, R.A.; Hsu, F.; et al. The UCSC Genome Browser Database: Update 2006. Nucleic Acids Res. 2006, 34, D590–D598. [Google Scholar] [CrossRef]
- Czipa, E.; Schiller, M.; Nagy, T.; Kontra, L.; Steiner, L.; Koller, J.; Pálné-Szén, O.; Barta, E. ChIPSummitDB: A ChIP-seq-based database of human transcription factor binding sites and the topological arrangements of the proteins bound to them. Database 2020, 2020, baz141. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Huang, X.; Ebert, D.; Mills, C.; Guo, X.; Thomas, P.D. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 2019, 14, 703–721. [Google Scholar] [CrossRef]
- Kinsella, R.J.; Kähäri, A.; Haider, S.; Zamora, J.; Proctor, G.; Spudich, G.; Almeida-King, J.; Staines, D.; Derwent, P.; Kerhornou, A.; et al. Ensembl BioMarts: A hub for data retrieval across taxonomic space. Database 2011, 2011, bar030. [Google Scholar] [CrossRef]
- Xie, K.; Ning, C.; Yang, A.; Zhang, Q.; Wang, D.; Fan, X. Resequencing Analyses Revealed Genetic Diversity and Selection Signatures during Rabbit Breeding and Improvement. Genes 2024, 15, 433. [Google Scholar] [CrossRef]
- Andrade, P.; Alves, J.M.; Pereira, P.; Rubin, C.J.; Silva, E.; Sprehn, C.G.; Enbody, E.; Afonso, S.; Faria, R.; Zhang, Y.; et al. Selection against domestication alleles in introduced rabbit populations. Nat. Ecol. Evol. 2024, 8, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- Magalhaes, V.; Abrantes, J.; Munoz-Pajares, A.J.; Esteves, P.J. Genetic diversity comparison of the DQA gene in European rabbit (Oryctolagus cuniculus) populations. Immunogenetics 2015, 67, 579–590. [Google Scholar] [CrossRef]
- Gage, M.J.; Surridge, A.K.; Tomkins, J.L.; Green, E.; Wiskin, L.; Bell, D.J.; Hewitt, G.M. Reduced heterozygosity depresses sperm quality in wild rabbits, Oryctolagus cuniculus. Curr. Biol. 2006, 16, 612–617. [Google Scholar] [CrossRef]
- Ziege, M.; Theodorou, P.; Jungling, H.; Merker, S.; Plath, M.; Streit, B.; Lerp, H. Population genetics of the European rabbit along a rural-to-urban gradient. Sci. Rep. 2020, 10, 2448. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Filtering Steps | Quality Filtering | Missingness and Allele Count | LD Pruning |
|---|---|---|---|
| Samples | 100 | 97 | 97 |
| Variants | 67,084,502 | 27,524,641 | 2,780,336 |
| SNPs | 50,253,306 | 21,863,765 | 1,825,434 |
| Indels | 18,882,395 | 5,660,876 | 954,902 |
| Others (mixed) | 1,352,355 | 0 | 0 |
| Multiallelic sites | 8,344,576 | 0 | 0 |
| Multiallelic SNP sites | 2,506,463 | 0 | 0 |
| Total in Genome | Name | Found in Selected | Fisher_Exact p |
|---|---|---|---|
| 9610 | Atf3 | 11 | 0.029718 |
| 1796 | Bcl6 | 4 | 0.02263 |
| 46,581 | CEBPB | 38 | 0.030488 |
| 5 | ESR1 | 1 | 0.002941 |
| 18,173 | NFIC | 21 | 0.003292 |
| 9848 | Rxra | 11 | 0.034464 |
| 2302 | ZNF143 | 7 | 5.07 × 10−4 |
| Gene ID | Per-Site Fst | Name | |
|---|---|---|---|
| >0.75 | >0.9 | ||
| ENSOCUG00000001839 | 72 | 22 | PPP2R3A |
| ENSOCUG00000001843 | 72 | 22 | MSL2 |
| ENSOCUG00000002636 | 9 | 0 | FHIP1A |
| ENSOCUG00000005429 | 9 | 0 | BCL6 |
| ENSOCUG00000005623 | 39 | 0 | SCN9A |
| ENSOCUG00000005958 | 2 | 1 | ATRNL1 |
| ENSOCUG00000006058 | 1 | 2 | POLR1B |
| ENSOCUG00000006802 | 11 | 0 | LCORL |
| ENSOCUG00000007266 | 3 | 0 | SCN1A |
| ENSOCUG00000008219 | 24 | 5 | POC1B |
| ENSOCUG00000008635 | 36 | 19 | novel |
| ENSOCUG00000010297 | 11 | 0 | SIPA1L2 |
| ENSOCUG00000010372 | 8 | 0 | MAP3K21 |
| ENSOCUG00000011072 | 132 | 61 | CFLAR |
| ENSOCUG00000014363 | 40 | 0 | PCNX2 |
| ENSOCUG00000015875 | 16 | 8 | RFTN1 |
| ENSOCUG00000016507 | 11 | 3 | ATP2B1 |
| ENSOCUG00000016916 | 58 | 38 | CEP85 |
| ENSOCUG00000016927 | 58 | 38 | SH3BGRL3 |
| ENSOCUG00000016938 | 58 | 38 | UBXN11 |
| ENSOCUG00000017729 | 42 | 0 | SCN7A |
| ENSOCUG00000032144 | 97 | 33 | novel, lncRNA |
| ENSOCUG00000034322 | 11 | 5 | novel, lncRNA |
| ENSOCUG00000034812 | 3 | 0 | novel |
| ENSOCUG00000034993 | 97 | 33 | novel, lncRNA |
| ENSOCUG00000038656 | 131 | 40 | novel, lncRNA |
| ENSOCUG00000039398 | 5 | 1 | novel |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fekete, Z.; Német, Z.; Ninausz, N.; Fehér, P.; Schiller, M.; Alnajjar, M.; Szenes, Á.; Nagy, T.; Stéger, V.; Kontra, L.; et al. Whole-Genome Sequencing-Based Population Genetic Analysis of Wild and Domestic Rabbit Breeds. Animals 2025, 15, 775. https://doi.org/10.3390/ani15060775
Fekete Z, Német Z, Ninausz N, Fehér P, Schiller M, Alnajjar M, Szenes Á, Nagy T, Stéger V, Kontra L, et al. Whole-Genome Sequencing-Based Population Genetic Analysis of Wild and Domestic Rabbit Breeds. Animals. 2025; 15(6):775. https://doi.org/10.3390/ani15060775
Chicago/Turabian StyleFekete, Zsófia, Zoltán Német, Nóra Ninausz, Péter Fehér, Mátyás Schiller, Maher Alnajjar, Áron Szenes, Tibor Nagy, Viktor Stéger, Levente Kontra, and et al. 2025. "Whole-Genome Sequencing-Based Population Genetic Analysis of Wild and Domestic Rabbit Breeds" Animals 15, no. 6: 775. https://doi.org/10.3390/ani15060775
APA StyleFekete, Z., Német, Z., Ninausz, N., Fehér, P., Schiller, M., Alnajjar, M., Szenes, Á., Nagy, T., Stéger, V., Kontra, L., & Barta, E. (2025). Whole-Genome Sequencing-Based Population Genetic Analysis of Wild and Domestic Rabbit Breeds. Animals, 15(6), 775. https://doi.org/10.3390/ani15060775